


Atorvastatin Can Modulate DNA Damage Repair in Endothelial Cells Exposed to Mitomycin C
 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
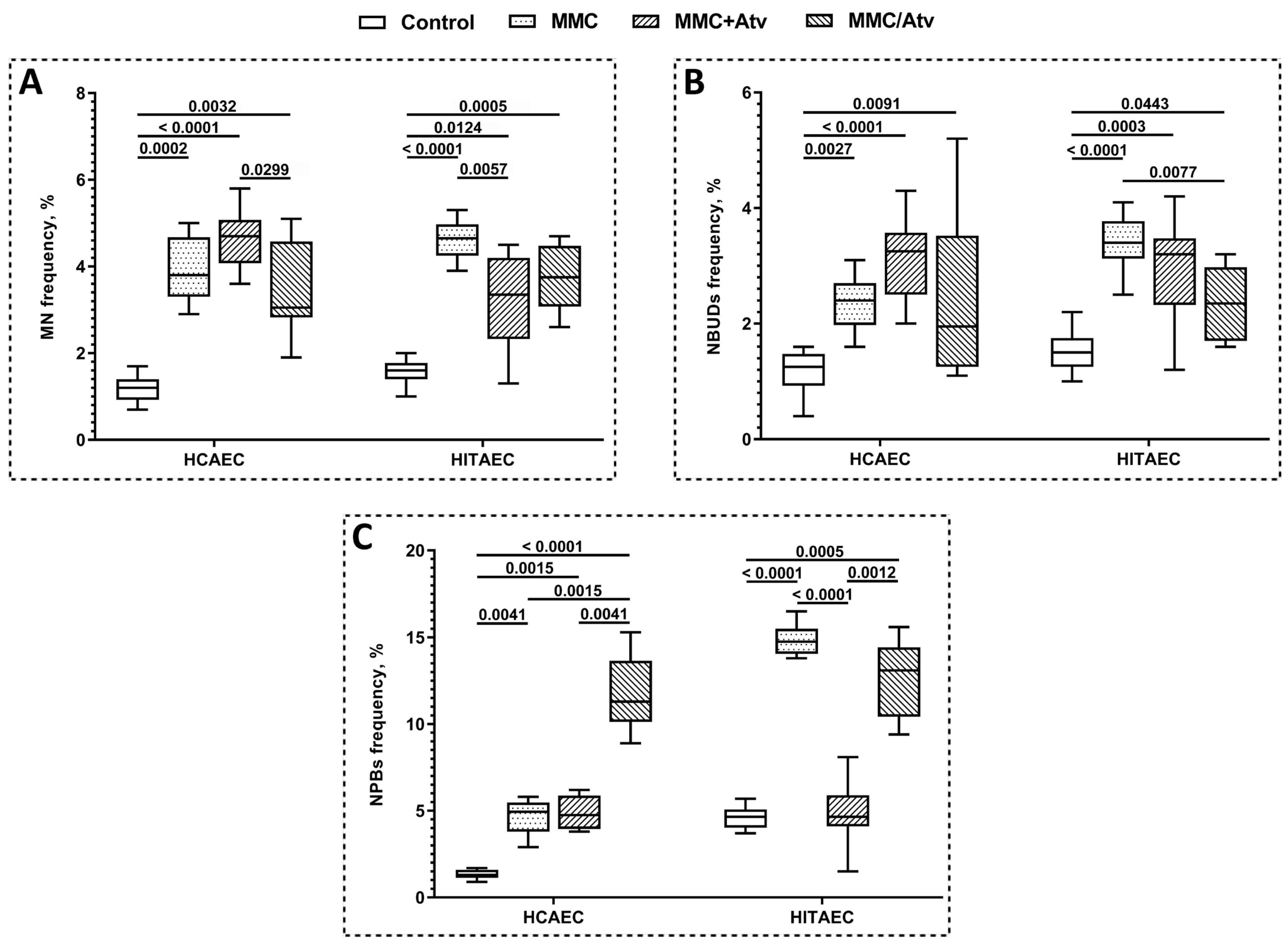
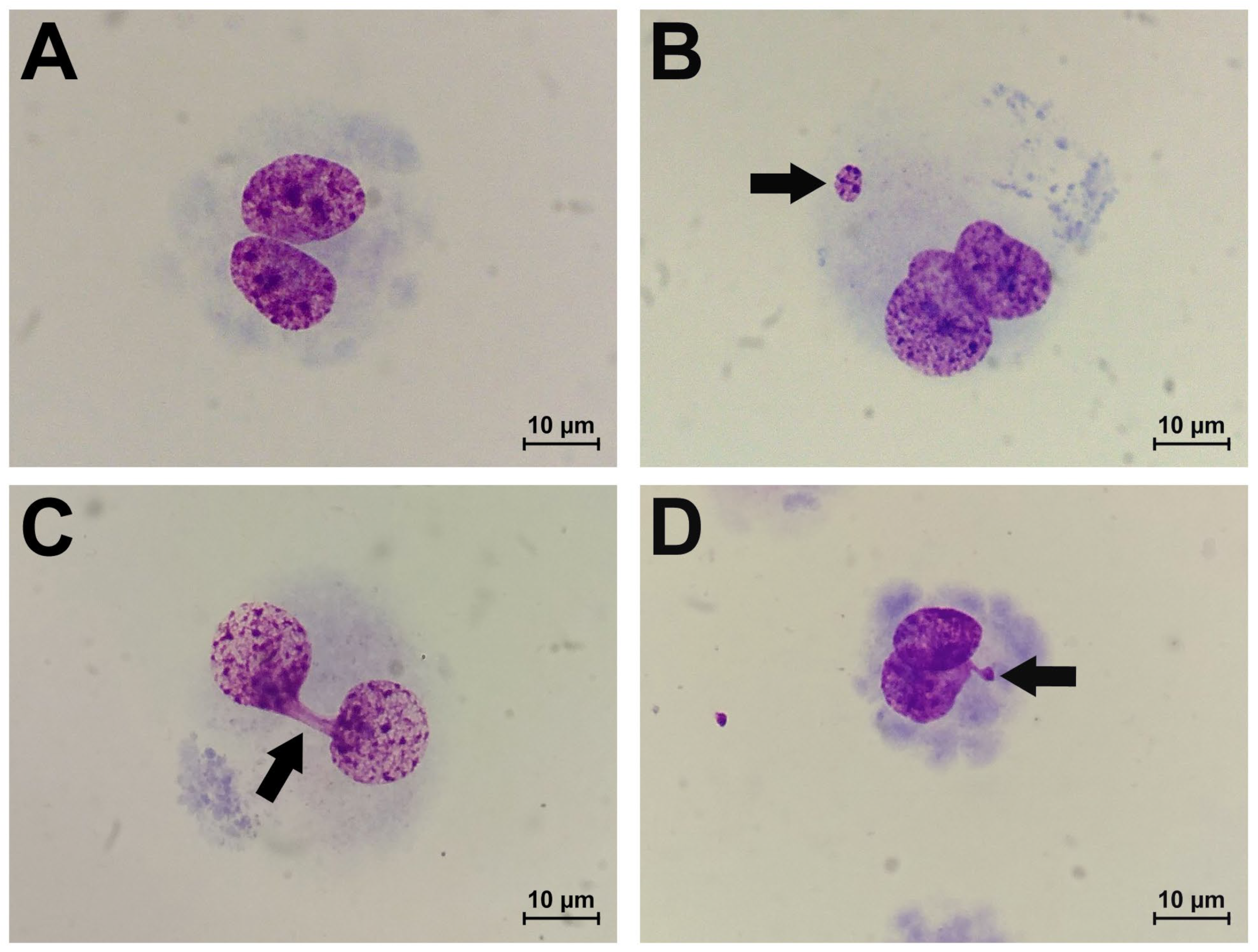
2.1. Results of Cytokinesis-Block Micronucleus Assay
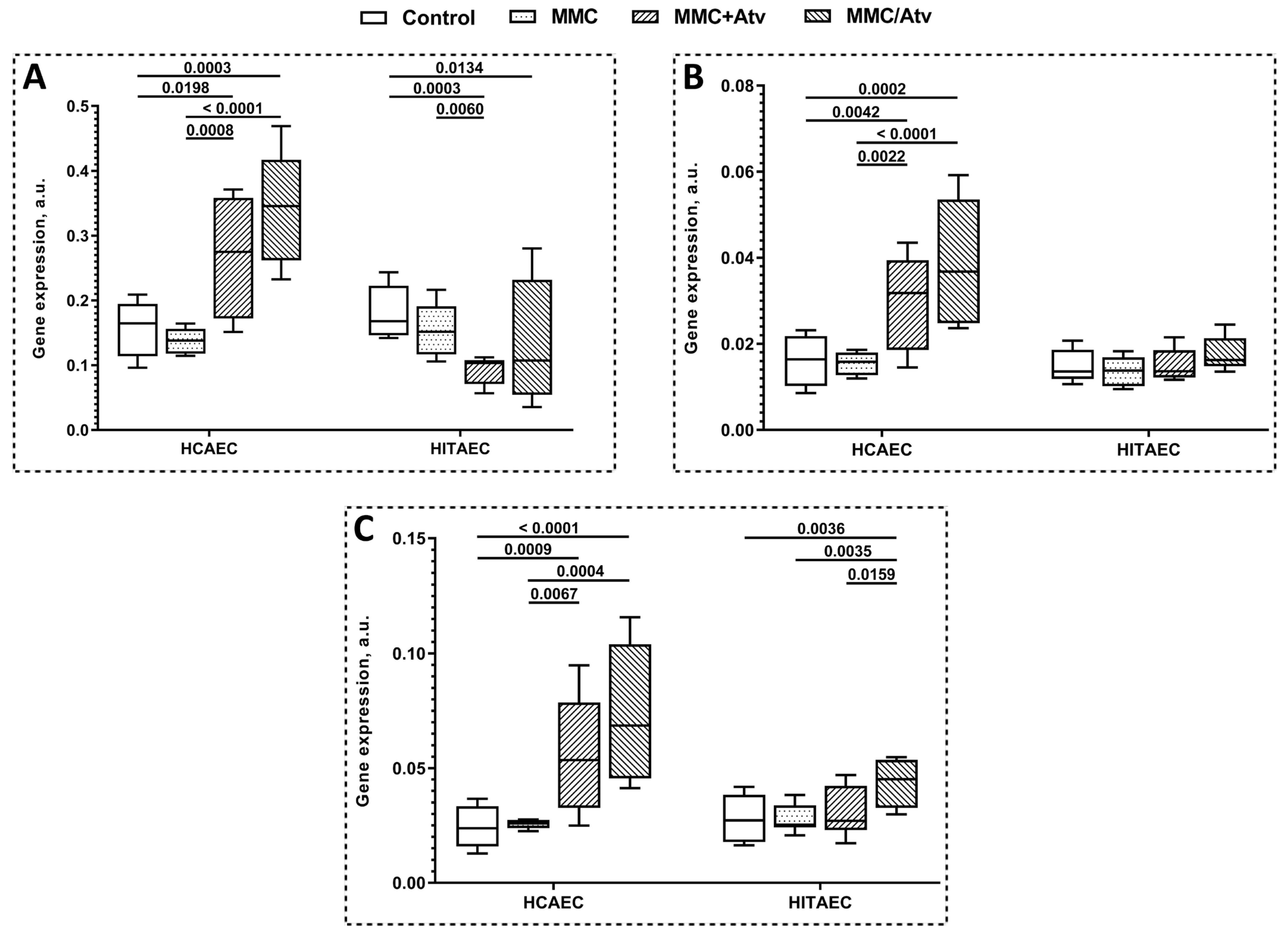
2.2. Results of Gene Expression Analysis
3. Discussion
4. Materials and Methods
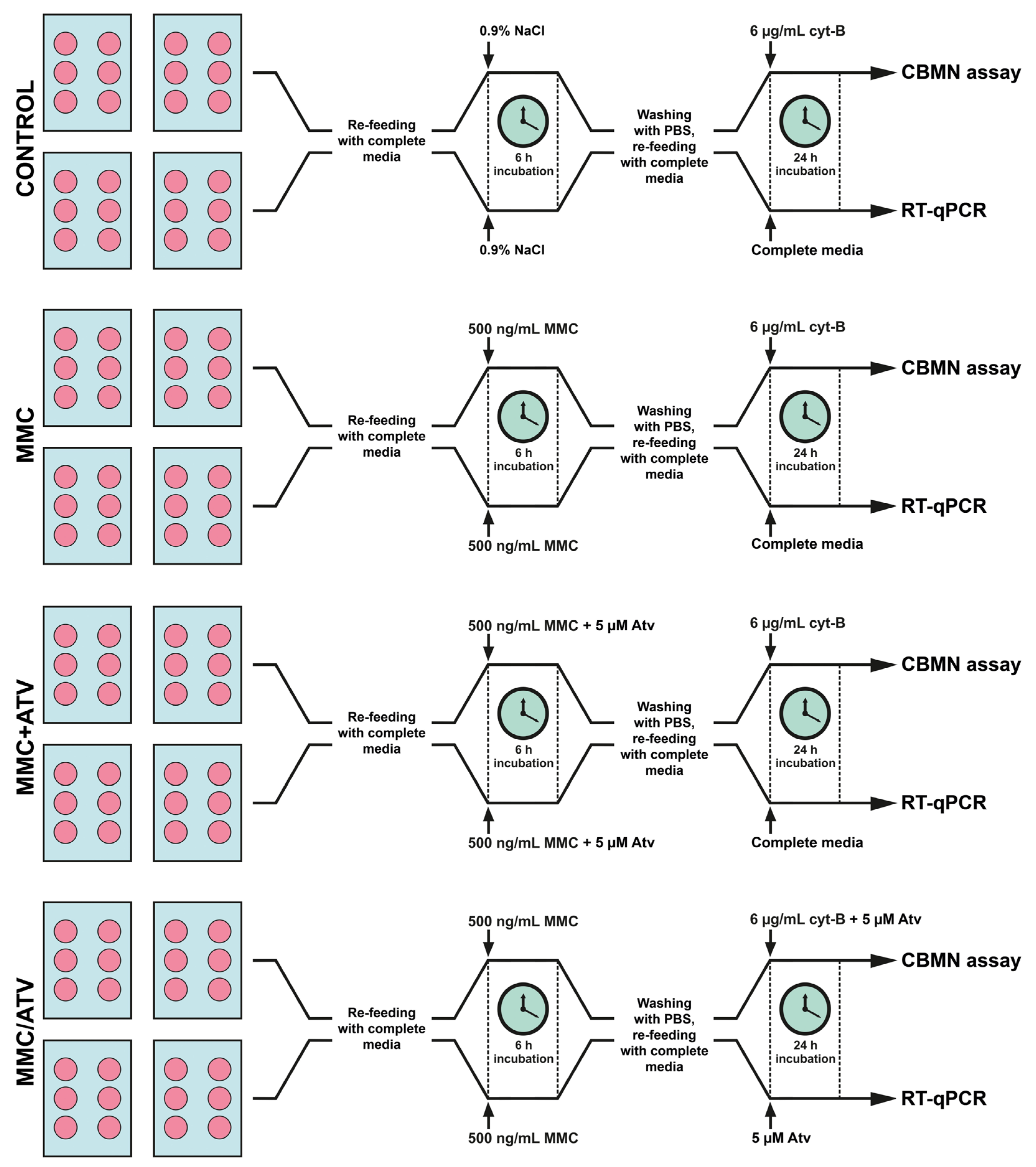
4.1. Cell Cultures and Laboratory Assays
4.1.1. Endothelial Cells Culture
4.1.2. Cytogenetic Analysis
4.1.3. RNA Extraction
4.1.4. Complementary DNA Synthesis
4.1.5. Gene Expression Analysis
4.2. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nakano, E.M.; Bains, H.S.; Hirai, F.E.; Portellinha, W.; Oliveira, M.; Nakano, K. Comparison of laser epithelial keratomileusis with and without mitomycin C for wavefront customized surface ablations. J. Refract. Surg. 2007, 23, S1021–S1028. [Google Scholar] [CrossRef]
- Pakravan, M.; Homayoon, N.; Shahin, Y.; Ali Reza, B.R. Trabeculectomy with mitomycin C versus Ahmed glaucoma implant with mitomycin C for treatment of pediatric aphakic glaucoma. J. Glaucoma 2007, 16, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Kersey, J.P.; Vivian, A.J. Mitomycin and amniotic membrane: A new method of reducing adhesions and fibrosis in strabismus surgery. Strabismus 2008, 16, 116–118. [Google Scholar] [CrossRef]
- Gupta, V.P.; Sanghi, S.; Rohatgi, J.; Dhaliwal, U. Outcomes of preoperative intrapterygial injection of mitomycin C for pterygium excision with and without inferior conjunctival flap. Oman J. Ophthalmol. 2019, 12, 171–176. [Google Scholar] [CrossRef]
- Al-Otaibi, W.A.; Alkhatib, M.H.; Wali, A.N. Cytotoxicity and apoptosis enhancement in breast and cervical cancer cells upon coadministration of mitomycin C and essential oils in nanoemulsion formulations. Biomed. Pharmacother. 2018, 106, 946–955. [Google Scholar] [CrossRef]
- Yurttas, C.; Hoffmann, G.; Tolios, A.; Haen, S.P.; Schwab, M.; Königsrainer, I.; Königsrainer, A.; Beckert, S.; Löffler, M.W. Systematic review of variations in hyperthermic intraperitoneal chemotherapy (HIPEC) for peritoneal metastasis from colorectal cancer. J. Clin. Med. 2018, 7, 567. [Google Scholar] [CrossRef] [PubMed]
- Faraj, K.; Chang, Y.H.; Rose, K.M.; Habermann, E.B.; Etzioni, D.A.; Blodgett, G.; Castle, E.P.; Humphreys, M.R.; Tyson Ii, M.D. Single-dose perioperative mitomycin-C versus thiotepa for low-grade noninvasive bladder cancer. Can. J. Urol. 2019, 26, 9922–9930. [Google Scholar] [PubMed]
