
Structure and Dynamics of Three Escherichia coli NfsB Nitro-Reductase Mutants Selected for Enhanced Activity with the Cancer Prodrug CB1954

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
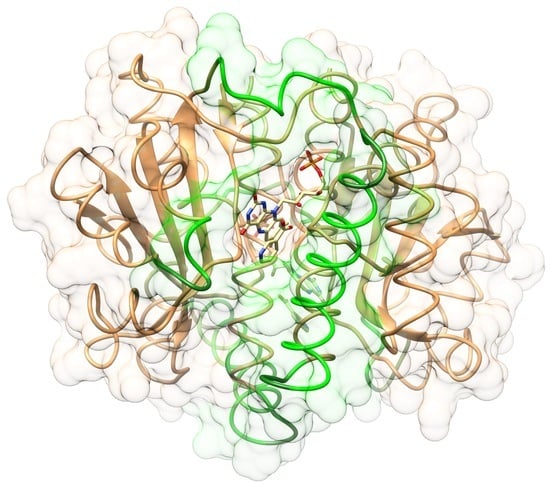
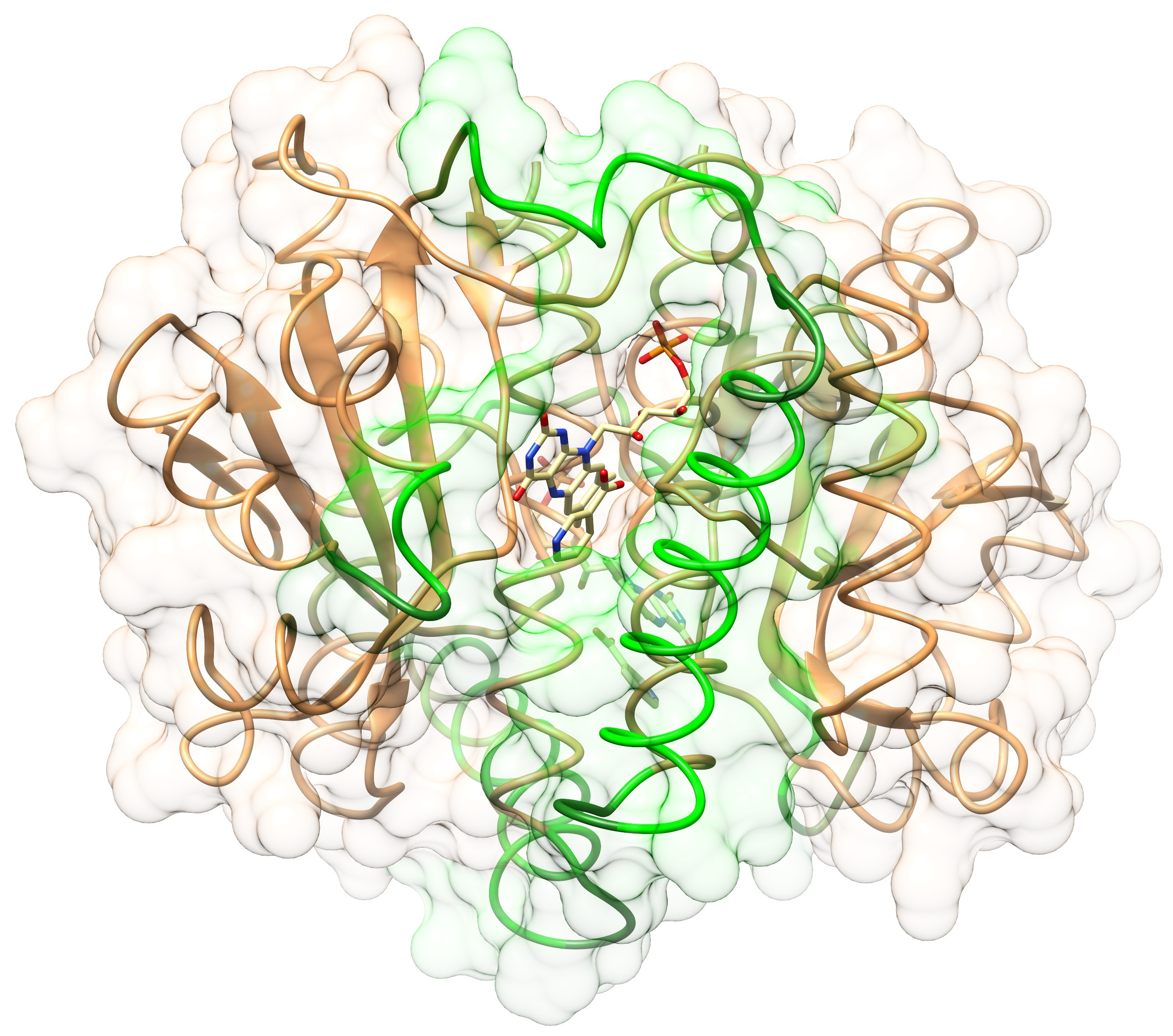
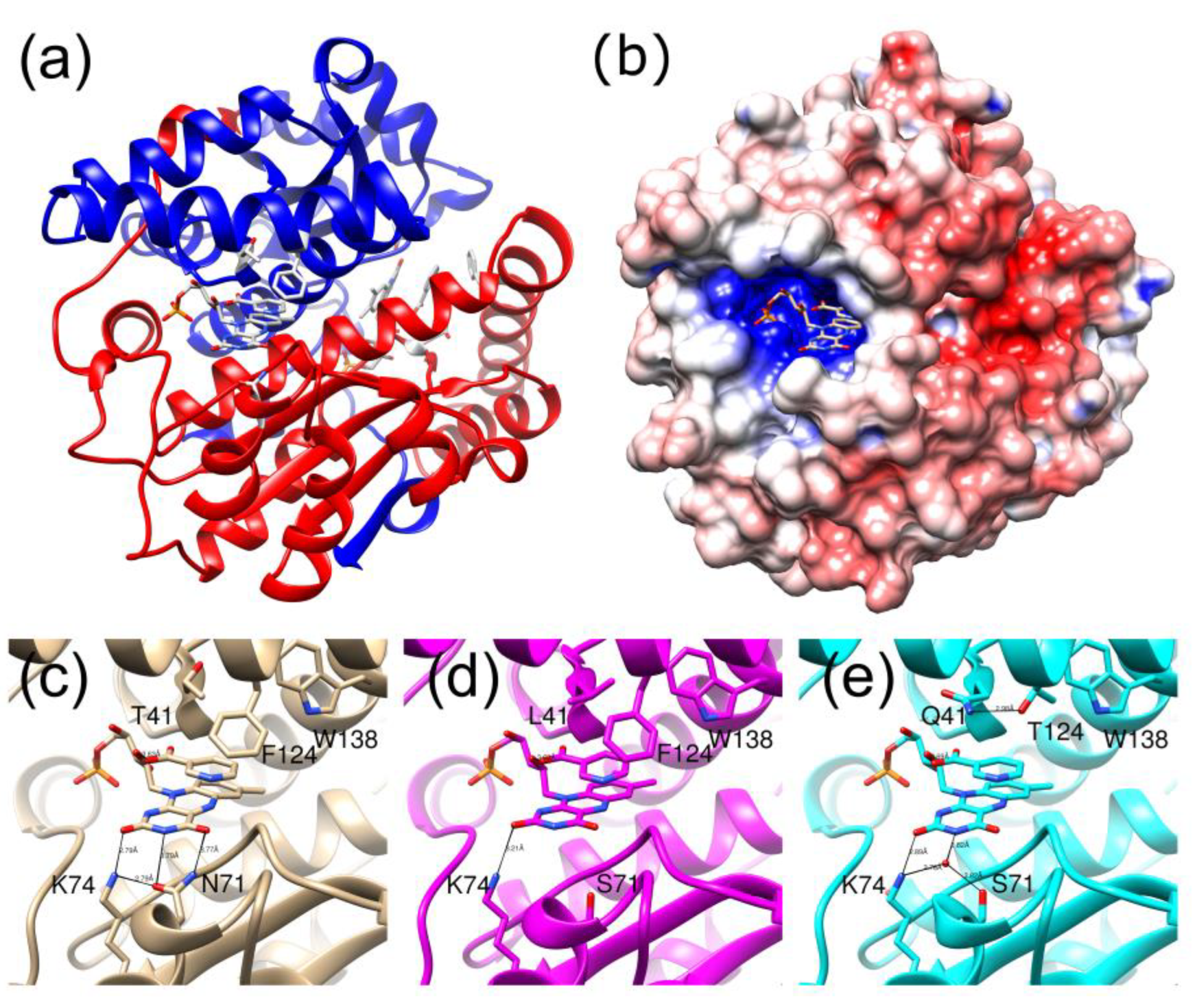
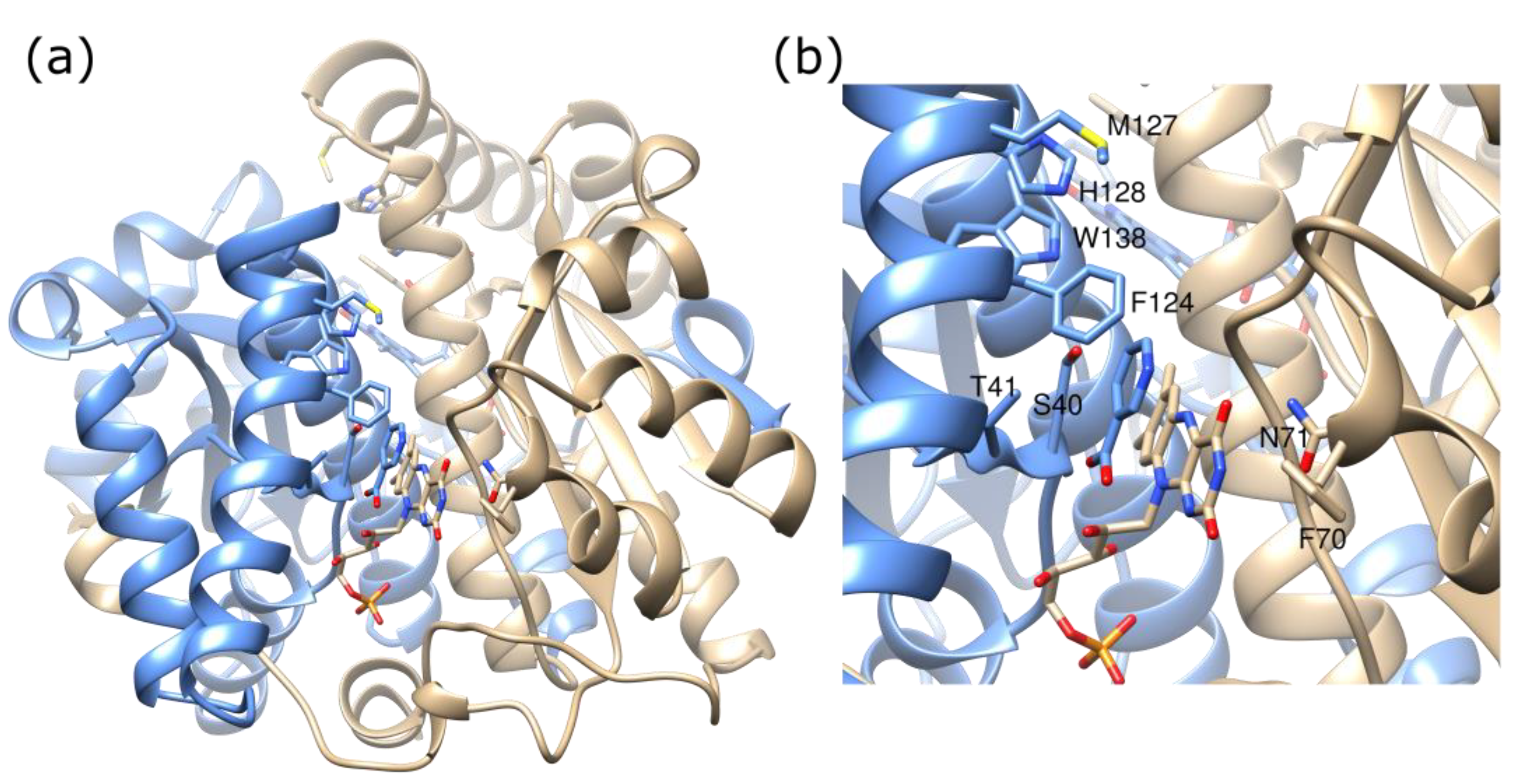
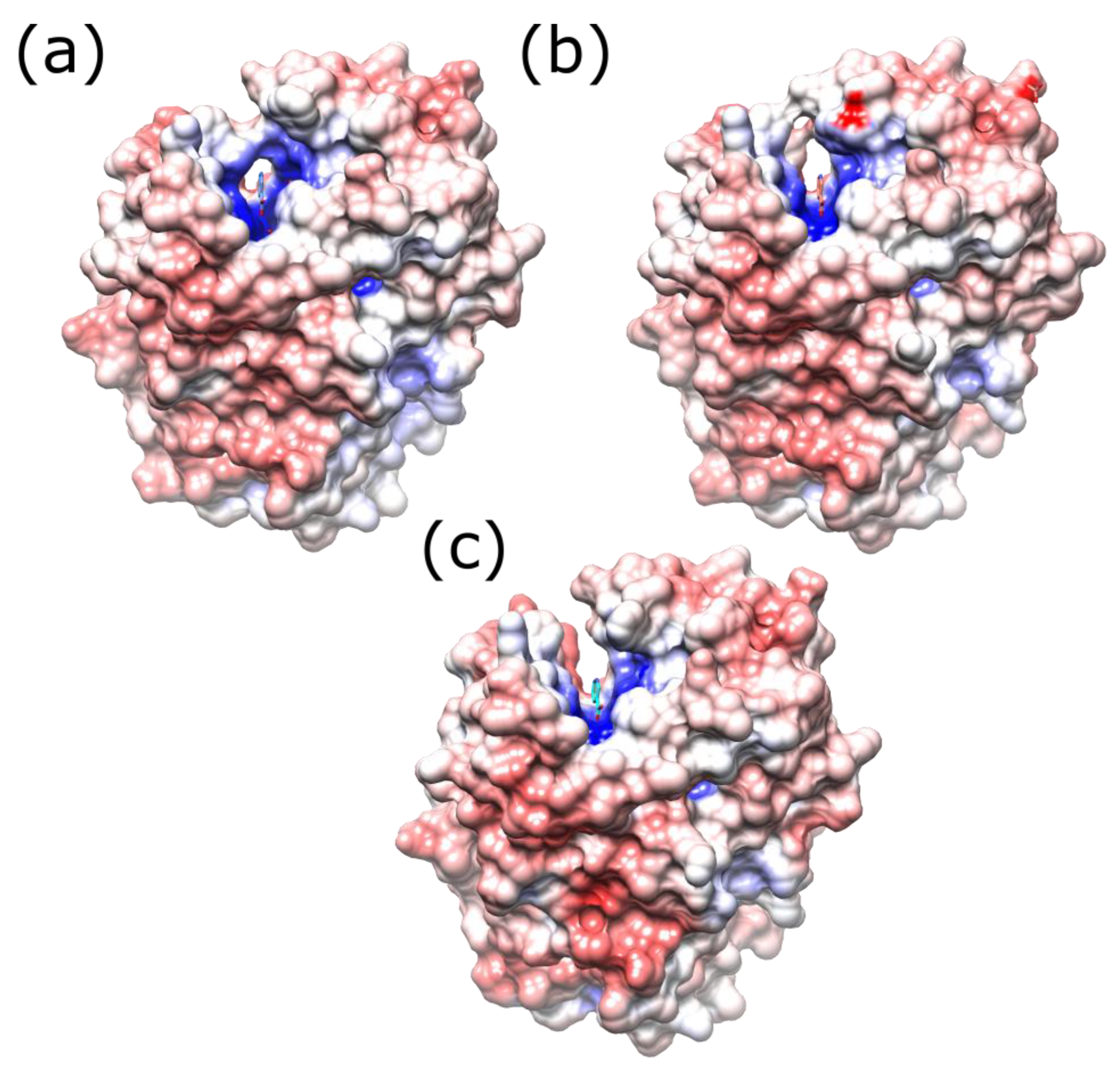
2.1. Structures of the T41L/N71S NfsB and T41Q/N71S/F124T NfsB Mutants
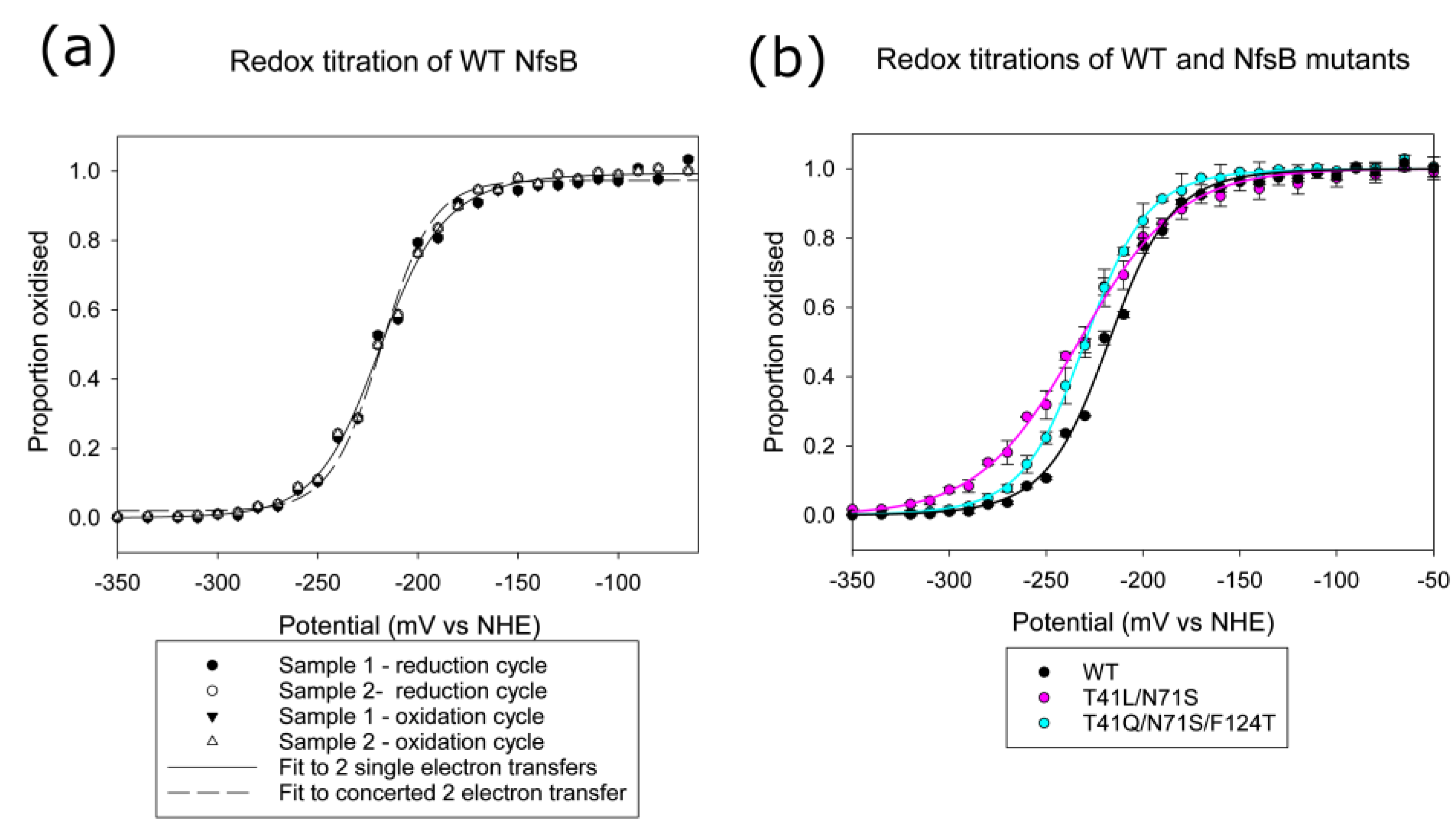
2.2. Redox Potentials of Wild Type, T41L/N71S and T41Q/ N71S/F124T NfsB
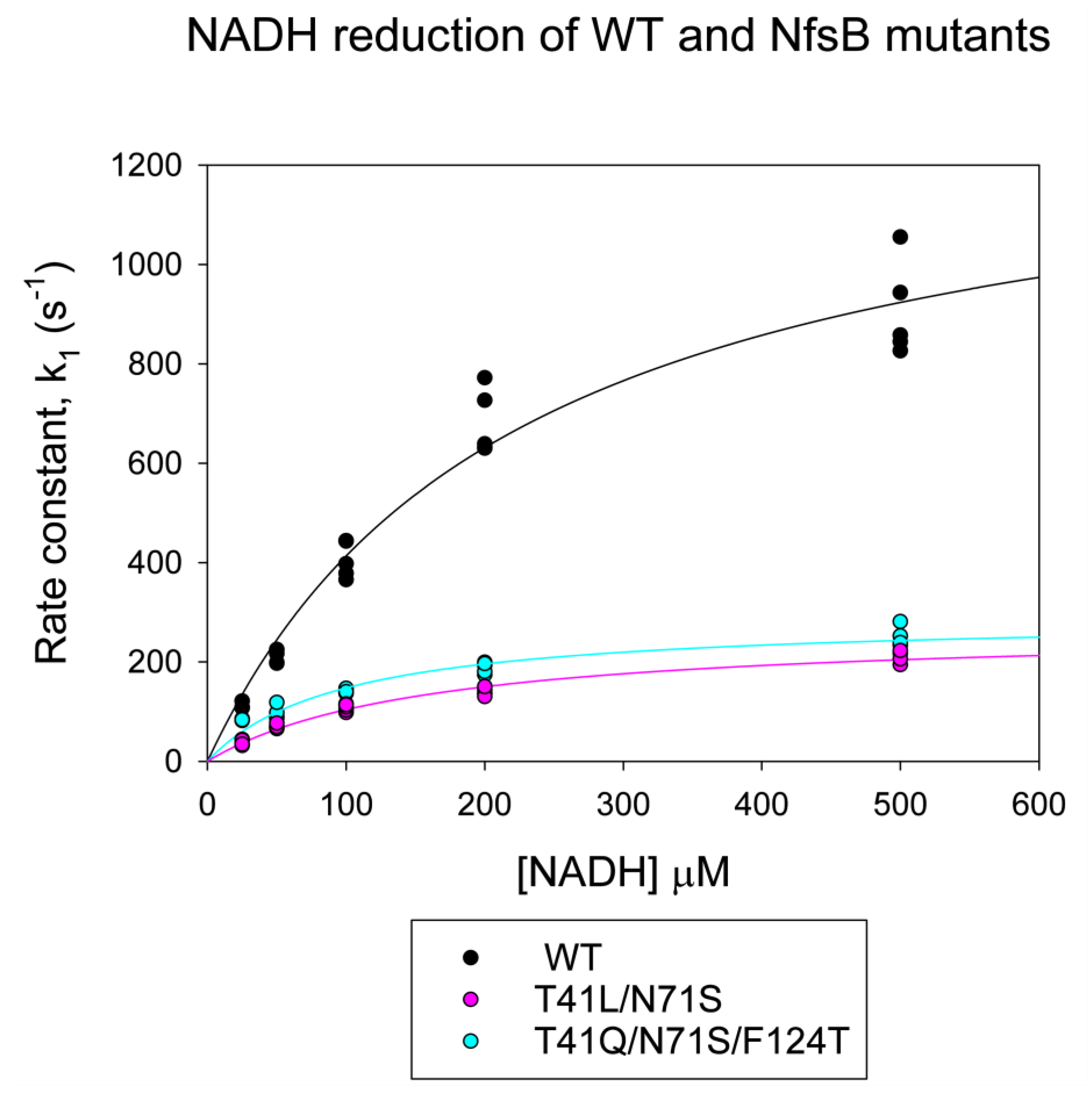
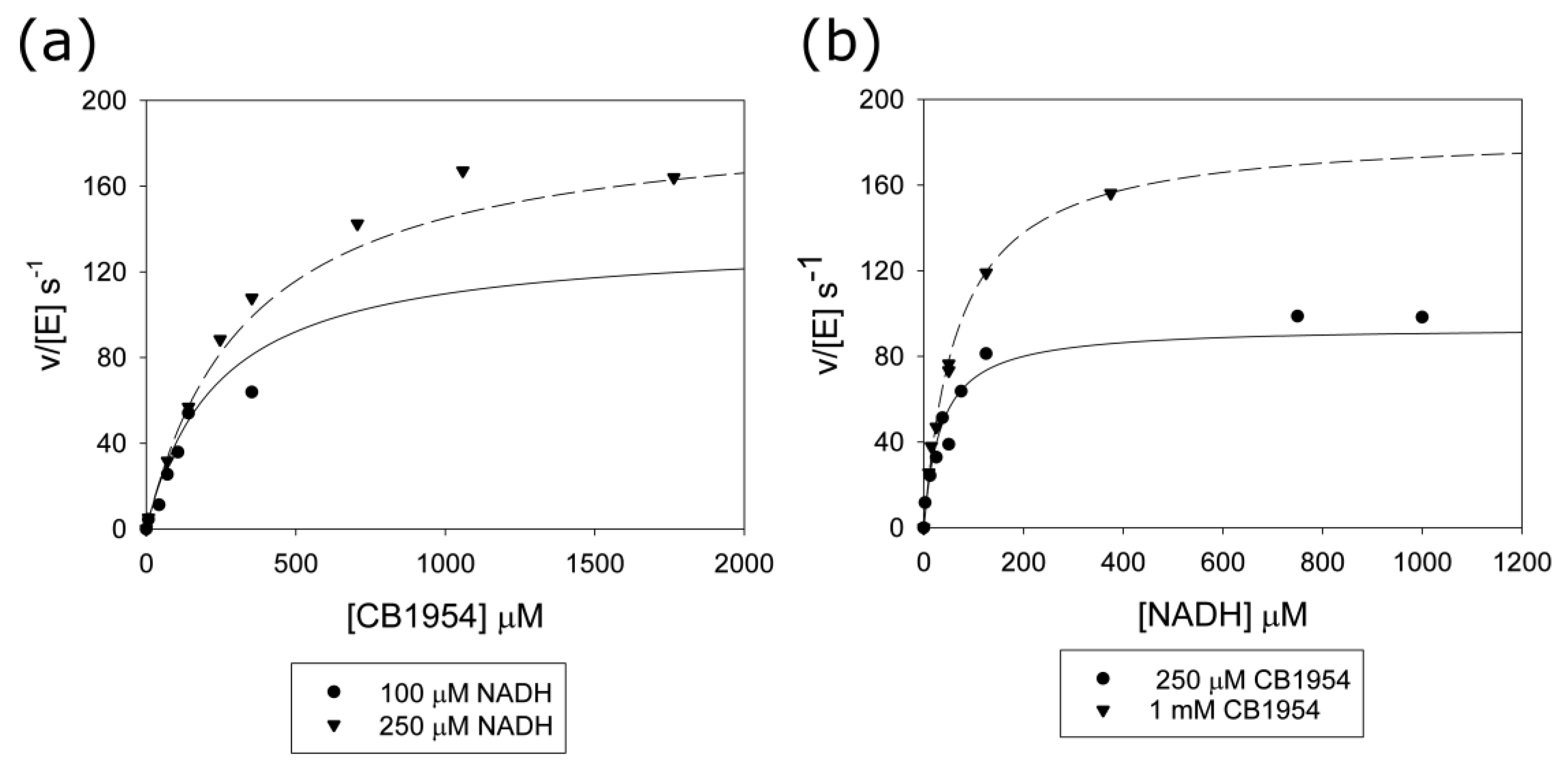
2.3. Stopped-Flow Kinetics
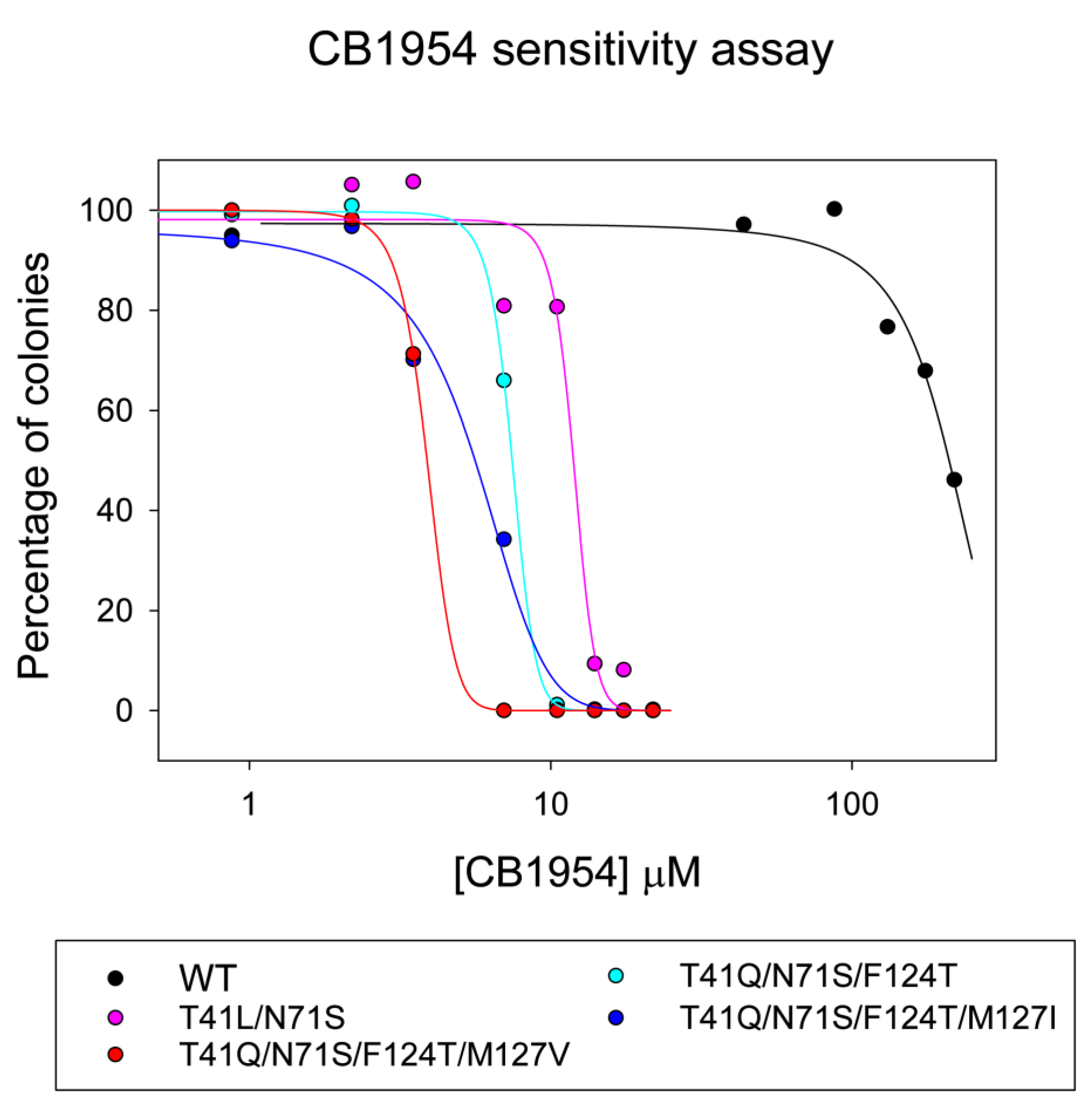
2.4. Quadruple Mutants
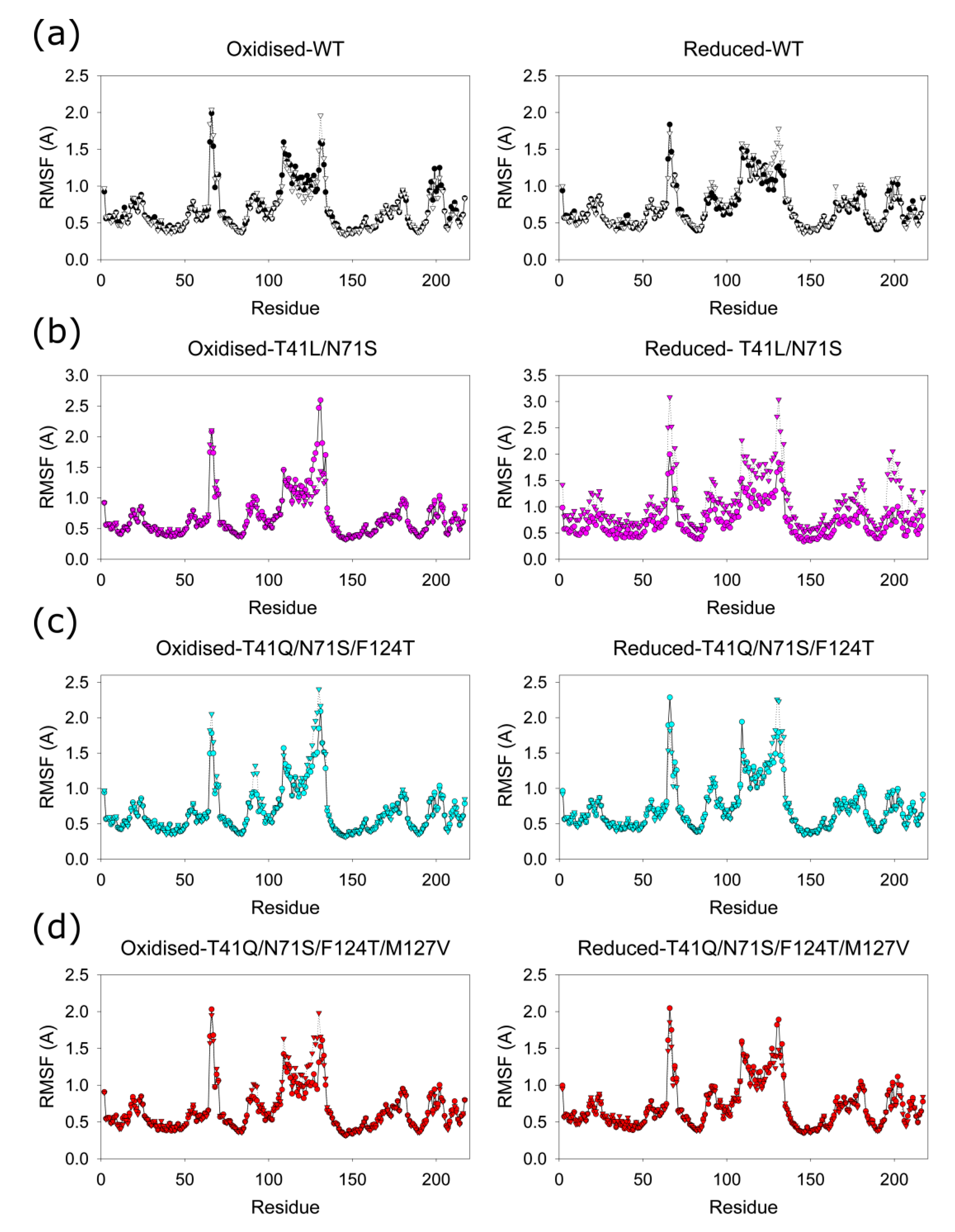
2.5. Molecular Dynamics (MD) Simulations
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Protein Crystallisation
4.3. Redox Potentiometry
4.4. Stopped-Flow Kinetics
4.5. Generation and Selection of Further Mutants
4.6. Analysis of Selected Phages
4.7. Steady-State Kinetic Enzyme Assays
4.8. Molecular Dynamics (MD) Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- McCalla, D.R.; Kaiser, C.; Green, M.H.L. Genetics of Nitrofurazone Resistance in Escherichia coli. J. Bacteriol. 1978, 133, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zenno, S.; Koike, H.; Tanokura, M.; Saigo, K. Gene cloning, purification, and characterization of NfsB, a minor oxygen-insensitive nitroreductase from Escherichia coli, similar in biochemical properties to FRase I, the major flavin reductase in Vibrio fischeri. J. Biochem. 1996, 120, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Anlezark, G.M.; Melton, R.G.; Sherwood, R.F.; Coles, B.; Friedlos, F.; Knox, R.J. The bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954)—I. Purification and properties of a nitroreductase enzyme from Escherichia coli—A potential enzyme for antibody-directed enzyme prodrug therapy (ADEPT). Biochem. Pharmacol. 1992, 44, 2289–2295. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; Green, R.M.; Ossor, A.; White, S.A.; Searle, P.F.; Wrighton, C.J.; Hyde, E.I. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 2005, 280, 13256–13264. [Google Scholar] [CrossRef] [PubMed]
