
Regulation of Cardiac Cav1.2 Channels by Calmodulin
 , , , and
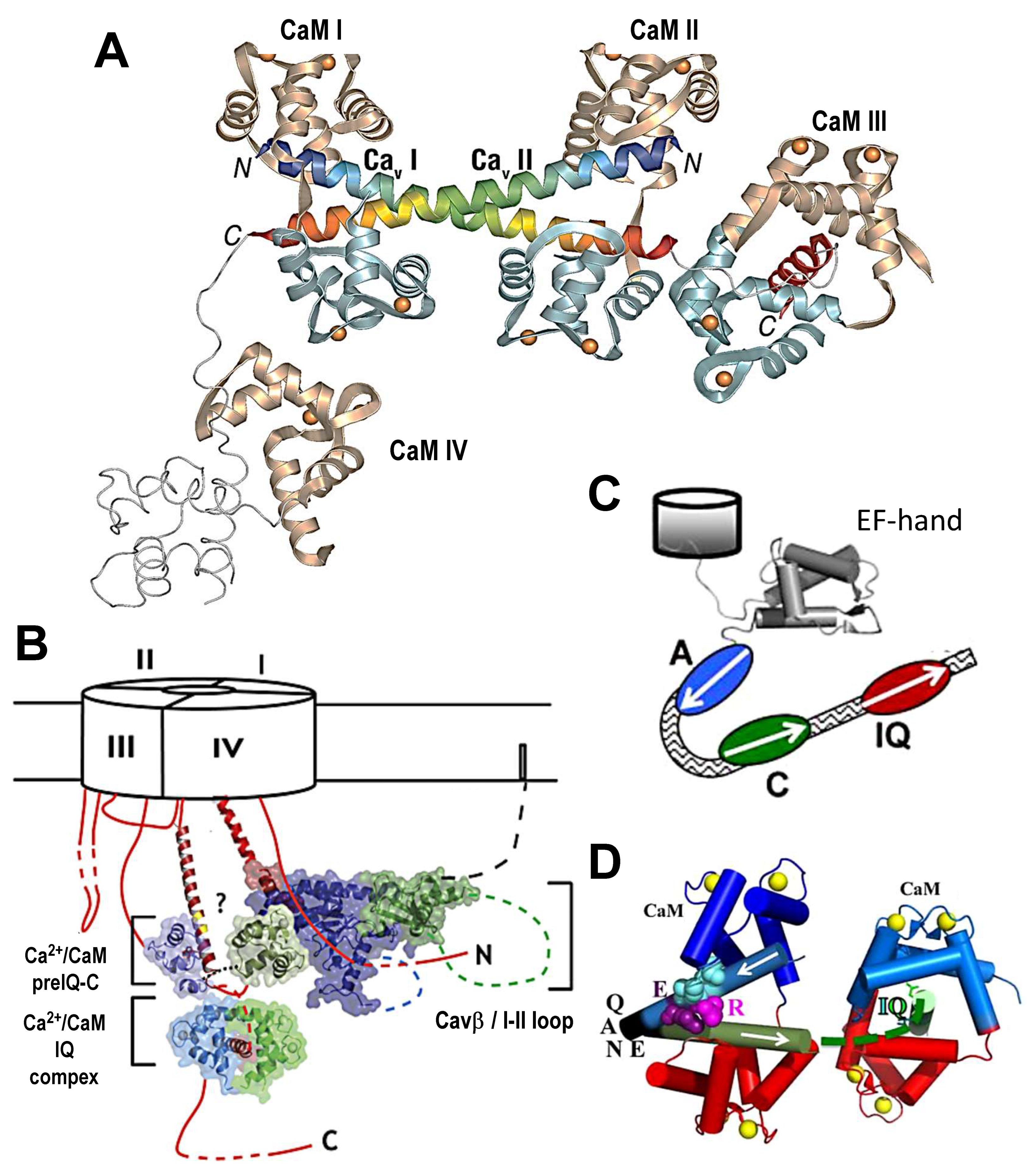
, , , and 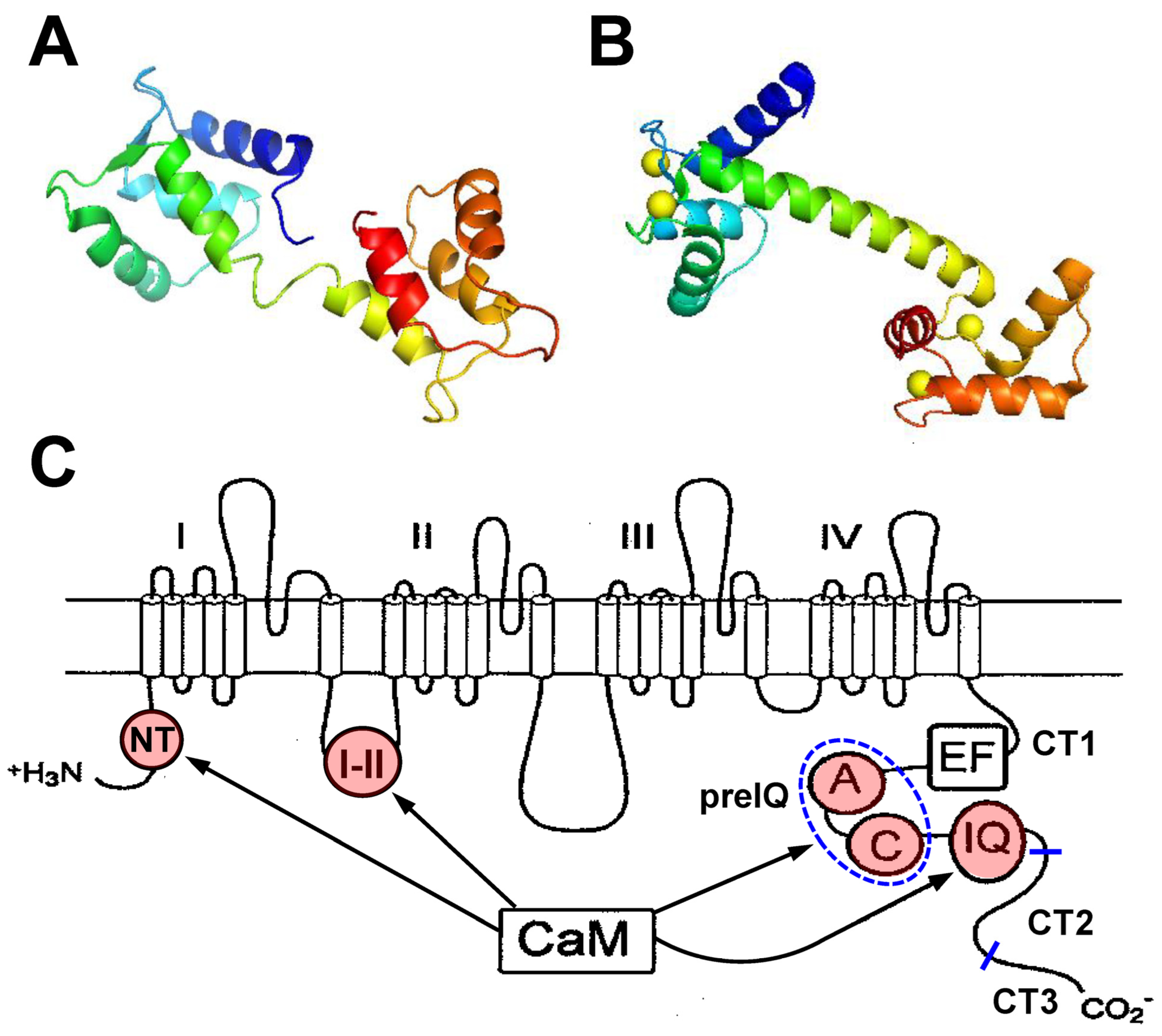{kind=link}
{kind=link}
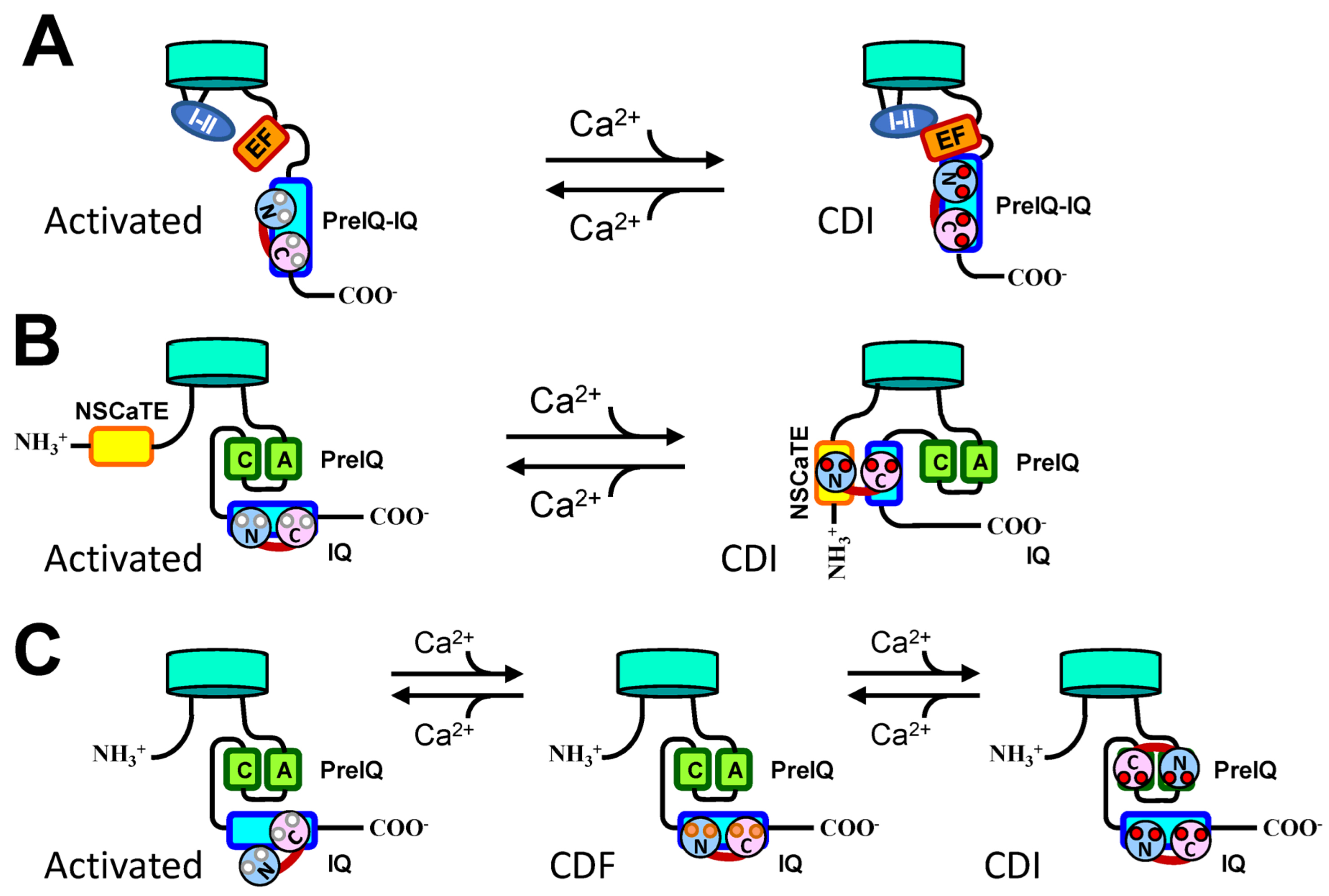{kind=link}
{kind=link}
Abstract
1. Introduction
2. Functional Studies
2.1. Rundown of Channels
2.1.1. History
2.1.2. How to Prevent the Rundown
2.2. Repriming of Channels
2.2.1. CaM Is Crucial for Channel Activity

2.2.2. ATP Is Another Crucial Factor
2.3. Slow Rundown of Channels
Contribution of CaMKII
2.4. Ca2+-Dependent Facilitation (CDF)
2.4.1. Overview of CDF
2.4.2. CaMKII Mediated CDF
2.4.3. CaMKII-Independent CDF
2.4.4. Biphasic Effects of CaM
2.4.5. Mutations That Affect CDF
2.4.6. Physiological Significance of CDF
2.5. Ca2+-Dependent Inactivation (CDI)
2.5.1. Overview of CDI
2.5.2. Dephosphorylation vs. Ca2+/CaM
2.5.3. Mutations That Affect CDI
2.5.4. Interactions of Domains within C-Terminal Tail of Cav1.2
2.5.5. C-Terminal Modulator of Cav1.3 and Cav1.4
2.5.6. Lobe Specific Effects of CaM
2.5.7. CaM-Dependent Inactivation
2.5.8. Additional Ca2+-Sensing Site
2.5.9. CaBP1-Mediated Regulation
3. Molecular Biological Studies
3.1. Ca2+/CaM Binding Site
3.2. ApoCaM Pre-Association Site
3.3. Models of the Conformation of CDF and CDI
3.4. Downstream Mechanism of CDI
3.5. CaMKII Phosphorylation Sites
3.6. Two CaM Hypothesis
3.7. ATP-Binding Site (s)
4. Structural Studies
5. Channel Clustering/Multimerization
6. Trafficking of Cav1.2 Channels
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kakiuchi, S.; Yamazaki, R. Calcium Dependent Phosphodiesterase Activity and Its Activating Factor (PAF) from Brain Studies on Cyclic 3’,5’-Nucleotide Phosphodiesterase (III). Biochem. Biophys. Res. Commun. 1970, 41, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Halling, D.B.; Liebeskind, B.J.; Hall, A.W.; Aldrich, R.W. Conserved Properties of Individual Ca2+-Binding Sites in Calmodulin. Proc. Natl. Acad. Sci. USA 2016, 113, E1216–E1225. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Ikura, M. Calmodulin in Action: Diversity in Target Recognition and Activation Mechanisms. Cell 2002, 108, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a Protein Linker and a Regulator of Adaptor/Scaffold Proteins. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.S.; Sack, J.S.; Greenhough, T.J.; Bugg, C.E.; Means, A.R.; Cook, W.J. Three-Dimensional Structure of Calmodulin. Nature 1985, 315, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FEBS J. 1997, 11, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-Type CaV1.2 Calcium Channels: From in Vitro Findings to in Vivo Function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef]
- Kovalevskaya, N.V.; Van De Waterbeemd, M.; Bokhovchuk, F.M.; Bate, N.; Bindels, R.J.M.; Hoenderop, J.G.J.; Vuister, G.W. Structural Analysis of Calmodulin Binding to Ion Channels Demonstrates the Role of Its Plasticity in Regulation. Pflugers Arch. 2013, 465, 1507–1519. [Google Scholar] [CrossRef]
- Sorensen, A.B.; Søndergaard, M.T.; Overgaard, M.T. Calmodulin in a Heartbeat. FEBS J. 2013, 280, 5511–5532. [Google Scholar] [CrossRef]
- Ben-Johny, M.; Yue, D.T. Calmodulin Regulation (Calmodulation) of Voltage-Gated Calcium Channels. J. Gen. Physiol. 2014, 143, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Pinggera, A.; Kaur, G.; Bock, G.; Tuluc, P. L-Type Ca2+ Channels in Heart and Brain. Wiley Interdiscip. Rev. Membr. Transp. Signal 2014, 3, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-Gated Calcium Channels and Their Auxiliary Subunits: Physiology and Pathophysiology and Pharmacology. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.B. L-Type Ca2+ Channel Regulation by Calmodulin and CaBP1. Biomolecules 2021, 11, 1811. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Byerly, L. The Calcium Channel. Trends Neurosci. 1983, 6, 189–193. [Google Scholar] [CrossRef]
- Kostyuk, P.G. Intracellular Perfusion of Nerve Cells and Its Effects on Membrane Currents. Physiol. Rev. 1984, 64, 435–454. [Google Scholar] [CrossRef]
- Mcdonald, T.F.; Pelzer, S.; Trautwein, W.; Pelzer, D.J. Regulation and Modulation of Calcium Channels in Cardiac, Skeletal, and Smooth Muscle Cells. Physiol. Rev. 1994, 74, 365–507. [Google Scholar] [CrossRef]
- Fedulova, S.A.; Kostyuk, P.G.; Veselovsky, N.S. Calcium Channels in the Somatic Membrane of the Rat Dorsal Root Ganglion Neurons, Effect of CAMP. Brain Res. 1981, 214, 210–214. [Google Scholar] [CrossRef]
- Chad, J.E.; Eckert, R. An Enzymatic Mechanism for Calcium Current Inactivation in Dialysed Helix Neurones. J. Physiol. 1986, 378, 31–51. [Google Scholar] [CrossRef]
- Armstrong, D.; Eckert, R. Voltage-Activated Calcium Channels That Must Be Phosphorylated to Respond to Membrane Depolarization. Proc. Natl. Acad. Sci. USA 1987, 84, 2518–2522. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Fozzard, H.A. Phosphorylation Restores Activity of L-Type Calcium Channels after Rundown in inside-out Patches from Rabbit Cardiac Cells. J. Physiol. 1992, 454, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Irisawa, H.; Kokubun, S. Modulation by Intracellular ATP and Cyclic AMP of the Slow Inward Current in Isolated Single Ventricular Cells of the Guinea-Pig. J. Physiol. 1983, 338, 321–337. [Google Scholar] [CrossRef]
- Kameyama, M.; Kameyama, A.; Takano, E.; Maki, M. Run-down of the Cardiac L-Type Ca2+ Channel: Partial Restoration of Channel Activity in Cell-Free Patches by Calpastatin. Pflugers Arch. 1998, 435, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, M.; Kameyama, A.; Nakayama, T.; Kaibara, M. Tissue Extract Recovers Cardiac Calcium Channels from “Run-Down”. Pflugers Arch. 1988, 412, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, A.; Yazawa, K.; Kaibara, M.; Ozono, K.; Kameyama, M. Run-down of the Cardiac Ca2+ Channel: Characterization and Restoration of Channel Activity by Cytoplasmic Factors. Pflugers Arch. 1997, 433, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Romanin, C.; Grösswagen, P.; Schindler, H. Calpastatin and Nucleotides Stabilize Cardiac Calcium Channel Activity in Excised Patches. Pflugers Arch. 1991, 418, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.Y.; Kameyama, A.; Kameyama, M. A Cytoplasmic Factor, Calpastatin and ATP Together Reverse Run-down of Ca2+ Channel Activity in Guinea-Pig Heart. J. Physiol. 1999, 514 Pt 3, 687–699. [Google Scholar] [CrossRef]
- Belles, B.; Hescheler, J.; Trautwein, W.; Blomgren, K.; Karlsson, J.O. A Possible Physiological Role of the Ca-Dependent Protease Calpain and Its Inhibitor Calpastatin on the Ca Current in Guinea Pig Myocytes. Pflugers Arch. 1988, 412, 554–556. [Google Scholar] [CrossRef]
- Horn, R.; Marty, A. Muscarinic Activation of Ionic Currents Measured by a New Whole-Cell Recording Method. J. Gen Physiol. 1988, 92, 145–159. [Google Scholar] [CrossRef]
- Hao, L.Y.; Kameyama, A.; Kuroki, S.; Takano, J.; Takano, E.; Maki, M.; Kameyama, M. Calpastatin Domain L Is Involved in the Regulation of L-Type Ca2+ Channels in Guinea Pig Cardiac Myocytes. Biochem. Biophys. Res. Commun. 2000, 279, 756–761. [Google Scholar] [CrossRef]