- Tung, S.Y.; Lin, C.T.; Chen, C.N.; Huang, W.S. Effect of mitomycin C on X-ray repair cross complementing group 1 expression and consequent cytotoxicity regulation in human gastric cancer cells. J. Cell. Biochem. 2019, 120, 8333–8342. [Google Scholar] [CrossRef]
- Hoey, B.M.; Butler, J.; Swallow, A.J. Reductive activation of mitomycin C. Biochemistry 1988, 27, 2608–2614. [Google Scholar] [CrossRef]
- Paz, M.M. Reductive activation of mitomycin C by thiols: Kinetics, mechanism, and biological implications. Chem. Res. Toxicol. 2009, 22, 1663–1668. [Google Scholar] [CrossRef]
- Rink, S.M.; Lipman, R.; Alley, S.C.; Hopkins, P.B.; Tomasz, M. Bending of DNA by the mitomycin C-induced, GpG intrastrand cross-link. Chem. Res. Toxicol. 1996, 9, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Park, S.J.; Ciccone, S.L.; Kim, C.R.; Lee, S.H. An in vivo analysis of MMC-induced DNA damage and its repair. Carcinogenesis 2006, 27, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, J.L.; Wishnok, J.S.; Tannenbaum, S.R. Nitric oxide-induced interstrand cross-links in DNA. Chem. Res. Toxicol. 2003, 16, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Colis, L.C.; Raychaudhury, P.; Basu, A.K. Mutational specificity of gamma-radiation-induced guanine-thymine and thymine-guanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta. Biochemistry 2008, 74, 8070–8079. [Google Scholar] [CrossRef]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by alpha, beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Davies, K.J.A.; Medeiros, M.H.; Di Mascio, P.; Wagner, J.R. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Kutikhin, A.G.; Tsepokina, A.V.; Shishkova, D.K.; Asanov, M.A.; Yuzhalin, A.E.; Minina, V.I.; Ponasenko, A.V. Mitomycin C induced genotoxic stress in endothelial cells is associated with differential expression of proinflammatory cytokines. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 858, 503252. [Google Scholar] [CrossRef] [PubMed]
- Sinitsky, M.Y.; Tsepokina, A.V.; Kutikhin, A.G.; Shishkova, D.K.; Ponasenko, A.V. The gene expression profile in endothelial cells exposed to mitomycin C. Biochem. Suppl. Ser. B Biomed. Chem. 2021, 15, 255–261. [Google Scholar] [CrossRef]
