
Adaptation of the Porcine Pituitary Transcriptome, Spliceosome and Editome during Early Pregnancy

 ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Overall Statistics of RNA-Seq Data Mapping
2.2. Differentially Expressed Genes
2.3. Long Noncoding RNA Identification and Cis-/Trans-Acting on Protein-Coding Genes
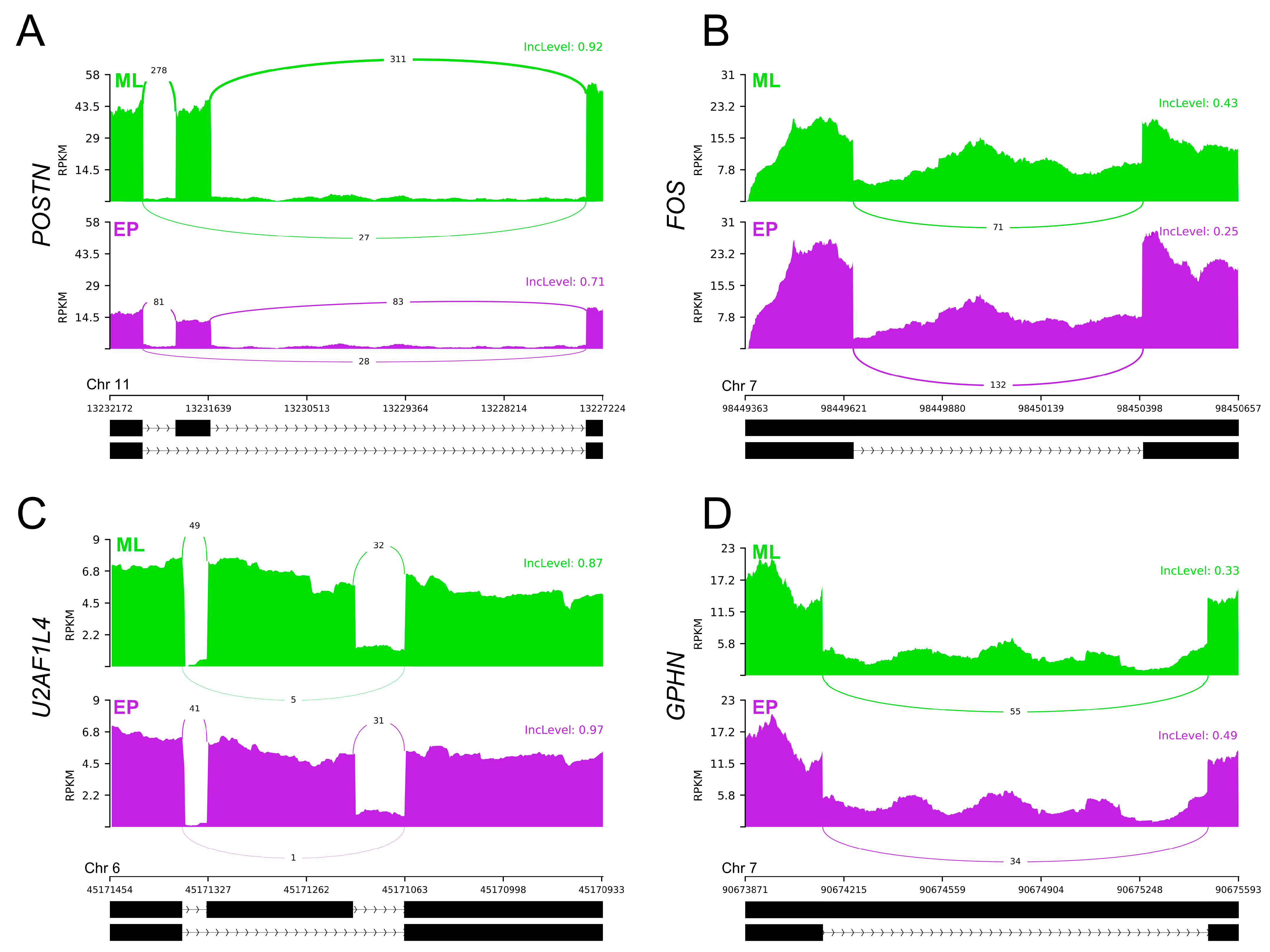
2.4. Differential Alternative Splicing Events of Differentially Expressed Genes
2.5. Single Nucleotide Variant Calling, Allele-Specific Expression Variations and RNA Editing
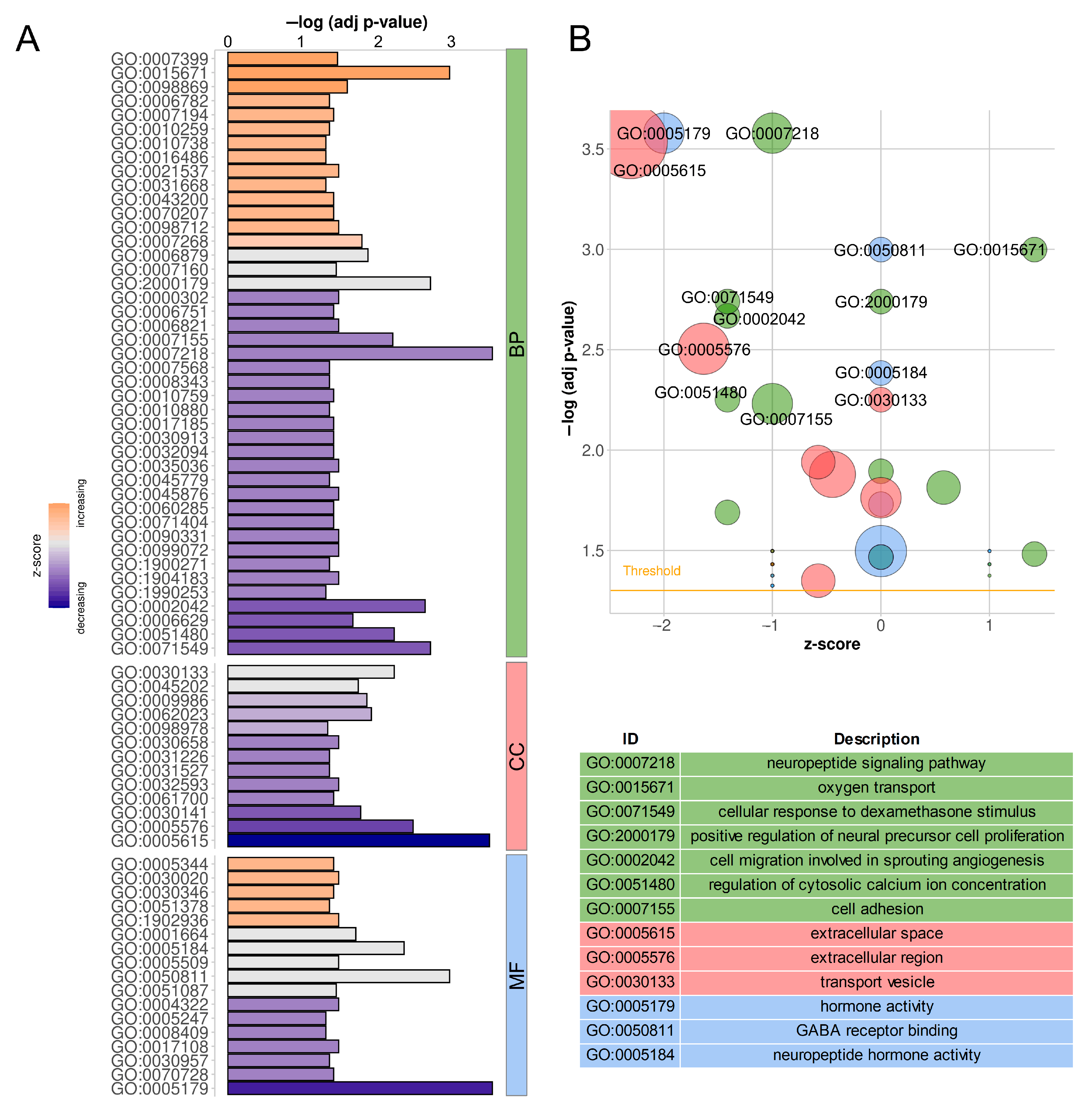
2.6. Functional Annotation of Target Protein-Coding Genes
2.7. Quantitative Real-Time PCR and PCR Validations
3. Discussion
3.1. Reception of the HPG Axis Signals—GnRH and Estrogens
3.2. Reception of Hypothalamic Neurotransmitters
3.3. Intracellular Signal Transduction—Secondary Messengers
3.4. Intracellular Signal Transduction—PI3K/AKT and MAPK Pathways
3.5. IL6 Signal Reception Modifications
3.6. Mechanisms Influencing the Course of Transcription and Translation Processes
3.7. Predicted Secretory Effects
4. Materials and Methods
4.1. Experimental Animals and Samples Collection
4.2. RNA Isolation, Library Preparation and High-Throughput Sequencing Procedure
4.3. Bioinformatic Analyses
4.3.1. Raw Reads Pre-Processing, Mapping to a Reference Genome and Differentially Expressed Genes Processing
4.3.2. Long Noncoding RNA Analyses
4.3.3. Differential Alternative Splicing Events Analysis
4.3.4. Single Nucleotide Variants’ Detection, Allele-Specific Expression Events’ Identification and RNA Editing Sites’ Analyses
4.3.5. Functional Annotation of Target Genes
4.4. Quantitative Real-Time PCR and PCR Validations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKAP11 | A-kinase anchoring protein 11 |
| ALDH3A2 | Aldehyde dehydrogenase 3 family member A2 |
| AQR | Aquarius intron-binding spliceosomal factor |
| ATRX | ATRX chromatin remodeler |
| BLTP2 | Bridge-like lipid transfer protein family member 2 |
| BMS1 | BMS1 ribosome biogenesis factor |
| BNIP5 | BCL2-interacting protein 5 |
| BTAF1 | B-TFIID TATA box-binding protein-associated factor 1 |
| C16orf72 | Chromosome 16 open reading frame 72 |