- Minobe, E.; Hao, L.Y.; Saud, Z.A.; Xu, J.J.; Kameyama, A.; Maki, M.; Jewell, K.K.; Parr, T.; Bardsley, R.G.; Kameyama, M. A Region of Calpastatin Domain L That Reprimes Cardiac L-Type Ca2+ Channels. Biochem. Biophys. Res. Commun. 2006, 348, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Hao, L.Y.; Kameyama, A.; Kameyama, M. Calmodulin Reverses Rundown of L-Type Ca2+ Channels in Guinea Pig Ventricular Myocytes. Am. J. Physiol. Cell Physiol. 2004, 287, C1717–C1724. [Google Scholar] [CrossRef] [PubMed]
- Saud, Z.A.; Minobe, E.; Wang, W.Y.; Han, D.Y.; Horiuchi, M.; Hao, L.Y.; Kameyama, M. Calpastatin Binds to a Calmodulin-Binding Site of Cardiac Cav1.2 Ca2+ Channels. Biochem. Biophys. Res. Commun. 2007, 364, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Minobe, E.; Asmara, H.; Saud, Z.A.; Kameyama, M. Calpastatin Domain L Is a Partial Agonist of the Calmodulin-Binding Site for Channel Activation in Cav1.2 Ca2+ Channels. J. Biol. Chem. 2011, 286, 39013–39022. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Wong, S.T.; Gallagher, D.; Li, B.; Storm, D.R.; Scheuer, T.; Catterall, W.A. Ca2+/Calmodulin Binds to and Modulates P/Q-Type Calcium Channels. Nature 1999, 399, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Scheuer, T.; Catterall, W.A. Ca2+/Calmodulin-Dependent Facilitation and Inactivation of P/Q-Type Ca2+ Channels. J. Neurosci. 2000, 20, 6830–6838. [Google Scholar] [CrossRef] [PubMed]
- Mouton, J.; Feltz, A.; Maulet, Y. Interactions of Calmodulin with Two Peptides Derived from the C-Terminal Cytoplasmic Domain of the Cav1.2 Ca2+ Channel Provide Evidence for a Molecular Switch Involved in Ca2+-Induced Inactivation. J. Biol. Chem. 2001, 276, 22359–22367. [Google Scholar] [CrossRef]
- Pitt, G.S.; Zühlke, R.D.; Hudmon, A.; Schulman, H.; Reuter, H.; Tsien, R.W. Molecular Basis of Calmodulin Tethering and Ca2+-Dependent Inactivation of L-Type Ca2+ Channels. J. Biol. Chem. 2001, 276, 30794–30802. [Google Scholar] [CrossRef]
- DeMaria, C.D.; Soong, T.W.; Alseikhan, B.A.; Alvania, R.S.; Yue, D.T. Calmodulin Bifurcates the Local Ca2+ Signal That Modulates P/Q-Type Ca2+ Channels. Nature 2001, 411, 484–489. [Google Scholar] [CrossRef]
- Zühlke, R.D.; Pitt, G.S.; Deisseroth, K.; Tsien, R.W.; Reuter, H. Calmodulin Supports Both Inactivation and Facilitation of L-Type Calcium Channels. Nature 1999, 399, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Zühlke, R.D.; Pitt, G.S.; Tsien, R.W.; Reuter, H. Ca2+ -Sensitive Inactivation and Facilitation of L-Type Ca2+ Channels Both Depend on Specific Amino Acid Residues in a Consensus Calmodulin-Binding Motif in the α1C Subunit. J. Biol. Chem. 2000, 275, 21121–21129. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.I.A.; Hell, J.W.; Shea, M.A. Thermodynamic Linkage between Calmodulin Domains Binding Calcium and Contiguous Sites in the C-Terminal Tail of CaV1.2. Biophys. Chem. 2011, 159, 172–187. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Turner, M.; Anderson, D.E.; Bartels, P.; Nieves-Cintron, M.; Coleman, A.M.; Henderson, P.B.; Man, K.N.M.; Tseng, P.; Yarovoy, V.; Bers, D.M.; et al. α-Actinin-1 Promotes Activity of the L-Type Ca2+ Channel Cav 1.2. EMBO J. 2020, 39, e102622. [Google Scholar] [CrossRef]
- Wu, X.; Bers, D.M. Free and Bound Intracellular Calmodulin Measurements in Cardiac Myocytes. Cell Calcium 2007, 41, 353–364. [Google Scholar] [CrossRef]
- Shao, D.; Zhao, M.; Xu, J.; Feng, R.; Guo, F.; Hu, H.; Sun, X.; Gao, Q.; He, G.; Sun, W.; et al. The Individual N- and C-Lobes of Calmodulin Tether to the Cav1.2 Channel and Rescue the Channel Activity from Run-down in Ventricular Myocytes of Guinea-Pig Heart. FEBS Lett. 2014, 588, 3855–3861. [Google Scholar] [CrossRef]
- Bartels, P.; Salveson, I.; Coleman, A.M.; Anderson, D.E.; Jeng, G.; Estrada-Tobar, Z.M.; Man, K.N.M.; Yu, Q.; Kuzmenkina, E.; Nieves-Cintron, M.; et al. Half-Calcified Calmodulin Promotes Basal Activity and Inactivation of the L-Type Calcium Channel CaV1.2. J. Biol. Chem. 2022, 298, 102701. [Google Scholar] [CrossRef]
- Adams, P.J.; Ben-Johny, M.; Dick, I.E.; Inoue, T.; Yue, D.T. Apocalmodulin Itself Promotes Ion Channel Opening and Ca2+ Regulation. Cell 2014, 159, 608–622. [Google Scholar] [CrossRef]
- Banerjee, R.; Yoder, J.B.; Yue, D.T.; Amzel, L.M.; Tomaselli, G.F.; Gabelli, S.B.; Ben-Johny, M. Bilobal Architecture Is a Requirement for Calmodulin Signaling to CaV1.3 Channels. Proc. Natl. Acad. Sci. USA 2018, 115, E3026–E3035. [Google Scholar] [CrossRef]
- Liu, X.; Yang, P.S.; Yang, W.; Yue, D.T. Enzyme-Inhibitor-like Tuning of Ca2+ Channel Connectivity with Calmodulin. Nature 2010, 463, 968–972. [Google Scholar] [CrossRef]
- Han, D.Y.; Minobe, E.; Wang, W.Y.; Guo, F.; Xu, J.J.; Hao, L.Y.; Kameyama, M. Calmodulin- and Ca2+-Dependent Facilitation and Inactivation of the Cav1.2 Ca2+ Channels in Guinea-Pig Ventricular Myocytes. J. Pharmacol. Sci. 2010, 112, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yamada, Y.; Fukao, M.; Shiratori, K.; Tsutsuura, M.; Tanimoto, K.; Tohse, N. The GK Domain of the Voltage-Dependent Calcium Channel β Subunit Is Essential for Binding to the α Subunit. Biochem. Biophys. Res. Commun. 2007, 360, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Papa, A.; Katchman, A.N.; Zakharov, S.I.; Roybal, D.; Hennessey, J.A.; Kushner, J.; Yang, L.; Chen, B.X.; Kushnir, A.; et al. Mechanism of Adrenergic CaV1.2 Stimulation Revealed by Proximity Proteomics. Nature 2020, 577, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, K.; Kameyama, A.; Yasui, K.; Li, J.M.; Kameyama, M. ATP Regulates Cardiac Ca2+ Channel Activity via a Mechanism Independent of Protein Phosphorylation. Pflugers Arch. 1997, 433, 557–562. [Google Scholar] [CrossRef]
- Liu, S.Y.; Xu, J.J.; Minobe, E.; Gao, Q.H.; Feng, R.; Zhao, M.M.; Guo, F.; Yang, L.; Hao, L.Y.; Kameyama, M. Nucleotides Maintain the Activity of Cav1.2 Channels in Guinea-Pig Ventricular Myocytes. Biochem. Biophys. Res. Commun. 2015, 460, 813–818. [Google Scholar] [CrossRef]
- O’Rourke, B.; Backx, P.H.; Marban, E. Phosphorylation-Independent Modulation of L-Type Calcium Channels by Magnesium-Nucleotide Complexes. Science 1992, 257, 245–248. [Google Scholar] [CrossRef]
- Feng, R.; Xu, J.; Minobe, E.; Kameyama, A.; Yang, L.; Yu, L.; Hao, L.; Kameyama, M. Adenosine Triphosphate Regulates the Activity of Guinea Pig Cav1.2 Channel by Direct Binding to the Channel in a Dose-Dependent Manner. Am. J. Physiol. Cell Physiol. 2014, 306, C856–C863. [Google Scholar] [CrossRef]
- Hao, L.Y.; Xu, J.J.; Minobe, E.; Kameyama, A.; Kameyama, M. Calmodulin Kinase II Activation Is Required for the Maintenance of Basal Activity of L-Type Ca2+ Channels in Guinea-Pig Ventricular Myocytes. J. Pharmacol. Sci. 2008, 108, 290–300. [Google Scholar] [CrossRef]
- Hao, L.Y.; Wang, W.Y.; Minobe, E.; Han, D.Y.; Xu, J.J.; Kameyama, A.; Kameyama, M. The Distinct Roles of Calmodulin and Calmodulin Kinase II in the Reversal of Run-down of L-Type Ca2+ Channels in Guinea-Pig Ventricular Myocytes. J. Pharmacol. Sci. 2009, 111, 416–425. [Google Scholar] [CrossRef]