- Sinitsky, M.; Sinitskaya, A.; Shishkova, D.; Tupikin, A.; Asanov, M.; Khutornaya, M.; Kabilov, M.; Ponasenko, A. Identification of Key Genes and Pathways in Genotoxic Stress Induced Endothelial Dysfunction: Results of Whole Transcriptome Sequencing. Biomedicines 2022, 10, 2067. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Sinitsky, M.Y.; Ponasenko, A.V. The role of mutagenesis in atherosclerosis. Complex Issues Cardiovasc. Dis. 2017, 6, 92–101. [Google Scholar] [CrossRef]
- Bertani, F.; Di Francesco, D.; Corrado, M.D.; Talmon, M.; Fresu, L.G.; Boccafoschi, F. Paracrine Shear-Stress-Dependent Signaling from Endothelial Cells Affects Downstream Endothelial Function and Inflammation. Int. J. Mol. Sci. 2021, 22, 13300. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.L.; Mountjoy-Venning, W.C.; Anjomshoa, M.; Banoub, J.A.; Yasin, Y.J. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Koh, K.K. Effects of statins on vascular wall: Vasomotor function, inflammation, and plaque stability. Cardiovasc. Res. 2000, 47, 648–657. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Casey, P.J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef]
- McTaggart, S.J. Isoprenylated proteins. Cell. Mol. Life Sci. 2006, 63, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liu, P.J.; Liao, J.K. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef]
- Fritz, G.; Kaina, B. Rho GTPases: Promising cellular targets for novel anticancer drugs. Curr. Cancer Drug Targets 2006, 6, 1–14. [Google Scholar] [PubMed]
- Fritz, G.; Henninger, C.; Huelsenbeck, J. Potential use of HMG-CoA reductase inhibitors (statins) as radioprotective agents. Br. Med. Bull. 2011, 97, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, V.; Albers, A.; Fritz, G. Lovastatin protects keratinocytes from DNA damage-related pro-apoptotic stress responses stimulated by anticancer therapeutics. Biochim. Biophys. Acta 2016, 1863, 1082–1092. [Google Scholar] [CrossRef]
- Efimova, E.V.; Ricco, N.; Labay, E.; Mauceri, H.J.; Flor, A.C.; Ramamurthy, A.; Sutton, H.G.; Weichselbaum, R.R.; Kron, S.J. HMG-CoA Reductase Inhibition Delays DNA Repair and Promotes Senescence After Tumor Irradiation. Mol. Cancer Ther. 2018, 17, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulakos, J.; Ye, L.Y.; Benzaquen, M.; Moore, M.J.; Kamel-Reid, S.; Freedman, M.H.; Yeger, H.; Penn, L.Z. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: Therapeutic implications. Clin. Cancer Res. 2001, 7, 158–167. [Google Scholar]