| C2CD5 | C2 calcium-dependent domain containing 5 |
| CACNA1D | Calcium voltage-gated channel subunit α1 D |
| CAMKK1 | Calcium/calmodulin-dependent protein kinase kinase 1 |
| CAVIN2 | Caveolae-associated protein 2 |
| CCP110 | Centriolar coiled-coil protein 110 |
| CD47 | Surface antigen CD47 |
| CDS1 | CDP-diacylglycerol synthase 1 |
| CELF4 | CUGBP elav-like family member 4 |
| CNST | Consortin, connexin-sorting protein |
| CPT1A | Carnitine palmitoyltransferase 1A |
| CUL* | Cullin *(4A, 7) |
| DERL3 | Derlin 3 |
| DGK*(E, G) | Diacylglycerol kinase *(ε, γ) |
| DISP1 | Dispatched RND transporter family member 1 |
| DMXL1 | Dmx like 1 |
| DNAJB4 | DnaJ heat shock protein family (Hsp40) member B4 |
| DUSP2 | Dual Specificity Phosphatase 2 |
| ELP1 | Elongator acetyltransferase complex subunit 1 |
| ESR2 | Estrogen receptor 2 |
| EXTL1 | Exostosin-likeglycosyltransferase 1 |
| FOS | Fos proto-oncogene, AP-1 transcription factor subunit |
| FSHB | Follicle-stimulating hormone subunit β |
| FZD3 | Frizzled class receptor 3 |
| GABARAPL1 | GABA type A receptor-associated protein-like1 |
| GABRA1 | GABA type A Receptor subunit α1 |
| GALP | Galanin-like peptide |
| GALT | Galactose-1-phosphate uridylyltransferase |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GMPS | Guanine monophosphate synthase |
| GNAL | G protein subunit α L |
| GnRHR | GnRH receptor |
| GPHN | Gephyrin |
| GRIK2 | Glutamate ionotropic receptor kainate type subunit 2 |
| GTF2I | General transcription factor IIi |
| HNRNPA3 | Heterogeneous nuclear ribonucleoprotein A3 |
| IGSF1 | Immunoglobulin superfamily member 1 |
| IL6 | Interleukin 6 |
| IL6R | Interleukin 6 receptor |
| IL6ST | Interleukin 6 cytokine family signal transducer |
| ISYNA1 | Inositol-3-phosphate synthase 1 |
| ITGAV | Integrin subunit alpha V |
| ITPR2 | Inositol-1,4,5-trisphosphate receptor type 2 |
| LDHD | Lactate dehydrogenase D |
| LHB | Luteinizing hormone subunit β |
| LSR | Lipolysis-stimulated lipoprotein receptor |
| MAGI1 | Membrane-associated guanylate kinase, WW and PDZ domain containing 1 |
| MAP1A | Microtubule-associated protein 1A |
| MAPK10 | Mitogen-activated protein kinase 10 |