- Wang, W.Y.; Hao, L.Y.; Minobe, E.; Saud, Z.A.; Han, D.Y.; Kameyama, M. CaMKII Phosphorylates a Threonine Residue in the C-Terminal Tail of Cav1.2 Ca2+ Channel and Modulates the Interaction of the Channel with Calmodulin. J. Physiol. Sci. 2009, 59, 283–290. [Google Scholar] [CrossRef]
- Lei, M.; Xu, J.; Gao, Q.; Minobe, E.; Kameyama, M.; Hao, L. PKA Phosphorylation of Cav1.2 Channel Modulates the Interaction of Calmodulin with the C Terminal Tail of the Channel. J. Pharmacol. Sci. 2018, 137, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Halling, D.B.; Aracena-Parks, P.; Hamilton, S.L. Regulation of Voltage-Gated Ca2+ Channels by Calmodulin. Sci. STKE 2006, 2006, er1. [Google Scholar] [CrossRef]
- Chapman, R.A.; Niedergerke, R. Interaction between Heart Rate and Calcium Concentration in the Control of Contractile Strength of the Frog Heart. J. Physiol. 1970, 211, 423–443. [Google Scholar] [CrossRef]
- Balcazar, D.; Regge, V.; Santalla, M.; Meyer, H.; Paululat, A.; Mattiazzi, A.; Ferrero, P. SERCA Is Critical to Control the Bowditch Effect in the Heart. Sci. Rep. 2018, 8, 12447. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Shimoni, Y. The Calcium and Frequency Dependence of the Slow Inward Current ‘Staircase’ in Frog Atrium. J. Physiol. 1981, 310, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Shimoni, Y. Voltage-Dependent Potentiation of the Slow Inward Current in Frog Atrium. J. Physiol. 1981, 310, 77–95. [Google Scholar] [CrossRef]
- Marban, E.; Tsien, R.W. Enhancement of Calcium Current during Digitalis Inotropy in Mammalian Heart: Positive Feed-Back Regulation by Intracellular Calcium? J. Physiol. 1982, 329, 589–614. [Google Scholar] [CrossRef]
- Lee, K.S. Potentiation of the Calcium-Channel Currents of Internally Perfused Mammalian Heart Cells by Repetitive Depolarization. Proc. Natl. Acad. Sci. USA 1987, 84, 3941–3945. [Google Scholar] [CrossRef]
- Fedida, D.; Noble, D.; Spindler, A.J. Mechanism of the Use Dependence of Ca2+ Current in Guinea-Pig Myocytes. J. Physiol. 1988, 405, 461–475. [Google Scholar] [CrossRef]
- Tseng, G.N. Calcium Current Restitution in Mammalian Ventricular Myocytes Is Modulated by Intracellular Calcium. Circ. Res. 1988, 63, 468–482. [Google Scholar] [CrossRef]
- Zygmunt, A.C.; Maylie, J. Stimulation-Dependent Facilitation of the High Threshold Calcium Current in Guinea-Pig Ventricular Myocytes. J. Physiol. 1990, 428, 653–671. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D.; Hess, P. Novel Mechanism of Voltage-Dependent Gating in L-Type Calcium Channels. Nature 1990, 346, 651–655. [Google Scholar] [CrossRef]
- Kleppisch, T.; Pedersen, K.; Strubing, C.; Bosse-Doenecke, E.; Flockerzi, V.; Hofmann, F.; Hescheler, J. Double-Pulse Facilitation of Smooth Muscle α1-Subunit Ca2+ Channels Expressed in CHO Cells. EMBO J. 1994, 13, 2502–2507. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.M.; Charnet, P.; Pye, J.M.; Nargeot, J. Augmentation of Cardiac Calcium Current by Flash Photolysis of Intracellular Caged-Ca2+ Molecules. Nature 1989, 341, 65–68. [Google Scholar] [CrossRef]
- Hadley, R.W.; Lederer, W.J. Ca2+ and Voltage Inactivate Ca2+ Channels in Guinea-Pig Ventricular Myocytes through Independent Mechanisms. J. Physiol. 1991, 444, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E.; Gurney, A.M. Ca2+-Dependent Block and Potentiation of L-Type Calcium Current in Guinea-Pig Ventricular Myocytes. J. Physiol. 1993, 466, 345–365. [Google Scholar] [CrossRef]
- Hirano, Y.; Hiraoka, M. Dual Modulation of Unitary L-Type Ca2+ Channel Currents by [Ca2+]i in Fura-2-Loaded Guinea-Pig Ventricular Myocytes. J. Physiol. 1994, 480, 449–463. [Google Scholar] [CrossRef]
- Yamaoka, K.; Seyama, I. Regulation of Ca Channel by Intracellular Ca2+ and Mg2+ in Frog Ventricular Cells. Pflugers Arch. 1996, 431, 305–317. [Google Scholar] [CrossRef]
- Nie, H.G.; Hao, L.Y.; Xu, J.J.; Minobe, E.; Kameyama, A.; Kameyama, M. Distinct Roles of CaM and Ca2+/CaM-Dependent Protein Kinase II in Ca2+-Dependent Facilitation and Inactivation of Cardiac L-Type Ca2+ Channels. J. Physiol. Sci. 2008, 046. [Google Scholar] [CrossRef]
- Anderson, M.E.; Braun, A.P.; Schulman, H.; Premack, B.A. Multifunctional Ca2+/Calmodulin-Dependent Protein Kinase Mediates Ca2+-Induced Enhancement of the L-Type Ca2+ Current in Rabbit Ventricular Myocytes. Circ. Res. 1994, 75, 854–861. [Google Scholar] [CrossRef]
- Xiao, R.P.; Cheng, H.; Lederer, W.J.; Suzuki, T.; Lakatta, E.G. Dual Regulation of Ca2+/Calmodulin-Dependent Kinase II Activity by Membrane Voltage and by Calcium Influx. Proc. Natl. Acad. Sci. USA 1994, 91, 9659–9663. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Bers, D.M. Ca-Dependent Facilitation of Cardiac Ca Current Is Due to Ca-Calmodulin-Dependent Protein Kinase. Am. J. Physiol. 1994, 267, H982–H993. [Google Scholar] [CrossRef] [PubMed]
- Hudmon, A.; Schulman, H.; Kim, J.; Maltez, J.M.; Tsien, R.W.; Pitt, G.S. CaMKII Tethers to L-Type Ca2+ Channels, Establishing a Local and Dedicated Integrator of Ca2+ Signals for Facilitation. J. Cell Biol. 2005, 171, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Abiria, S.A.; Colbran, R.J. CaMKII Associates with CaV1.2 L-Type Calcium Channels via Selected β Subunits to Enhance Regulatory Phosphorylation. J. Neurochem. 2010, 112, 150–161. [Google Scholar] [CrossRef]
- Erxleben, C.; Liao, Y.; Gentile, S.; Chin, D.; Gomez-Alegria, C.; Mori, Y.; Birnbaumer, L.; Armstrong, D.L. Cyclosporin and Timothy Syndrome Increase Mode 2 Gating of CaV1.2 Calcium Channels through Aberrant Phosphorylation of S6 Helices. Proc. Natl. Acad. Sci. USA 2006, 103, 3932–3937. [Google Scholar] [CrossRef]
- Lee, T.S.; Karl, R.; Moosmang, S.; Lenhardt, P.; Klugbauer, N.; Hofmann, F.; Kleppisch, T.; Welling, A. Calmodulin Kinase II Is Involved in Voltage-Dependent Facilitation of the L-Type Cav1.2 Calcium Channel: Identification of the Phosphorylation Sites. J. Biol. Chem. 2006, 281, 25560–25567. [Google Scholar] [CrossRef]
- Grueter, C.E.; Abiria, S.A.; Dzhura, I.; Wu, Y.; Ham, A.J.L.; Mohler, P.J.; Anderson, M.E.; Colbran, R.J. L-Type Ca2+ Channel Facilitation Mediated by Phosphorylation of the Beta Subunit by CaMKII. Mol. Cell 2006, 23, 641–650. [Google Scholar] [CrossRef]
- Blaich, A.; Welling, A.; Fischer, S.; Wegener, J.W.; Köstner, K.; Hofmann, F.; Moosmang, S. Facilitation of Murine Cardiac L-Type Cav1.2 Channel Is Modulated by Calmodulin Kinase II-Dependent Phosphorylation of S1512 and S1570. Proc. Natl. Acad. Sci. USA 2010, 107, 10285–10289. [Google Scholar] [CrossRef]
- Brandmayr, J.; Poomvanicha, M.; Domes, K.; Ding, J.; Blaich, A.; Wegener, J.W.; Moosmang, S.; Hofmann, F. Deletion of the C-Terminal Phosphorylation Sites in the Cardiac β-Subunit Does Not Affect the Basic β-Adrenergic Response of the Heart and the Cav1.2 Channel. J. Biol. Chem. 2012, 287, 22584–22592. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Liu, Y.; Zhang, J.; Zheng, X.; Sun, X.; Lei, S.; Kang, Z.; Chen, X.; Lei, M.; et al. Distinct Roles of Calmodulin and Ca2+/Calmodulin-Dependent Protein Kinase II in Isopreterenol-Induced Cardiac Hypertrophy. Biochem. Biophys. Res. Commun. 2020, 526, 960–966. [Google Scholar] [CrossRef]