- Green, N.K.; Youngs, D.J.; Searle, P.F.; Young, L.S.; Kerr, D.J.; Neoptolemos, J.P. Nitroreductase mediated prodrug killing in colorectal and pancreatic cancer cell lines by gene therapy. Cancer Gene Ther. 1995, 2, 247. [Google Scholar]
- Searle, P.F.; Weedon, S.J.; McNeish, I.; Gilligan, M.G.; Ford, M.J.; Friedlos, F.; Springer, C.J.; Young, L.S.; Kerr, D.J. Sensitisation of human ovarian cancer cells to killing by the prodrug CB1954 following retroviral or adenoviral transfer of the E. coli nitroreductase gene. Cancer Gene Ther. 1997, 4, 304. [Google Scholar]
- Djeha, A.H.; Hulme, A.; Dexter, M.T.; Mountain, A.; Young, L.S.; Searle, P.F.; Kerr, D.J.; Wrighton, C.J. Expression of E. coli B nitroreductase in established human tumour xenografts in mice results in potent anti-tumour and bystander effects upon systemic administration of the prodrug CB1954. Cancer Gene Ther. 2000, 7, 721–731. [Google Scholar] [CrossRef]
- Palmer, D.H.; Mautner, V.; Mirza, D.; Oliff, S.; Gerritsen, W.; van der Sijp, J.R.M.; Hubscher, S.; Reynolds, G.; Bonney, S.; Rajaratnam, R.; et al. Virus-directed enzyme prodrug therapy: Intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J. Clin. Oncol. 2004, 22, 1546–1552. [Google Scholar] [CrossRef]
- Patel, P.; Young, J.G.; Mautner, V.; Ashdown, D.; Bonney, S.; Pineda, R.G.; Collins, S.I.; Searle, P.F.; Hull, D.; Peers, E.; et al. A Phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954. Mol. Ther. 2009, 17, 1292–1299. [Google Scholar] [CrossRef]
- Chen, M.J.; Green, N.K.; Reynolds, G.M.; Flavell, J.R.; Mautner, V.; Kerr, D.J.; Young, L.S.; Searle, P.F. Enhanced efficacy of Escherichia coli nitroreductase/CB1954 prodrug activation gene therapy using an E1B-55K-deleted oncolytic adenovirus vector. Gene Ther. 2004, 11, 1126–1136. [Google Scholar] [CrossRef]