- Graaf, M.R.; Richel, D.J.; van Noorden, C.J.; Guchelaar, H.J. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat. Rev. 2004, 30, 609–641. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Liu, C.; Rashid, A.; Hopmans, S.N.; Tsiani, E.; Schultz, C.; Farrell, T.; Singh, G.; Wright, J.; Tsakiridis, T. Lovastatin sensitizes lung cancer cells to ionizing radiation: Modulation of molecular pathways of radioresistance and tumor suppression. J. Thorac. Oncol. 2011, 6, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Zheng, L.; Lee, K.J.; Kim, D.H.; Kim, C.S.; Cai, D.Q.; Wu, Z.; Qin, J.W.; Yu, Y.H.; Kim, S.K. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell. Death Dis. 2013, 4, e518. [Google Scholar] [CrossRef]
- Fritz, G. Targeting the mevalonate pathway for improved anticancer therapy. Curr. Cancer Drug Targets 2009, 9, 626–638. [Google Scholar] [CrossRef]
- Osmak, M. Statins and cancer: Current and future prospects. Cancer Lett. 2012, 324, 1–12. [Google Scholar] [CrossRef]
- Jani, J.P.; Specht, S.; Stemmler, N.; Blanock, K.; Singh, S.V.; Gupta, V.; Katoh, A. Metastasis of B16F10 mouse melanoma inhibited by lovastatin, an inhibitor of cholesterol biosynthesis. Invasion Metastasis 1993, 13, 314–324. [Google Scholar]
- Hamalukic, M.; Huelsenbeck, J.; Schad, A.; Wirtz, S.; Kaina, B.; Fritz, G. Rac1-regulated endothelial radiation response stimulates extravasation and metastasis that can be blocked by HMG-CoA reductase inhibitors. PLoS ONE 2011, 6, e26413. [Google Scholar] [CrossRef]
- Gnad, R.; Aktories, K.; Kaina, B.; Fritz, G. Inhibition of protein isoprenylation impairs rho-regulated early cellular response to genotoxic stress. Mol. Pharmacol. 2000, 58, 1389–1397. [Google Scholar] [CrossRef]
- Gnad, R.; Kaina, B.; Fritz, G. Rho GTPases are involved in the regulation of NF-kB by genotoxic stress. Exp. Cell Res. 2001, 264, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Nübel, T.; Dippold, W.; Kaina, B.; Fritz, G. Ionizing radiation-induced E-selectin gene expression and tumor cell adhesion is inhibited by lovastatin and all-trans retinoic acid. Carcinogenesis 2004, 25, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Damrot, J.; Nübel, T.; Epe, B.; Roos, W.P.; Kaina, B.; Fritz, G. Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. Br. J. Pharmacol. 2006, 149, 988–997. [Google Scholar] [CrossRef]