- Zheng, X.; Su, F.; Kang, Z.; Li, J.; Zhang, C.; Zhang, Y.; Hao, L. Analysis of Therapeutic Targets of A Novel Peptide Athycaltide-1 in the Treatment of Isoproterenol-Induced Pathological Myocardial Hypertrophy. Cardiovasc. Ther. 2022, 2022, 2715084. [Google Scholar] [CrossRef]
- Van Petegem, F.; Chatelain, F.C.; Minor, D.L. Insights into Voltage-Gated Calcium Channel Regulation from the Structure of the CaV1.2 IQ Domain-Ca2+/Calmodulin Complex. Nat. Struct. Mol. Biol. 2005, 12, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Song, L.S.; Zhu, W.Z.; Chakir, K.; Wang, W.; Wu, C.; Wang, Y.; Xiao, R.P.; Chen, S.R.W.; Cheng, H. Calmodulin Regulation of Excitation-Contraction Coupling in Cardiac Myocytes. Circ. Res. 2003, 92, 659–667. [Google Scholar] [CrossRef]
- Wu, Y.; Dzhura, I.; Colbran, R.J.; Anderson, M.E. Calmodulin Kinase and a Calmodulin-Binding ‘IQ’ Domain Facilitate L-Type Ca2+ Current in Rabbit Ventricular Myocytes by a Common Mechanism. J. Physiol. 2001, 535, 679–687. [Google Scholar] [CrossRef]
- Rodney, G.G.; Williams, B.Y.; Strasburg, G.M.; Beckingham, K.; Hamilton, S.L. Regulation of RYR1 Activity by Ca2+ and Calmodulin. Biochemistry 2000, 39, 7807–7812. [Google Scholar] [CrossRef]
- Guo, F.; Minobe, E.; Yazawa, K.; Asmara, H.; Bai, X.Y.; Han, D.Y.; Hao, L.Y.; Kameyama, M. Both N- and C-Lobes of Calmodulin Are Required for Ca2+-Dependent Regulations of CaV1.2 Ca2+ Channels. Biochem. Biophys. Res. Commun. 2010, 391, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ghosh, S.; Nunziato, D.A.; Pitt, G.S. Identification of the Components Controlling Inactivation of Voltage-Gated Ca2+ Channels. Neuron 2004, 41, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Dick, I.E.; Tadross, M.R.; Liang, H.; Tay, L.H.; Yang, W.; Yue, D.T. A Modular Switch for Spatial Ca2+ Selectivity in the Calmodulin Regulation of CaV Channels. Nature 2008, 451, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Anderson, D.E.; Hell, J.W.; Ames, J.B. Calmodulin Promotes a Ca2+-Dependent Conformational Change in the C-Terminal Regulatory Domain of CaV 1.2. FEBS Lett. 2022, 596, 2974–2985. [Google Scholar] [CrossRef]
- Maier, L.S.; Ziolo, M.T.; Bossuyt, J.; Persechini, A.; Mestril, R.; Bers, D.M. Dynamic Changes in Free Ca-Calmodulin Levels in Adult Cardiac Myocytes. J. Mol. Cell Cardiol. 2006, 41, 451–458. [Google Scholar] [CrossRef]
- Zhao, M.; Feng, R.; Shao, D.; Liu, S.; Lei, M.; Wang, H.; Sun, X.; Guo, F.; Hu, H.; Kameyama, M.; et al. Mg2+-Dependent Facilitation and Inactivation of L-Type Ca2+ Channels in Guinea Pig Ventricular Myocytes. J. Pharmacol. Sci. 2015, 129, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.Z.; DeMaria, C.D.; Yue, D.T. Calmodulin Is the Ca2+ Sensor for Ca2+ -Dependent Inactivation of L-Type Calcium Channels. Neuron 1999, 22, 549–558. [Google Scholar] [CrossRef]
- Zühlke, R.D.; Reuter, H. Ca2+-Sensitive Inactivation of L-Type Ca2+ Channels Depends on Multiple Cytoplasmic Amino Acid Sequences of the Alpha1C Subunit. Proc. Natl. Acad. Sci. USA 1998, 95, 3287–3294. [Google Scholar] [CrossRef] [PubMed]
- Young Kim, E.; Findeisen, F.; Minor, D.L. Calmodulin Interactions with Cav1 and Cav2 Voltage-Gated Calcium Channel IQ Domains. In Handbook of Metalloproteins; John Wiley: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Ben-Johny, M.; Dick, I.E.; Sang, L.; Limpitikul, W.B.; Kang, P.W.; Niu, J.; Banerjee, R.; Yang, W.; Babich, J.S.; Issa, J.B.; et al. Towards a Unified Theory of Calmodulin Regulation (Calmodulation) of Voltage-Gated Calcium and Sodium Channels. Curr. Mol. Pharmacol. 2015, 8, 188–205. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Souza, I.A.; Zamponi, G.W. A Novel Calmodulin Site in the Cav1.2 N-Terminus Regulates Calcium-Dependent Inactivation. Pflugers Arch. 2014, 466, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Rumpf, C.H.; Van Petegem, F.; Arant, R.J.; Findeisen, F.; Cooley, E.S.; Isacoff, E.Y.; Minor, D.L. Multiple C-Terminal Tail Ca2+/CaMs Regulate Cav1.2 Function but Do Not Mediate Channel Dimerization. EMBO J. 2010, 29, 3924–3938. [Google Scholar] [CrossRef]
- Ross, J.; Miura, T.; Kambayashi, M.; Eising, G.P.; Ryu, K.H. Adrenergic Control of the Force-Frequency Relation. Circulation 1995, 92, 2327–2332. [Google Scholar] [CrossRef]
- Brehm, P.; Eckert, R. Calcium Entry Leads to Inactivation of Calcium Channel in Paramecium. Science 1978, 202, 1203–1206. [Google Scholar] [CrossRef]
- Brehm, P.; Eckert, R.; Tillotson, D. Calcium-mediated Inactivation of Calcium Current in Paramecium. J. Physiol. 1980, 306, 193–203. [Google Scholar] [CrossRef]
- Tillotson, D. Inactivation of Ca Conductance Dependent on Entry of Ca Ions in Molluscan Neurons. Proc. Natl. Acad. Sci. USA 1979, 76, 1497–1500. [Google Scholar] [CrossRef]
- Eckert, R.; Chad, J.E. Inactivation of Ca Channels. Prog. Biophys. Mol. Biol. 1984, 44, 215–267. [Google Scholar] [CrossRef] [PubMed]
- Taiakina, V.; Boone, A.N.; Fux, J.; Senatore, A.; Weber-Adrian, D.; Guillemette, J.G.; Spafford, J.D. The Calmodulin-Binding, Short Linear Motif, NSCaTE Is Conserved in L-Type Channel Ancestors of Vertebrate Cav1.2 and Cav1.3 Channels. PLoS ONE 2013, 8, e61765. [Google Scholar] [CrossRef]
- Christel, C.; Lee, A. Ca2+-Dependent Modulation of Voltage-Gated Ca2+ Channels. Biochim. Biophys. Acta 2012, 1820, 1243–1252. [Google Scholar] [CrossRef]
- Sang, L.; Vieira, D.C.O.; Yue, D.T.; Ben-Johny, M.; Dick, I.E. The Molecular Basis of the Inhibition of CaV1 Calcium-Dependent Inactivation by the Distal Carboxy Tail. J. Biol. Chem. 2021, 296, 100502. [Google Scholar] [CrossRef]
- Zhou, J.; Olcese, R.; Qin, N.; Noceti, F.; Birnbaumer, L.; Stefani, E. Feedback Inhibition of Ca2+ Channels by Ca2+ Depends on a Short Sequence of the C Terminus That Does Not Include the Ca2+ -Binding Function of a Motif with Similarity to Ca2+ -Binding Domains. Proc. Natl. Acad. Sci. USA 1997, 94, 2301–2305. [Google Scholar] [CrossRef]
- Qin, N.; Olcese, R.; Bransby, M.; Lin, T.; Birnbaumer, L. Ca2+-Induced Inhibition of the Cardiac Ca2+ Channel Depends on Calmodulin. Proc. Natl. Acad. Sci. USA 1999, 96, 2435–2438. [Google Scholar] [CrossRef]
- Soldatov, N.M.; Zühlke, R.D.; Bouron, A.; Reuter, H. Molecular Structures Involved in L-Type Calcium Channel Inactivation. Role of the Carboxyl-Terminal Region Encoded by Exons 40-42 in α1C Subunit in the Kinetics and Ca2+ Dependence of Inactivation. J. Biol. Chem. 1997, 272, 3560–3566. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, S.F.; Dittmer, P.J.; Youn, D.H.; Dell’Acqua, M.L.; Sather, W.A. Localized Calcineurin Confers Ca2+-Dependent Inactivation on Neuronal L-Type Ca2+ Channels. J. Neurosci. 2012, 32, 15328–15337. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, S.F.; Dell’Acqua, M.L.; Sather, W.A. AKAP79/150 Anchoring of Calcineurin Controls Neuronal L-Type Ca2+ Channel Activity and Nuclear Signaling. Neuron 2007, 55, 261–275. [Google Scholar] [CrossRef]