| MARCH8 | Membrane-associated ring-CH-type finger 8 |
| MIGA1 | Mitoguardin 1 |
| MKNK1 | MAPK-interacting serine/threonine kinase 1 |
| MMADHC | Metabolism of cobalamin-associated D |
| MGRN1 | Mahogunin ring finger 1 |
| MTMR7 | Myotubularin-related protein 7 |
| NDUFA10 | NADH:ubiquinone oxidoreductase subunit A10 |
| NELFCD | Negative elongation factor complex member C/D |
| NRN1 | Neuritin 1 |
| OCIAD2 | OCIA domain containing 2 |
| PABPN1 | Poly(A)-binding protein nuclear 1 |
| PARG | Poly(ADP-ribose) glycohydrolase |
| PDE5A | Phosphodiesterase 5A |
| PEMT | Phosphatidylethanolamine N-methyltransferase |
| PI4K2B | Phosphatidylinositol 4-kinase type 2β |
| PLC*(B4, G1) | Phospholipase C *(β4, γ1) |
| POMC | Proopiomelanocortin |
| POSTN | Periostin |
| PPIA | Peptidylprolyl isomerase A |
| PPIL2 | Peptidylprolyl isomerase-like2 |
| PPP3CB | Protein phosphatase 3 catalytic subunit β |
| PRPF3 | Pre-mRNA processing factor 3 |
| PTPRN2 | Protein tyrosine phosphatase receptor type N2 |
| RBMX | RNA-binding motif protein X-linked |
| RBP4 | Retinol-binding protein 4 |
| RNMT | RNA guanine-7 methyltransferase |
| ROBO1 | Roundabout guidance receptor 1 |
| RTN4 | Reticulon 4 |
| SEPTIN2 | Septin 2 |
| SRSF* | Serine- and arginine-rich splicing factor *(4, 5, 7) |
| SIL1 | SIL1 nucleotide exchange factor |
| SLC*(1A2, 9B2) | Solute carrier family *(1 member 2, 9 member B2) |
| STAM2 | Signal transducing adaptor molecule 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| SUCLA2 | Succinate-CoA ligase ADP-forming subunit β |
| SYTL3 | Synaptotagmin-like3 |
| TAOK2 | TAO kinase 2 |
| TENM3 | Teneurin transmembrane protein 3 |
| TMED3 | Transmembrane P24 trafficking protein 3 |
| TMF1 | TATA element modulatory factor 1 |
| U*(1–6) | U*(1, 2, 4, 5, 6) small nuclear RNA |
| U2AF1L4 | U2 small nuclear RNA auxiliary factor 1-like4 |
| UBA6 | Ubiquitin-like modifier activating enzyme 6 |
| URGCP | Upregulator of cell proliferation |
| VGF | VGF nerve growth factor inducible |
| ZKSCAN5 | Zinc finger with KRAB and SCAN domains 5 |
| ZNF664 | Zinc-finger protein 664 |
References
- Dorton, A. The Pituitary Gland: Embryology, Physiology, and Pathophysiology. Neonatal Netw. 2000, 19, 9–17. [Google Scholar] [CrossRef]