- Ran, X.Z.; Ran, X.; Zong, Z.W.; Liu, D.Q.; Xiang, G.M.; Su, Y.P.; Zheng, H.E. Protective effect of atorvastatin on radiation-induced vascular endothelial cell injury in vitro. J. Radiat. Res. 2010, 51, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Huelsenbeck, J.; Fritz, G. Mevalonate pathway inhibitors affect anticancer drug-induced cell death and DNA damage response of human sarcoma cells. Cancer Lett. 2011, 304, 60–69. [Google Scholar] [CrossRef]
- Bourgier, C.; Haydont, V.; Milliat, F.; François, A.; Holler, V.; Lasser, P.; Bourhis, J.; Mathé, D.; Vozenin-Brotons, M.C. Inhibition of Rho kinase modulates radiation induced fibrogenic phenotype in intestinal smoothmuscle cells through alteration of the cytoskeleton and connective tissue growth factor expression. Gut 2005, 54, 336–343. [Google Scholar] [CrossRef]
- Haydont, V.; Bourgier, C.; Pocard, M.; Lusinchi, A.; Aigueperse, J.; Mathé, D.; Bourhis, J.; Vozenin-Brotons, M.C. Pravastatin inhibits the Rho/CCN2/extracellular matrix cascade in human fibrosis explants and improves radiation-induced intestinal fibrosis in rats. Clin. Cancer Res. 2007, 13, 5331–5340. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Gorenne, I.; Mercer, J.; Figg, N.; Littlewood, T.; Bennett, M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ. Res. 2008, 103, 717–725. [Google Scholar] [CrossRef]
- Sultan, S.; D’Souza, A.; Zabetakis, I.; Lordan, R.; Tsoupras, A.; Kavanagh, E.P.; Hynes, N. Chapter 6—Statins: Rationale, Mode of Action, and Side Effects. In The Impact of Nutrition and Statins on Cardiovascular Diseases; Zabetakis, I., Lordan, R., Tsoupras, A., Eds.; Academic Press: San Diego, CA, USA, 2019; Volume 3, pp. 171–200. [Google Scholar]
- Patel, K.K.; Sehgal, V.S.; Kashfi, K. Molecular targets of statins and their potential side effects: Not all the glitter is gold. Eur. J. Pharmacol. 2022, 922, 174906. [Google Scholar] [CrossRef]
- Razavi, A.C.; Mehta, A.; Sperling, L.S. Statin therapy for the primary prevention of cardiovascular disease: Pros. Atherosclerosis 2022, 356, 41–45. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e563–e595. [Google Scholar] [CrossRef]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef]