- Hulme, J.T.; Westenforoek, R.E.; Scheuer, T.; Catterall, W.A. Phosphorylation of Serine 1928 in the Distal C-Terminal Domain of Cardiac CaV1.2 Channels during Beta1-Adrenergic Regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 16574–16579. [Google Scholar] [CrossRef]
- Dittmer, P.J.; Dell’Acqua, M.L.; Sather, W.A. Ca2+/Calcineurin-Dependent Inactivation of Neuronal L-Type Ca2+ Channels Requires Priming by AKAP-Anchored Protein Kinase A. Cell Rep. 2014, 7, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.; Wang, Y.; Jones, L.; Perez-Reyes, E.; Wei, X.; Soong, T.W.; Snutch, T.P.; Yue, D.T. Essential Ca2+-Binding Motif for Ca2+-Sensitive Inactivation of L-Type Ca2+ Channels. Science 1995, 270, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, N.M.; Oz, M.; O’Brien, K.A.; Abernethy, D.R.; Morad, M. Molecular Determinants of L-Type Ca2+ Channel Inactivation. Segment Exchange Analysis of the Carboxyl-Terminal Cytoplasmic Motif Encoded by Exons 40-42 of the Human α1C Subunit Gene. J. Biol. Chem. 1998, 273, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.Z.; Lee, J.S.; Mulle, J.G.; Wang, V.; De Leon, M.; Yue, D.T. Critical Determinants of Ca2+-Dependent Inactivation within an EF-Hand Motif of L-Type Ca2+ Channels. Biophys. J. 2000, 78, 1906–1920. [Google Scholar] [CrossRef]
- Zhou, H.; Kim, S.A.; Kirk, E.A.; Tippens, A.L.; Sun, H.; Haeseleer, F.; Lee, A. Ca2+-Binding Protein-1 Facilitates and Forms a Postsynaptic Complex with Cav1.2 (L-Type) Ca2+ Channels. J. Neurosci. 2004, 24, 4698–4708. [Google Scholar] [CrossRef]
- Fallon, J.L.; Baker, M.R.; Xiong, L.; Loy, R.E.; Yang, G.; Dirksen, R.T.; Hamilton, S.L.; Quiocho, F.A. Crystal Structure of Dimeric Cardiac L-Type Calcium Channel Regulatory Domains Bridged by Ca2+·Calmodulins. Proc. Natl. Acad. Sci. USA 2009, 106, 5135–5140. [Google Scholar] [CrossRef]
- Erickson, M.G.; Liang, H.; Mori, M.X.; Yue, D.T. FRET Two-Hybrid Mapping Reveals Function and Location of L-Type Ca2+ Channel CaM Preassociation. Neuron 2003, 39, 97–107. [Google Scholar] [CrossRef]
- Poomvanicha, M.; Wegener, J.W.; Blaich, A.; Fischer, S.; Domes, K.; Moosmang, S.; Hofmann, F. Facilitation and Ca2+-Dependent Inactivation Are Modified by Mutation of the Cav1.2 Channel IQ Motif. J. Biol. Chem. 2011, 286, 26702–26707. [Google Scholar] [CrossRef]
- Blaich, A.; Pahlavan, S.; Tian, Q.; Oberhofer, M.; Poomvanicha, M.; Lenhardt, P.; Domes, K.; Wegener, J.W.; Moosmang, S.; Ruppenthal, S.; et al. Mutation of the Calmodulin Binding Motif IQ of the L-Type Cav1.2 Ca2+ Channel to EQ Induces Dilated Cardiomyopathy and Death. J. Biol. Chem. 2012, 287, 22616–22625. [Google Scholar] [CrossRef]
- Crotti, L.; Spazzolini, C.; Tester, D.J.; Ghidoni, A.; Baruteau, A.E.; Beckmann, B.M.; Behr, E.R.; Bennett, J.S.; Bezzina, C.R.; Bhuiyan, Z.A.; et al. Calmodulin Mutations and Life-Threatening Cardiac Arrhythmias: Insights from the International Calmodulinopathy Registry. Eur. Heart J. 2019, 40, 2964–2975. [Google Scholar] [CrossRef]
- Su, J.; Gao, Q.; Yu, L.; Sun, X.; Feng, R.; Shao, D.; Yuan, Y.; Zhu, Z.; Sun, X.; Kameyama, M.; et al. The LQT-Associated Calmodulin Mutant E141G Induces Disturbed Ca2+-Dependent Binding and a Flickering Gating Mode of the CaV1.2 Channel. Am. J. Physiol. Cell Physiol. 2020, 318, C991–C1004. [Google Scholar] [CrossRef]
- Minobe, E.; Mori, M.X.; Kameyama, M. Calmodulin and ATP Support Activity of the Cav1.2 Channel through Dynamic Interactions with the Channel. J. Physiol. 2017, 595, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Crump, S.M.; Andres, D.A.; Sievert, G.; Satin, J. The Cardiac L-Type Calcium Channel Distal Carboxy Terminus Autoinhibition Is Regulated by Calcium. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H455–H464. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.; Gao, Q.; Xu, J.; Minobe, E.; Zhu, T.; Kameyama, M. A New Interaction between Proximal and Distal C-Terminus of Cav1.2 Channels. J. Pharmacol. Sci. 2017, 133, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Lieb, A.; Ortner, N.; Striessnig, J. C-Terminal Modulatory Domain Controls Coupling of Voltage-Sensing to Pore Opening in Cav1.3 L-Type Ca2+ Channels. Biophys. J. 2014, 106, 1467–1475. [Google Scholar] [CrossRef]
- Scharinger, A.; Eckrich, S.; Vandael, D.H.; Schönig, K.; Koschak, A.; Hecker, D.; Kaur, G.; Lee, A.; Sah, A.; Bartsch, D.; et al. Cell-Type-Specific Tuning of Cav1.3 Ca2+-Channels by a C-Terminal Automodulatory Domain. Front. Cell Neurosci. 2015, 9, 309. [Google Scholar] [CrossRef]
- Kuzmenkina, E.; Novikova, E.; Jangsangthong, W.; Matthes, J.; Herzig, S. Single-Channel Resolution of the Interaction between C-Terminal CaV1.3 Isoforms and Calmodulin. Biophys. J. 2019, 116, 836–846. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Baumann, L.; Cuny, H.; Eckert, C.; Griessmeier, K.; Biel, M. Switching off Calcium-Dependent Inactivation in L-Type Calcium Channels by an Autoinhibitory Domain. Proc. Natl. Acad. Sci. USA 2006, 103, 15657–15662. [Google Scholar] [CrossRef]
- Singh, A.; Hamedinger, D.; Hoda, J.C.; Gebhart, M.; Koschak, A.; Romanin, C.; Striessnig, J. C-Terminal Modulator Controls Ca2+-Dependent Gating of Ca v1.4 L-Type Ca2+ Channels. Nat. Neurosci. 2006, 9, 1108–1116. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, Z.; Geng, J.; Liu, M.; Liu, N.; Li, P.; Hong, W.; Yue, S.; Jiang, H.; Ge, H.; et al. Cytosolic Peptides Encoding CaV1 C-Termini Downregulate the Calcium Channel Activity-Neuritogenesis Coupling. Commun. Biol. 2022, 5, 484. [Google Scholar] [CrossRef]
- Singh, A.; Gebhart, M.; Fritsch, R.; Sinnegger-Brauns, M.J.; Poggiani, C.; Hoda, J.C.; Engel, J.; Romanin, C.; Striessnig, J.; Koschak, A. Modulation of Voltage- and Ca2+-Dependent Gating of Cav1.3 L-Type Calcium Channels by Alternative Splicing of a C-Terminal Regulatory Domain. J. Biol. Chem. 2008, 283, 20733–20744. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Bolz, H.J.; Koschak, A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 Voltage-Gated L-Type Ca2+ Channels. Pflugers Arch. 2010, 460, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Bock, G.; Gebhart, M.; Scharinger, A.; Jangsangthong, W.; Busquet, P.; Poggiani, C.; Sartori, S.; Mangoni, M.E.; Sinnegger-Brauns, M.J.; Herzig, S.; et al. Functional Properties of a Newly Identified C-Terminal Splice Variant of Cav1.3 L-Type Ca2+ Channels. J. Biol. Chem. 2011, 286, 42736–42748. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yang, Y.; Ge, L.; Liu, M.; Colecraft, H.M.; Liu, X. Cooperative and Acute Inhibition by Multiple C-Terminal Motifs of L-Type Ca2+ Channels. eLife 2017, 6, e21989. [Google Scholar] [CrossRef]
- Alseikhan, B.A.; DeMaria, C.D.; Colecraft, H.M.; Yue, D.T. Engineered Calmodulins Reveal the Unexpected Eminence of Ca2+ Channel Inactivation in Controlling Heart Excitation. Proc. Natl. Acad. Sci. USA 2002, 99, 17185–17190. [Google Scholar] [CrossRef]
- Liang, H.; DeMaria, C.D.; Erickson, M.G.; Mori, M.X.; Alseikhan, B.A.; Yue, D.T. Unified Mechanisms of Ca2+ Regulation across the Ca2+ Channel Family. Neuron 2003, 39, 951–960. [Google Scholar] [CrossRef]