- Hu, L.Q.; Yu, C.Z.; Jiang, Y.Y.; Han, J.Y.; Li, Z.R.; Browne, P.; Race, P.R.; Knox, R.J.; Searle, P.F.; Hyde, E.I. Nitroaryl phosphoramides as novel prodrugs for E. coli nitroreductase activation in enzyme prodrug therapy. J. Med. Chem. 2003, 46, 4818–4821. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Knox, R.J.; Hobbs, S.M.; Jenkins, T.C.; Mauger, A.B.; Melton, R.G.; Burke, P.J.; Connors, T.A.; Hart, I.R. Investigation of alternative prodrugs for use with E. coli nitroreductase in ‘suicide gene’ approaches to cancer therapy. Gene Ther. 1996, 3, 1143–1150. [Google Scholar] [PubMed]
- Friedlos, F.; Denny, W.A.; Palmer, B.D.; Springer, C.J. Mustard prodrugs for activation by Escherichia coli nitroreductase in gene-directed enzyme prodrug therapy. J. Med. Chem. 1997, 40, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Mauger, A.B.; Burke, P.J.; Somani, H.H.; Friedlos, F.; Knox, R.J. Self-Immolative Prodrugs—Candidates for Antibody-Directed Enzyme Prodrug Therapy in Conjunction with a Nitroreductase Enzyme. J. Med. Chem. 1994, 37, 3452–3458. [Google Scholar] [CrossRef]
- Hay, M.P.; Atwell, G.J.; Wilson, W.R.; Pullen, S.M.; Denny, W.A. Structure-activity relationships for 4-nitrobenzyl carbamates of 5-aminobenz[e]indoline minor groove alkylating agents as prodrugs for GDEPT in conjunction with E. coli nitroreductase. J. Med. Chem. 2003, 46, 2456–2466. [Google Scholar] [CrossRef]
- Curado, S.; Anderson, R.M.; Jungblut, B.; Mumm, J.; Schroeter, E.; Stainier, D.Y.R. Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. Dev. Dyn. 2007, 236, 1025–1035. [Google Scholar] [CrossRef]
- Kaya, F.; Mannioui, A.; Chesneau, A.; Sekizar, S.; Maillard, E.; Ballagny, C.; Houel-Renault, L.; DuPasquier, D.; Bronchain, O.; Holtzmann, I.; et al. Live Imaging of Targeted Cell Ablation in Xenopus: A New Model to Study Demyelination and Repair. J. Neurosci. 2012, 32, 12885–12895. [Google Scholar] [CrossRef]