- Vaughan, C.J.; Gotto, A.M.; Basson, C.T. The evolving role of statins in the management of atherosclerosis. J. Am. Coll. Cardiol. 2000, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Zieman, S.; Go, A.S.; Fortmann, S.P.; Wenger, N.K.; Fleg, J.L.; Radziszewska, B.; Stone, N.J.; Zoungas, S.; Gurwitz, J.H. Statins for Primary Prevention in Older Adults-Moving Toward Evidence-Based Decision-Making. J. Am. Geriatr. Soc. 2018, 66, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ikeda, U.; Shimpo, M.; Shimada, K. Direct effects of statins on cells primarily involved in atherosclerosis. Hypertens. Res. 2000, 23, 187–192. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Bellosta, S.; Ferri, N.; Bernini, F.; Paoletti, R.; Corsini, A. Non-lipid-related effects of statins. Ann. Med. 2000, 32, 164–176. [Google Scholar] [CrossRef]
- Ikeda, U.; Shimada, K. Pleiotropic effects of statins on the vascular tissue. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2001, 1, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Dulak, J.; Loboda, A.; Jazwa, A.; Zagorska, A.; Dörler, J.; Alber, H.; Dichtl, W.; Weidinger, F.; Frick, M.; Jozkowicz, A. Atorvastatin affects several angiogenic mediators in human endothelial cells. Endothelium 2005, 12, 233–241. [Google Scholar] [CrossRef]
- Douglas, G.; Channon, K.M. The pathogenesis of atherosclerosis. Medicine 2014, 42, 480–484. [Google Scholar] [CrossRef]
- Jose, M.A.; Anandkumar, S.; Narmadha, M.P.; Sandeep, M. Comparative effect of atorvastatin with other statins in patients of hyperlipidemia. Indian J. Pharmacol. 2012, 44, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Shaghaghi, Z.; Alvandi, M.; Farzipour, S.; Dehbanpour, M.R.; Nosrati, S. A review of effects of atorvastatin in cancer therapy. Med. Oncol. 2022, 40, 27. [Google Scholar] [CrossRef] [PubMed]
- Cilla, D.D., Jr.; Whitfield, L.R.; Gibson, D.M.; Sedman, A.J.; Posvar, E.L. Multiple-dose pharmacokinetics, pharmacodynamics and safety of atorvastatin, an inhibitor of HMG-CoA reductase in healthy subjects. Clin. Pharmacol. Ther. 1996, 60, 687–695. [Google Scholar] [CrossRef]
- Tsioufis, K.; Castellano Vázquez, J.M.; Sykara, G.; Mondello Malvestiti, F.; van Vugt, J. Real-world Evidence for Adherence and Persistence with Atorvastatin Therapy. Cardiol. Ther. 2021, 10, 445–464. [Google Scholar] [CrossRef]
- Harangi, M.; Seres, I.; Varga, Z.; Emri, G.; Szilvássy, Z.; Paragh, G.; Remenyik, E. Atorvastatin effect on high-density lipoprotein-associated paraoxonase activity and oxidative DNA damage. Eur. J. Clin. Pharmacol. 2004, 60, 685–691. [Google Scholar] [CrossRef]
- Gundapaneni, K.K.; Shyamala, N.; Galimudi, R.K.; Sahu, S.K.; Hanumanth, S.R. Therapeutic Effects of Atorvastatin on Genetic Damage in Coronary Artery Disease. J. Clin. Diagn. Res. 2016, 10, 28–30. [Google Scholar] [CrossRef]