- Tadross, M.R.; Dick, I.E.; Yue, D.T. Mechanism of Local and Global Ca2+ Sensing by Calmodulin in Complex with a Ca2+ Channel. Cell 2008, 133, 1228–1240. [Google Scholar] [CrossRef]
- Hye, Y.P.; Kim, S.A.; Korlach, J.; Rhoades, E.; Kwok, L.W.; Zipfel, W.R.; Waxham, M.N.; Webb, W.W.; Pollack, L. Conformational Changes of Calmodulin upon Ca2+ Binding Studied with a Microfluidic Mixer. Proc. Natl. Acad. Sci. USA 2008, 105, 542–547. [Google Scholar] [CrossRef]
- Limpitikul, W.B.; Greenstein, J.L.; Yue, D.T.; Dick, I.E.; Winslow, R.L. A Bilobal Model of Ca2+-Dependent Inactivation to Probe the Physiology of L-Type Ca2+ Channels. J. Gen. Physiol. 2018, 150, 1688–1701. [Google Scholar] [CrossRef]
- Hoang, J.; Prosser, R.S. Conformational Selection and Functional Dynamics of Calmodulin: A 19F Nuclear Magnetic Resonance Study. Biochemistry 2014, 53, 5727–5736. [Google Scholar] [CrossRef]
- Gsponer, J.; Christodoulou, J.; Cavalli, A.; Bui, J.M.; Richter, B.; Dobson, C.M.; Vendruscolo, M. A Coupled Equilibrium Shift Mechanism in Calmodulin-Mediated Signal Transduction. Structure 2008, 16, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Westerlund, A.M.; Delemotte, L. Effect of Ca2+ on the Promiscuous Target-Protein Binding of Calmodulin. PLoS Comput. Biol. 2018, 14, e1006072. [Google Scholar] [CrossRef] [PubMed]
- Putkey, J.A.; Kleerekoper, Q.; Gaertner, T.R.; Waxham, M.N. A New Role for IQ Motif Proteins in Regulating Calmodulin Function. J. Biol. Chem. 2003, 278, 49667–49670. [Google Scholar] [CrossRef] [PubMed]
- Moradi, F.; Copeland, E.N.; Baranowski, R.W.; Scholey, A.E.; Stuart, J.A.; Fajardo, V.A. Calmodulin-Binding Proteins in Muscle: A Minireview on Nuclear Receptor Interacting Protein, Neurogranin, and Growth-Associated Protein 43. Int. J. Mol. Sci. 2020, 21, 1016. [Google Scholar] [CrossRef]
- Caprara, G.A.; Morabito, C.; Perni, S.; Navarra, R.; Guarnieri, S.; Mariggiò, M.A. Evidence for Altered Ca2+ Handling in Growth Associated Protein 43-Knockout Skeletal Muscle. Front. Physiol. 2016, 7, 493. [Google Scholar] [CrossRef]
- Pang, C.; Crump, S.M.; Jin, L.; Correll, R.N.; Finlin, B.S.; Satin, J.; Andres, D.A. Rem GTPase Interacts with the Proximal CaV1.2 C-Terminus and Modulates Calcium-Dependent Channel Inactivation. Channels 2010, 4, 192–202. [Google Scholar] [CrossRef]
- Tseng, P.Y.; Henderson, P.B.; Hergarden, A.C.; Patriarchi, T.; Coleman, A.M.; Lillya, M.W.; Montagut-Bordas, C.; Lee, B.; Hell, J.W.; Horne, M.C. α-Actinin Promotes Surface Localization and Current Density of the Ca2+ Channel CaV1.2 by Binding to the IQ Region of the A1 Subunit. Biochemistry 2017, 56, 3669–3681. [Google Scholar] [CrossRef]
- Haeseleer, F.; Sokal, I.; Verlinde, C.L.M.J.; Erdjument-Bromage, H.; Temps, P.; Pronin, A.N.; Benovic, J.L.; Fariss, R.N.; Palczewski, K. Five Members of a Novel Ca2+-Binding Protein (CABP) Subfamily with Similarity to Calmodulin. J. Biol. Chem. 2000, 275, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, K.; McCoy, K.L.; Lee, A. Molecular Mechanism for Divergent Regulation of Cav1.2 Ca2+ Channels by Calmodulin and Ca2+-Binding Protein-1. J. Biol. Chem. 2005, 280, 29612–29619. [Google Scholar] [CrossRef]
- Findeisen, F.; Minor, D.L. Disruption of the IS6-AID Linker Affects Voltage-Gated Calcium Channel Inactivation and Facilitation. J. Gen. Physiol. 2009, 133, 327–343. [Google Scholar] [CrossRef]
- Findeisen, F.; Minor, D.L. Structural Basis for the Differential Effects of CaBP1 and Calmodulin on CaV1.2 Calcium-Dependent Inactivation. Structure 2010, 18, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, F.; Rumpf, C.H.; Minor, D.L. Apo States of Calmodulin and CaBP1 Control CaV1 Voltage-Gated Calcium Channel Function through Direct Competition for the IQ Domain. J. Mol. Biol. 2013, 425, 3217–3234. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Benmocha, A.; Sasson, Y.; Sachyani, D.; Almagor, L.; Lee, A.; Hirsch, J.A.; Dascal, N. Competitive and Non-Competitive Regulation of Calcium-Dependent Inactivation in CaV1.2 L-Type Ca2+ Channels by Calmodulin and Ca2+-Binding Protein 1. J. Biol. Chem. 2013, 288, 12680–12691. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.S.; Alseikhan, B.A.; Hiel, H.; Grant, L.; Mori, M.X.; Yang, W.; Fuchs, P.A.; Yue, D.T. Switching of Ca2+-Dependent Inactivation of Cav1.3 Channels by Calcium Binding Proteins of Auditory Hair Cells. J. Neurosci. 2006, 26, 10677–10689. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Meyer, A.C.; Calin-Jageman, I.; Neef, J.; Haeseleer, F.; Moser, T.; Lee, A. Ca2+-Binding Proteins Tune Ca2+-Feedback to Cav1.3 Channels in Mouse Auditory Hair Cells. J. Physiol. 2007, 585, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Ivanina, T.; Blumenstein, Y.; Shistik, E.; Barzilai, R.; Dascal, N. Modulation of L-Type Ca2+ Channels by Gβγ and Calmodulin via Interactions with N and C Termini of α1C. J. Biol. Chem. 2000, 275, 39846–39854. [Google Scholar] [CrossRef] [PubMed]
- Pate, P.; Mochca-Morales, J.; Wu, Y.; Zhang, J.Z.; Rodney, G.G.; Serysheva, I.I.; Williams, B.Y.; Anderson, M.E.; Hamilton, S.L. Determinants for Calmodulin Binding on Voltage-Dependent Ca2+ Channels. J. Biol. Chem. 2000, 275, 39786–39792. [Google Scholar] [CrossRef]
- Tang, W.; Halling, D.B.; Black, D.J.; Pate, P.; Zhang, J.Z.; Pedersen, S.; Altschuld, R.A.; Hamilton, S.L. Apocalmodulin and Ca2+ Calmodulin-Binding Sites on the CaV1.2 Channel. Biophys. J. 2003, 85, 1538–1547. [Google Scholar] [CrossRef]
- Anderson, M.E. Ca2+-Dependent Regulation of Cardiac L-Type Ca2+ Channels: Is a Unifying Mechanism at Hand? J. Mol. Cell Cardiol. 2001, 33, 639–650. [Google Scholar] [CrossRef]
- Jurado, L.A.; Chockalingam, P.S.; Jarrett, H.W. Apocalmodulin. Physiol. Rev. 1999, 79, 661–682. [Google Scholar] [CrossRef]
- Saimi, Y.; Kung, C. Calmodulin as an Ion Channel Subunit. Annu. Rev. Physiol. 2002, 64, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Chi, X.; Wei, J.; Zhou, G.; Huang, G.; Zhang, L.; Wang, R.; Lei, J.; Chen, S.R.W.; Yan, N. Modulation of Cardiac Ryanodine Receptor 2 by Calmodulin. Nature 2019, 572, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.W.; Westerlund, A.M.; Shi, J.; White, K.M.F.; Dou, A.K.; Cui, A.H.; Silva, J.R.; Delemotte, L.; Cui, J. Calmodulin Acts as a State-Dependent Switch to Control a Cardiac Potassium Channel Opening. Sci. Adv. 2020, 6, eabd6798. [Google Scholar] [CrossRef]
- Romanin, C.; Gamsjaeger, R.; Kahr, H.; Schaufler, D.; Carlson, O.; Abernethy, D.R.; Soldatov, N.M. Ca2+ Sensors of L-Type Ca2+ Channel. FEBS Lett. 2000, 487, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of Calmodulin with Voltage-Gated Ca2+ Channels Revealed by FRET in Single Living Cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef]
- Lian, L.Y.; Myatt, D.; Kitmitto, A. Apo Calmodulin Binding to the L-Type Voltage-Gated Calcium Channel Cav1.2 IQ Peptide. Biochem. Biophys. Res. Commun. 2007, 353, 565–570. [Google Scholar] [CrossRef]
- Black, D.J.; Halling, D.B.; Mandich, D.V.; Pedersen, S.E.; Altschuld, R.A.; Hamilton, S.L. Calmodulin Interactions with IQ Peptides from Voltage-Dependent Calcium Channels. Am. J. Physiol. Cell Physiol. 2005, 288, C669–C676. [Google Scholar] [CrossRef][Green Version]