- Sharrock, A.V.; Mulligan, T.S.; Hall, K.R.; Williams, E.M.; White, D.T.; Zhang, L.; Emmerich, K.; Matthews, F.; Nimmagadda, S.; Washington, S.; et al. NTR 2.0: A rationally engineered prodrug-converting enzyme with substantially enhanced efficacy for targeted cell ablation. Nat. Methods 2022, 19, 205–215. [Google Scholar] [CrossRef]
- Hannink, N.; Rosser, S.J.; French, C.E.; Basran, A.; Murray, J.A.; Nicklin, S.; Bruce, N.C. Phytodetoxification of TNT by transgenic plants expressing a bacterial nitroreductase. Nat. Biotechnol. 2001, 19, 1168–1172. [Google Scholar] [CrossRef]
- Panz, K.; Miksch, K. Phytoremediation of explosives (TNT, RDX, HMX) by wild-type and transgenic plants. J. Environ. Manag. 2012, 113, 85–92. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; White, S.A.; Grove, J.I.; Searle, P.F.; Wrighton, C.W.; Hyde, E.I. Kinetic and structural characterisation of Escherichia coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J. Mol. Biol. 2007, 368, 481–492. [Google Scholar] [CrossRef]
- Jaberipour, M.; Vass, S.O.; Guise, C.P.; Grove, J.I.; Knox, R.J.; Hu, L.Q.; Hyde, E.I.; Searle, P.F. Testing double mutants of the enzyme nitroreductase for enhanced cell sensitisation to prodrugs: Effects of combining beneficial single mutations. Biochem. Pharmacol. 2010, 79, 102–111. [Google Scholar] [CrossRef]
- Guise, C.P.; Grove, J.I.; Hyde, E.I.; Searle, P.F. Direct positive selection for improved nitroreductase variants using SOS triggering of bacteriophage lambda lytic cycle. Gene Ther. 2007, 14, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Linwu, S.W.; Wu, C.A.; Peng, F.C.; Wang, A.H.J. Structure-based development of bacterial nitroreductase against nitrobenzodiazepine-induced hypnosis. Biochem. Pharmacol. 2012, 83, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Yang, J.; Zhou, Y.; Yang, Q. Structural basis of Escherichia coli nitroreductase NfsB triple mutants engineered for improved activity and regioselectivity toward the prodrug CB1954. Process Biochem. 2015, 50, 1760–1766. [Google Scholar] [CrossRef]