- Donmez-Altuntas, H.; Bayram, F.; Coskun-Demirkalp, A.N.; Baspınar, O.; Kocer, D.; Toth, P.P. Therapeutic effects of statins on chromosomal DNA damage of dyslipidemic patients. Exp. Biol. Med. 2019, 244, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Sims, F.H. A comparison of coronary and internal mammary arteries and implications of the results in the etiology of atherosclerosis. Am. Heart J. 1983, 105, 560–566. [Google Scholar] [CrossRef]
- Aboyans, V.; Lacroix, P.; Criqui, M.H. Large and small vessels atherosclerosis: Similarities and differences. Prog. Cardiovasc. Dis. 2007, 50, 112–125. [Google Scholar] [CrossRef]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Gajski, G.; Garaj-Vrhovac, V.; Orescanin, V. Cytogenetic status and oxidative DNA-damage induced by atorvastatin in human peripheral blood lymphocytes: Standard and Fpg-modified comet assay. Toxicol. Appl. Pharmacol. 2008, 231, 85–93. [Google Scholar] [CrossRef]
- Dessy, C.; Ferron, O. Pathophysiological roles of nitric oxide: In the heart and the coronary vasculature, Antiinflamm. Antiallergy Agents Med. Chem. 2004, 3, 207–216. [Google Scholar] [CrossRef]
- Berbee, M.; Fu, Q.; Boerma, M.; Pathak, R.; Zhou, D.; Kumar, K.S.; Hauer-Jensen, M. Reduction of radiation-induced vascular nitrosative stress by the vitamin E analog γ-tocotrienol: Evidence of a role for tetrahydrobiopterin. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Berbee, M.; Fu, Q.; Boerma, M.; Sree Kumar, K.; Loose, D.S.; Hauer-Jensen, M. Mechanisms underlying the radioprotective properties of γ-tocotrienol: Comparative gene expression profiling in tocol-treated endothelial cells. Genes Nutr. 2012, 7, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Haaf, T.; Raderschall, E.; Reddy, G.; Ward, D.C.; Radding, C.M.; Golub, E.I. Sequestration of mammalian Rad51-recombination protein into micronuclei. J. Cell Biol. 1999, 144, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. The in vitro micronucleus technique. Mutat. Res. 2000, 455, 81–95. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.V.; Stancu, C.S.; Simionescu, M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009, 35, 191–203. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indicator | HCAEC | HITAEC | ||||
|---|---|---|---|---|---|---|
| MMC+Atv | MMC/Atv | Positive Control | MMC+Atv | MMC/Atv | Positive Control | |