- Sun, W.; Feng, R.; Hu, H.; Guo, F.; Gao, Q.; Shao, D.; Yin, D.; Wang, H.; Sun, X.; Zhao, M.; et al. The Ca2+-Dependent Interaction of Calpastatin Domain L with the C-Terminal Tail of the Cav1.2 Channel. FEBS Lett. 2014, 588, 665–671. [Google Scholar] [CrossRef]
- Black, D.J.; Persechini, A. In Calmodulin-IQ Domain Complexes, the Ca2+-Free and Ca2+-Bound Forms of the Calmodulin C-Lobe Direct the N-Lobe to Different Binding Sites. Biochemistry 2011, 50, 10061–10068. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, S.; Chen, S.; Zhang, J.; Shen, Y.; Su, J.; He, G.; Feng, R.; Shao, D.; Hao, L. Calmodulin Mutant in Central Linker Reduces the Binding Affinity with PreIQ and IQ While Interacting with CaV1.2 Channels. Biochem. Biophys. Res. Commun. 2020, 526, 78–84. [Google Scholar] [CrossRef]
- Asmara, H.; Minobe, E.; Saud, Z.A.; Kameyama, M. Interactions of Calmodulin with the Multiple Binding Sites of Cav1.2 Ca2+ Channels. J. Pharmacol. Sci. 2010, 112, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Few, A.P. Calcium Channel Regulation and Presynaptic Plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Vogel, H.J. Structural Basis for the Regulation of L-Type Voltage-Gated Calcium Channels: Interactions between the N-Terminal Cytoplasmic Domain and Ca2+-Calmodulin. Front. Mol. Neurosci. 2012, 5, 38. [Google Scholar] [CrossRef]
- Benmocha, A.; Almagor, L.; Oz, S.; Hirsch, J.A.; Dascal, N. Characterization of the Calmodulin-Binding Site in the N Terminus of CaV1.2. Channels 2009, 3, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Guggenheimer, A.B.; Almagor, L.; Tsemakhovich, V.; Tripathy, D.R.; Hirsch, J.A.; Dascal, N. Interactions between N and C Termini of α1C Subunit Regulate Inactivation of CaV1.2 L-Type Ca2+ Channel. Channels 2016, 10, 55–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gauberg, J.; Elkhatib, W.; Smith, C.L.; Singh, A.; Senatore, A. Divergent Ca2+/Calmodulin Feedback Regulation of CaV1 and CaV2 Voltage-Gated Calcium Channels Evolved in the Common Ancestor of Placozoa and Bilateria. J. Biol. Chem. 2022, 298. [Google Scholar] [CrossRef] [PubMed]
- Kawaji, K.; Minobe, E.; Mori, M.X.; Kameyama, M. Two Calmodulin Binding Site Model for the Regulation of Cav1.2 Channel. J. Physiol. Sci. 2016, 66, 76. [Google Scholar]
- Minobe, E.; Mori, M.X.; Kameyama, M. Cav1.2 Channel Inactivation Induced by Two Molecules of Calmodulin. J. Physiol. Sci. 2019, 69, 260. [Google Scholar]
- Ben-Johny, M.; Yang, P.S.; Bazzazi, H.; Yue, D.T. Dynamic Switching of Calmodulin Interactions Underlies Ca2+ Regulation of CaV1.3 Channels. Nat. Commun. 2013, 4, 1717. [Google Scholar] [CrossRef]
- Patton, D.E.; West, J.W.; Catterall, W.A.; Goldin, A.L. A Peptide Segment Critical for Sodium Channel Inactivation Functions as an Inactivation Gate in a Potassium Channel. Neuron 1993, 11, 967–974. [Google Scholar] [CrossRef]
- Shirokov, R.; Levis, R.; Shirokova, N.; Ríos, E. Ca2+-Dependent Inactivation of Cardiac L-Type Ca2+ Channels Does Not Affect Their Voltage Sensor. J. Gen. Physiol. 1993, 102, 1005–1030. [Google Scholar] [CrossRef]
- Zong, S.; Zhou, J.; Tanabe, T. Molecular Determinants of Calcium-Dependent Inactivation in Cardiac L-Type Calcium Channels. Biochem. Biophys. Res. Commun. 1994, 201, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Imredy, J.P.; Yue, D.T. Mechanism of Ca2+-Sensitive Inactivation of L-Type Ca2+ Channels. Neuron 1994, 12, 1301–1318. [Google Scholar] [CrossRef] [PubMed]
- Cens, T.; Restituito, S.; Galas, S.; Charnet, P. Voltage and Calcium Use the Same Molecular Determinants to Inactivate Calcium Channels. J. Biol. Chem. 1999, 274, 5483–5490. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F.; Tsien, R.W. The Timothy Syndrome Mutation Differentially Affects Voltage- and Calcium-Dependent Inactivation of CaV1.2 L-Type Calcium Channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Tadross, M.R.; Yue, D.T. Systematic Mapping of the State Dependence of Voltage- and Ca2+ dependent Inactivation Using Simple Open-Channel Measurements. J. Gen. Physiol. 2010, 135, 217–227. [Google Scholar] [CrossRef]
- Tadross, M.R.; Ben-Johny, M.; Yue, D.T. Molecular Endpoints of Ca2+/Calmodulin- and Voltage-Dependent Inactivation of Cav1.3 Channels. J. Gen. Physiol. 2010, 135, 197–215. [Google Scholar] [CrossRef]
- Raybaud, A.; Dodier, Y.; Bissonnette, P.; Simoes, M.; Bichet, D.G.; Sauvé, R.; Parent, L. The Role of the GX9GX3G Motif in the Gating of High Voltage-Activated Ca2+ Channels. J. Biol. Chem. 2006, 281, 39424–39436. [Google Scholar] [CrossRef]
- Almagor, L.; Chomsky-Hecht, O.; Ben-Mocha, A.; Hendin-Barak, D.; Dascal, N.; Hirsch, J.A. The Role of a Voltage-Dependent Ca2+ Channel Intracellular Linker: A Structure-Function Analysis. J. Neurosci. 2012, 32, 7602–7613. [Google Scholar] [CrossRef]
- Dick, I.E.; Joshi-Mukherjee, R.; Yang, W.; Yue, D.T. Arrhythmogenesis in Timothy Syndrome Is Associated with Defects in Ca2+-Dependent Inactivation. Nat. Commun. 2016, 7, 10370. [Google Scholar] [CrossRef]
- Limpitikul, W.B.; Dick, I.E.; Ben-Johny, M.; Yue, D.T. An Autism-Associated Mutation in CaV1.3 Channels Has Opposing Effects on Voltage- and Ca2+ -Dependent Regulation. Sci. Rep. 2016, 6, 27235. [Google Scholar] [CrossRef] [PubMed]
- Abderemane-Ali, F.; Findeisen, F.; Rossen, N.D.; Minor, D.L. A Selectivity Filter Gate Controls Voltage-Gated Calcium Channel Calcium-Dependent Inactivation. Neuron 2019, 101, 1134–1149.e3. [Google Scholar] [CrossRef] [PubMed]
- Babich, O.; Matveev, V.; Harris, A.L.; Shirokov, R. Ca2+-Dependent Inactivation of CaV1.2 Channels Prevents Gd3+ Block: Does Ca2+ Block the Pore of Inactivated Channels? J. Gen. Physiol. 2007, 129, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Cens, T.; Rousset, M.; Leyris, J.P.; Fesquet, P.; Charnet, P. Voltage- and Calcium-Dependent Inactivation in High Voltage-Gated Ca2+ Channels. Prog. Biophys. Mol. Biol. 2006, 90, 104–117. [Google Scholar] [CrossRef]
- Babich, O.; Isaev, D.; Shirokov, R. Role of Extracellular Ca2+ in Gating of CaV1.2 Channels. J. Physiol. 2005, 565, 709–715. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Li, Z.; Yan, C.; Lu, S.; Dong, M.; Yan, N. Structure of the Voltage-Gated Calcium Channel Cav1.1 Complex. Science 2015, 350, aad2395. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the Voltage-Gated Calcium Channel Cav1.1 at 3.6 Å Resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Pitt, G.S. Calmodulin and CaMKII as Molecular Switches for Cardiac Ion Channels. Cardiovasc. Res. 2007, 73, 641–647. [Google Scholar] [CrossRef]
- Zhang, R.; Dzhura, I.; Grueter, C.E.; Thiel, W.; Colbran, R.J.; Anderson, M.E. A Dynamic α-β Inter-Subunit Agonist Signaling Complex Is a Novel Feedback Mechanism for Regulating L-Type Ca2+ Channel Opening. FASEB J. 2005, 19, 1573–1575. [Google Scholar] [CrossRef]
- Mori, M.X.; Erickson, M.G.; Yue, D.T. Functional Stoichiometry and Local Enrichment of Calmodulin Interacting with Ca2+ Channels. Science 2004, 304, 432–435. [Google Scholar] [CrossRef]