- Bai, J.; Zhou, Y.; Chen, Q.; Yang, Q.; Yang, J. Altering the regioselectivity of a nitroreductase in the synthesis of arylhydroxylamines by structure-based engineering. ChemBioChem 2015, 16, 1219–1225. [Google Scholar] [CrossRef]
- Williams, E.M.; Rich, M.H.; Mowday, A.M.; Ashoorzadeh, A.; Copp, J.N.; Guise, C.P.; Anderson, R.F.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; et al. Engineering Escherichia coli NfsB To Activate a Hypoxia-Resistant Analogue of the PET Probe EF5 To Enable Non-Invasive Imaging during Enzyme Prodrug Therapy. Biochemistry 2019, 58, 3700–3710. [Google Scholar] [CrossRef]
- Jarrom, D.; Jaberipour, M.; Guise, C.P.; Daff, S.; White, S.A.; Searle, P.F.; Hyde, E.I. Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 2009, 48, 7665–7672. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Skelly, J.V.; Neidle, S. Crystal Structure of FMN-Dependent Nitroreductase from Escherichia coli B: A Prodrug-Activating Enzyme. J. Med. Chem. 2000, 43, 3624–3631. [Google Scholar] [CrossRef]
- Lovering, A.L.; Hyde, E.I.; Searle, P.F.; Scott, A.W. The structure of Escherichia coli nitroreductase complexed with nicotinic acid: Three crystal forms at 1.7 Å, 1.8 Å and 2.4 Å resolution. J. Mol. Biol. 2001, 309, 203–213. [Google Scholar] [CrossRef]
- Johansson, E.; Parkinson, G.N.; Denny, W.A.; Neidle, S. Studies on the Nitroreductase Prodrug-Activating System. Crystal Structures of Complexes with the Inhibitor Dicoumarol and Dinitrobenzamide Prodrugs and of the Enzyme Active Form. J. Med. Chem. 2003, 46, 4009–4020. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.A.; Koder, R.L.; Miller, A.F.; Rodgers, D.W. Structures of nitroreductase in three states—Effects of inhibitor binding and reduction. J. Biol. Chem. 2002, 277, 11513–11520. [Google Scholar] [CrossRef] [PubMed]