| MN frequency, % | 4.70 ± 0.80 | 3.05 ± 1.40 | 3.80 ± 1.35 | 3.35 ± 1.75 | 3.75 ± 1.30 | 4.65 ± 6.50 |
| NBUDs frequency, % | 3.25 ± 1.05 | 1.95 ± 2.05 | 2.40 ± 1.65 | 3.20 ± 1.10 | 2.35 ± 1.15 | 3.40 ± 0.60 |
| NPBs frequency, % | 4.75 ± 1.85 | 11.3 ± 3.25 | 4.95 ± 1.65 | 1.65 ± 1.85 | 13.1 ± 3.60 | 14.75 ± 1.20 |
| DDB1 expression, a.u. | 0.275 ± 0.018 | 0.346 ± 0.012 | 0.138 ± 0.033 | 0.103 ± 0.003 | 0.107 ± 0.013 | 0.152 ± 0.063 |
| ERCC4 expression, a.u. | 0.032 ± 0.002 | 0.037 ± 0.003 | 0.016 ± 0.005 | 0.014 ± 0.001 | 0.016 ± 0.001 | 0.014 ± 0.006 |
| ERCC5 expression, a.u. | 0.053 ± 0.003 | 0.069 ± 0.006 | 0.026 ± 0.003 | 0.027 ± 0.002 | 0.045 ± 0.002 | 0.025 ± 0.007 |
| Gene | Assay ID | Reporter/Quencher | Assay Design | Amplicon Length |
|---|---|---|---|---|
| HPRT1 | Hs02800695_m1 | FAM/MGB-NFQ | Probe spans exons | 82 |
| GAPDH | Hs02758991_g1 | FAM/MGB-NFQ | Probe spans exons | 93 |
| B2M | Hs00187842_m1 | FAM/MGB-NFQ | Probe spans exons | 64 |
| DDB1 | Hs01096550_m1 | FAM/MGB-NFQ | Probe spans exons | 75 |
| ERCC4 | Hs01063530_m1 | FAM/MGB-NFQ | Probe spans exons | 64 |
| ERCC5 | Hs01557031_m1 | FAM/MGB-NFQ | Probe spans exons | 71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinitsky, M.; Asanov, M.; Sinitskaya, A.; Shishkova, D.; Khutornaya, M.; Minina, V.; Ponasenko, A. Atorvastatin Can Modulate DNA Damage Repair in Endothelial Cells Exposed to Mitomycin C. Int. J. Mol. Sci. 2023, 24, 6783. https://doi.org/10.3390/ijms24076783
Sinitsky M, Asanov M, Sinitskaya A, Shishkova D, Khutornaya M, Minina V, Ponasenko A. Atorvastatin Can Modulate DNA Damage Repair in Endothelial Cells Exposed to Mitomycin C. International Journal of Molecular Sciences. 2023; 24(7):6783. https://doi.org/10.3390/ijms24076783
Chicago/Turabian StyleSinitsky, Maxim, Maxim Asanov, Anna Sinitskaya, Daria Shishkova, Maria Khutornaya, Varvara Minina, and Anastasia Ponasenko. 2023. "Atorvastatin Can Modulate DNA Damage Repair in Endothelial Cells Exposed to Mitomycin C" International Journal of Molecular Sciences 24, no. 7: 6783. https://doi.org/10.3390/ijms24076783
APA StyleSinitsky, M., Asanov, M., Sinitskaya, A., Shishkova, D., Khutornaya, M., Minina, V., & Ponasenko, A. (2023). Atorvastatin Can Modulate DNA Damage Repair in Endothelial Cells Exposed to Mitomycin C. International Journal of Molecular Sciences, 24(7), 6783. https://doi.org/10.3390/ijms24076783