- Xiong, L.; Kleerekoper, Q.K.; He, R.; Putkey, J.A.; Hamilton, S.L. Sites on Calmodulin That Interact with the C-Terminal Tail of Cav1.2 Channel. J. Biol. Chem. 2005, 280, 7070–7079. [Google Scholar] [CrossRef] [PubMed]
- Johny, M.B.; Yue, D.N.; Yue, D.T. A Novel FRET-Based Assay Reveals 1:1 Stoichiometry of Apocalmodulin Binding Across Voltage-Gated Ca and Na Ion Channels. Biophys. J. 2012, 102, 125a–126a. [Google Scholar] [CrossRef]
- Findeisen, F.; Minor, D.L. Progress in the Structural Understanding of Voltage-Gated Calcium Channel (CaV) Function and Modulation. Channels 2010, 4, 459–474. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Guo, F.; Zhu, T.; Shao, D.; Feng, R.; Yin, D.; Sun, X.; Hu, H.; Hwang, A.; Minobe, E.; et al. Lobe-Related Concentration- and Ca2+-Dependent Interactions of Calmodulin with C- and N-Terminal Tails of the CaV1.2 Channel. J. Physiol. Sci. 2013, 63, 345–353. [Google Scholar] [CrossRef]
- Shao, D.; Xu, J.; Zhao, M.; Sun, Y.; Minobe, E.; He, G.; Feng, R.; Hu, H.; Sun, X.; Guo, F.; et al. The Lobe-Specific Interaction of Calmodulin with the Cardiac CaV1.2 Channel. J. Physiol. Sci. 2014, 64, 177. [Google Scholar]
- Ben-Johny, M.; Yue, D.N.; Yue, D.T. Detecting Stoichiometry of Macromolecular Complexes in Live Cells Using FRET. Nat. Commun. 2016, 7, 13709. [Google Scholar] [CrossRef] [PubMed]
- Chakouri, N.; Diaz, J.; Yang, P.S.; Ben-Johny, M. CaV Channels Reject Signaling from a Second CaM in Eliciting Ca2+-Dependent Feedback Regulation. J. Biol. Chem. 2020, 295, 14948–14962. [Google Scholar] [CrossRef]
- Fallon, J.L.; Halling, D.B.; Hamilton, S.L.; Quiocho, F.A. Structure of Calmodulin Bound to the Hydrophobic IQ Domain of the Cardiac Cav1.2 Calcium Channel. Structure 2005, 13, 1881–1886. [Google Scholar] [CrossRef]
- Navedo, M.F.; Cheng, E.P.; Yuan, C.; Votaw, S.; Molkentin, J.D.; Scott, J.D.; Santana, L.F. Increased Coupled Gating of L-Type Ca2+ Channels during Hypertension and Timothy Syndrome. Circ. Res. 2010, 106, 748–756. [Google Scholar] [CrossRef]
- Findeisen, F.; Tolia, A.; Arant, R.; Kim, E.Y.; Isacoff, E.Y.; Minor, D.L. Calmodulin Overexpression Does Not Alter Cav1.2 Function or Oligomerization State. Channels 2011, 5, 320–324. [Google Scholar] [CrossRef][Green Version]
- Dixon, R.E.; Moreno, C.M.; Yuan, C.; Opitz-Araya, X.; Binder, M.D.; Navedo, M.F.; Santana, L.F. Graded Ca2+/Calmodulin-Dependent Coupling of Voltage-Gated CaV1.2 Channels. Elife 2015, 4, e05608. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.W.; Hannigan, K.I.; Ghosh, D.; Xu, B.; Del Villar, S.G.; Xiang, Y.K.; Dickson, E.J.; Navedo, M.F.; Dixon, R.E. β-Adrenergic-Mediated Dynamic Augmentation of Sarcolemmal CaV1.2 Clustering and Co-Operativity in Ventricular Myocytes. J. Physiol. 2019, 597, 2139–2162. [Google Scholar] [CrossRef]
- Simms, B.A.; Zamponi, G.W. Trafficking and Stability of Voltage-Gated Calcium Channels. Cell. Mol. Life Sci. 2012, 69, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zamponi, G.W. Trafficking of Neuronal Calcium Channels. Neuronal Signal 2017, 1, NS20160003. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.; Dixon, R.E. Mechanisms and Regulation of Cardiac CaV1.2 Trafficking. Int. J. Mol. Sci 2021, 22, 5927. [Google Scholar] [CrossRef]
- Béguin, P.; Ng, Y.J.A.; Krause, C.; Mahalakshmi, R.N.; Mei, Y.N.; Hunziker, W. RGK Small GTP-Binding Proteins Interact with the Nucleotide Kinase Domain of Ca2+-Channel β-Subunits via an Uncommon Effector Binding Domain. J. Biol. Chem. 2007, 282, 11509–11520. [Google Scholar] [CrossRef]
- Béguin, P.; Mahalakshmi, R.N.; Nagashima, K.; Cher, D.H.K.; Kuwamura, N.; Yamada, Y.; Seino, Y.; Hunziker, W. Roles of 14-3-3 and Calmodulin Binding in Subcellular Localization and Function of the Small G-Protein Rem2. Biochem. J. 2005, 390, 67–75. [Google Scholar] [CrossRef]
- Béguin, P.; Mahalakshmi, R.N.; Nagashima, K.; Cher, D.H.K.; Takahashi, A.; Yamada, Y.; Seino, Y.; Hunziker, W. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J. Cell Sci. 2005, 118 Pt 9, 1923–1934. [Google Scholar] [CrossRef][Green Version]
- Béguin, P.; Mahalakshmi, R.N.; Nagashima, K.; Cher, D.H.K.; Ikeda, H.; Yamada, Y.; Seino, Y.; Hunziker, W. Nuclear Sequestration of β-Subunits by Rad and Rem Is Controlled by 14-3-3 and Calmodulin and Reveals a Novel Mechanism for Ca2+ Channel Regulation. J. Mol. Biol. 2006, 355, 34–46. [Google Scholar] [CrossRef]
- Béguin, P.; Nagashima, K.; Gonoi, T.; Shibasaki, T.; Takahashi, K.; Kashima, Y.; Ozaki, N.; Geering, K.; Iwanaga, T.; Seino, S. Regulation of Ca2+ Channel Expression at the Cell Surface by the Small G-Protein Kir/Gem. Nature 2001, 411, 701–706. [Google Scholar] [CrossRef]
- Wang, H.G.; George, M.S.; Kim, J.; Wang, C.; Pitt, G.S. Ca2+/Calmodulin Regulates Trafficking of CaV1.2 Ca2+ Channels in Cultured Hippocampal Neurons. J. Neurosci. 2007, 27, 9086–9093. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ravindran, A.; Qi, Z.L.; Harry, J.B.; Abrahimi, P.; Kobrinsky, E.; Soldatov, N.M. Calmodulin-Dependent Gating of Cav1.2 Calcium Channels in the Absence of Cavβ Subunits. Proc. Natl. Acad. Sci. USA 2008, 105, 8154–8159. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Bunemann, M.; Gerhardstein, B.L.; Ma, H.; Hosey, M.M. Role of the C Terminus of the α1C (CaV1.2) Subunit in Membrane Targeting of Cardiac L-Type Calcium Channels. J. Biol. Chem. 2000, 275, 25436–25444. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, B.; Marger, F.; Wall-Lacelle, S.; Schneider, T.; Klein, H.; Sauvé, R.; Parent, L. Molecular Determinants of the CaVβ-Induced Plasma Membrane Targeting of the CaV1.2 Channel. J. Biol. Chem. 2010, 285, 22853–22863. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.D.; Dai, S.; Tseng, P.Y.; Malik, Z.; Nguyen, M.; Matt, L.; Schnizler, K.; Shephard, A.; Mohapatra, D.P.; Tsuruta, F.; et al. Competition between α-Actinin and Ca2+-Calmodulin Controls Surface Retention of the L-Type Ca2+ Channel CaV1.2. Neuron 2013, 78, 483–497. [Google Scholar] [CrossRef]
- Persechini, A.; Stemmer, P.M. Calmodulin is a Limiting Factor in the Cell. Trends Cardiovasc. Med. 2002, 12, 32–37. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kameyama, M.; Minobe, E.; Shao, D.; Xu, J.; Gao, Q.; Hao, L. Regulation of Cardiac Cav1.2 Channels by Calmodulin. Int. J. Mol. Sci. 2023, 24, 6409. https://doi.org/10.3390/ijms24076409
Kameyama M, Minobe E, Shao D, Xu J, Gao Q, Hao L. Regulation of Cardiac Cav1.2 Channels by Calmodulin. International Journal of Molecular Sciences. 2023; 24(7):6409. https://doi.org/10.3390/ijms24076409
Chicago/Turabian StyleKameyama, Masaki, Etsuko Minobe, Dongxue Shao, Jianjun Xu, Qinghua Gao, and Liying Hao. 2023. "Regulation of Cardiac Cav1.2 Channels by Calmodulin" International Journal of Molecular Sciences 24, no. 7: 6409. https://doi.org/10.3390/ijms24076409
APA StyleKameyama, M., Minobe, E., Shao, D., Xu, J., Gao, Q., & Hao, L. (2023). Regulation of Cardiac Cav1.2 Channels by Calmodulin. International Journal of Molecular Sciences, 24(7), 6409. https://doi.org/10.3390/ijms24076409