- Pitsawong, W.; Haynes, C.A.; Koder, R.L.; Rodgers, D.W.; Miller, A.F. Mechanism-Informed Refinement Reveals Altered Substrate-Binding Mode for Catalytically Competent Nitroreductase. Structure 2017, 25, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Sasaki, H.; Kobori, T.; Zenno, S.; Saigo, K.; Murphy, M.E.P.; Adman, E.T.; Tanokura, M. 1.8 angstrom crystal structure of the major NAD(P)H: FMN oxidoreductase of a bioluminescent bacterium, Vibrio fischeri: Overall structure, cofactor and substrate-analog binding, and comparison with related flavoproteins. J. Mol. Biol. 1998, 280, 259–273. [Google Scholar] [CrossRef]
- Koder, R.L.; Haynes, C.A.; Rodgers, M.E.; Rodgers, D.W.; Miller, A.F. Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductases. Biochemistry 2002, 41, 14197–14205. [Google Scholar] [CrossRef]
- Draper, R.D.; Ingraham, L.L. A potentiometric study of the flavin semiquinone equilibrium. Arch. Biochem. Biophys. 1968, 125, 802–808. [Google Scholar] [CrossRef]
- Zenno, S.; Koike, H.; Kumar, A.N.; Jayaraman, R.; Tanokura, M.; Saigo, K. Biochemical characterization of NfsA, the Escherichia coli major nitroreductase exhibiting a high amino acid sequence homology to Frp, a Vibrio harveyi flavin oxidoreductase. J. Bacteriol. 1996, 178, 4508–4514. [Google Scholar] [CrossRef]
- Koder, R.L.; Miller, A.F. Steady-state kinetic mechanism, stereospecificity, substrate and inhibitor specificity of Enterobacter cloacae nitroreductase. Biochim Biophys Acta (BBA) Protein Struct. Mol. Enzymol. 1998, 1387, 395–405. [Google Scholar] [CrossRef]
- Christofferson, A.J. Asymmetric ligand binding in homodimeric Enterobacter cloacae nitroreductase yields the Michaelis complex for nitroaromatic substrates. J. Mol. Model. 2020, 26, 28. [Google Scholar] [CrossRef]
- Day, M.A.; Jarrom, D.; Christofferson, A.J.; Graziano, A.E.; Anderson, J.L.R.; Searle, P.F.; Hyde, E.I.; White, S.A. The structures of E. coli NfsA bound to the antibiotic nitrofurantoin; to 1,4-benzoquinone and to FMN. Biochem. J. 2021, 478, 2601–2617. [Google Scholar] [CrossRef]