- Arendt, D. The evolution of cell types in animals: Emerging principles from molecular studies. Nat. Rev. Genet. 2008, 9, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Pawson, A.J.; McNeilly, A.S. The pituitary effects of GnRH. Anim. Reprod. Sci. 2005, 88, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, H.D.; Henricks, D.M.; Handlin, D.L. Plasma estrogen, progesterone and luteinizing hormone prior to estrus and during early pregnancy in pigs. Endocrinology 1972, 91, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Ziecik, A.; Tilton, J.E.; Weigl, R.; Williams, G.L. Plasma luteinizing hormone during pregnancy in the pig. Anim. Reprod. Sci. 1983, 5, 213–218. [Google Scholar] [CrossRef]
- Ziecik, A.J.; Przygrodzka, E.; Jalali, B.M.; Kaczmarek, M.M. Regulation of the porcine corpus luteum during pregnancy. Reproduction 2018, 156, R57–R67. [Google Scholar] [CrossRef]
- Wiesak, T.; Hunter, M.G.; Foxcroft, G.R. Ovarian follicular development during early pregnancy in the pig. Anim. Reprod. Sci. 1992, 29, 17–24. [Google Scholar] [CrossRef]
- Peltoniemi, O.A.T.; Easton, B.G.; Love, R.J.; Klupiec, C.; Evans, G. Effect of chronic treatment with a GnRH agonist (Goserelin) on LH secretion and early pregnancy in gilts. Anim. Reprod. Sci. 1995, 40, 121–133. [Google Scholar] [CrossRef]
- Cassar, G. Hormonal control of pig reproduction. In Proceedings of the London Swine Conference—Tools of the Trade, London, ON, Canada, 1–2 April 2009; pp. 137–139. [Google Scholar]
- Van De Wiel, D.F.M.; Erkens, J.; Koops, W.; Vos, E.; Van Landeghem, A.A.J. Periestrous and Midluteal Time Courses of Circulating LH, FSH, Prolactin, Estradiol-17β and Progesterone in the Domestic Pig. Biol. Reprod. 1981, 24, 223–233. [Google Scholar] [CrossRef]
- Anderson, L.L. Pigs. Reproduction in Farm Animals; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2016; pp. 182–191. [Google Scholar]
- Carr, B.R.; Parker, C.R.; Madden, J.D.; MacDonald, P.C.; Porter, J.C. Maternal plasma adrenocorticotropin and cortisol relationships throughout human pregnancy. Am. J. Obstet. Gynecol. 1981, 139, 416–422. [Google Scholar] [CrossRef]