- Sviatenko, L.K.; Gorb, L.; Leszczynski, J. NTO Degradation by Nitroreductase: A DFT Study. J. Phys. Chem. B 2022, 126, 5991–6006. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, A.; Wilkie, J. Mechanism of CB1954 reduction by Escherichia coli nitroreductase. Biochem. Soc. Trans. 2009, 37, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.I.; Lovering, A.L.; Guise, C.; Race, P.R.; Wrighton, C.J.; White, S.A.; Hyde, E.I.; Searle, P.F. Generation of Escherichia coli nitroreductase mutants conferring improved cell sensitization to the prodrug CB1954. Cancer Res. 2003, 63, 5532–5537. [Google Scholar] [PubMed]
- Akiva, E.; Copp, J.N.; Tokuriki, N.; Babbitt, P.C. Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Natl. Acad. Sci. USA 2017, 114, E9549–E9558. [Google Scholar] [CrossRef] [PubMed]
- Sharrock, A.V.; McManaway, S.P.; Rich, M.H.; Mumm, J.S.; Hermans, I.F.; Tercel, M.; Pruijn, F.B.; Ackerley, D.F. Engineering the Escherichia coli Nitroreductase NfsA to Create a Flexible Enzyme-Prodrug Activation System. Front. Pharmacol. 2021, 12, 701456. [Google Scholar] [CrossRef]
- White, S.A.; Christofferson, A.J.; Grainger, A.I.; Day, M.A.; Jarrom, D.; Graziano, A.E.; Searle, P.F.; Hyde, E.I. The 3D-structure, kinetics and dynamics of the E. coli nitroreductase NfsA with NADP+ provide glimpses of its catalytic mechanism. FEBS Lett. 2022, 596, 2425–2440. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to Measure and Predict the Molar Absorption-Coefficient of a Protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Bailey, S. The Ccp4 Suite—Programs for Protein Crystallography. Acta Crystallogr. Biol. Crystallogr. Sect. D. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Mccoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Winn, M.D.; Isupov, M.N.; Murshudov, G.N. Use of TLS parameters to model anisotropic displacemnets in macromolecular refinement. Acta. Crystallogr. Sect. D Struct. Biol. 2001, 57, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Struct. Biol. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Uski, V.; Lebedev, A.; Winn, M.; Ballard, C. Distributed computing for macromolecular crystallography. Acta Cryst. D Struct. Biol. 2018, 74, 143–151. [Google Scholar] [CrossRef]
- Grosse-Kunstleve, R.W.; Sauter, N.K.; Moriarty, N.W.; Adams, P.D. The Computational Crystallography Toolbox: Crystallographic algorithms in a reusable software framework. J. Appl. Crystallogr. 2002, 35, 126–136. [Google Scholar] [CrossRef]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.I.; Cisneros, G.A.; Cruzeiro, V.W.D.; Darden, T.A.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, M.A.; Christofferson, A.J.; Anderson, J.L.R.; Vass, S.O.; Evans, A.; Searle, P.F.; White, S.A.; Hyde, E.I. Structure and Dynamics of Three Escherichia coli NfsB Nitro-Reductase Mutants Selected for Enhanced Activity with the Cancer Prodrug CB1954. Int. J. Mol. Sci. 2023, 24, 5987. https://doi.org/10.3390/ijms24065987
Day MA, Christofferson AJ, Anderson JLR, Vass SO, Evans A, Searle PF, White SA, Hyde EI. Structure and Dynamics of Three Escherichia coli NfsB Nitro-Reductase Mutants Selected for Enhanced Activity with the Cancer Prodrug CB1954. International Journal of Molecular Sciences. 2023; 24(6):5987. https://doi.org/10.3390/ijms24065987
Chicago/Turabian StyleDay, Martin A., Andrew J. Christofferson, J. L. Ross Anderson, Simon O. Vass, Adam Evans, Peter F. Searle, Scott A. White, and Eva I. Hyde. 2023. "Structure and Dynamics of Three Escherichia coli NfsB Nitro-Reductase Mutants Selected for Enhanced Activity with the Cancer Prodrug CB1954" International Journal of Molecular Sciences 24, no. 6: 5987. https://doi.org/10.3390/ijms24065987
APA StyleDay, M. A., Christofferson, A. J., Anderson, J. L. R., Vass, S. O., Evans, A., Searle, P. F., White, S. A., & Hyde, E. I. (2023). Structure and Dynamics of Three Escherichia coli NfsB Nitro-Reductase Mutants Selected for Enhanced Activity with the Cancer Prodrug CB1954. International Journal of Molecular Sciences, 24(6), 5987. https://doi.org/10.3390/ijms24065987