- Atkinson, H.C.; Waddell, B.J. The hypothalamic-pituitary-adrenal axis in rat pregnancy and lactation: Circadian variation and interrelationship of plasma adrenocorticotropin and corticosterone. Endocrinology 1995, 136, 512–520. [Google Scholar] [CrossRef]
- Jiang, Q.; Wong, A.O.L. Signal transduction mechanisms for autocrine/paracrine regulation of somatolactin-α secretion and synthesis in carp pituitary cells by somatolactin-α and -β. AJP Endocrinol. Metab. 2013, 304, E176–E186. [Google Scholar] [CrossRef] [PubMed]
- Peillon, F.; Le Dafniet, M.; Pagesy, P.; Yuan Li, J.; Benlot, C.; Brandi, A.M.; Joubert, D. Neuropeptides of Anterior Pituitary Origin: Autocrine or Paracrine Functions? Pathol. Res. Pract. 1991, 187, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, X.; Ko, W.K.W.; Wong, A.O.L. Evidence for a novel intrapituitary autocrine/paracrine feedback loop regulating growth hormone synthesis and secretion in grass carp pituitary cells by functional interactions between gonadotrophs and somatotrophs. Endocrinology 2004, 145, 5548–5559. [Google Scholar] [CrossRef] [PubMed]
- Bilezikjian, L.M.; Blount, A.L.; Leal, A.M.O.; Donaldson, C.J.; Fischer, W.H.; Vale, W.W. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Mol. Cell. Endocrinol. 2004, 225, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Arzt, E.; Stalla, G.K. Cytokines: Autocrine and Paracrine Roles in the Anterior Pituitary. Neuroimmunomodulation 1996, 3, 28–34. [Google Scholar] [CrossRef]
- Ing, N.H. Steroid Hormones Regulate Gene Expression Posttranscriptionally by Altering the Stabilities of Messenger RNAs. Biol. Reprod. 2005, 72, 1290–1296. [Google Scholar] [CrossRef]
- Siawrys, G.; Kaminski, T.; Smolinska, N.; Przala, J. Expression of leptin and long form of leptin receptor genes and proteins in pituitary of cyclic and pregnant pigs. J. Physiol. Pharmacol. 2007, 58, 845–857. [Google Scholar]
- Kiezun, M.; Smolinska, N.; Maleszka, A.; Dobrzyn, K.; Szeszko, K.; Kaminski, T. Adiponectin expression in the porcine pituitary during the estrous cycle and its effect on LH and FSH secretion. Am. J. Physiol. Metab. 2014, 307, E1038–E1046. [Google Scholar] [CrossRef]
- Kisielewska, K.; Rytelewska, E.; Gudelska, M.; Kiezun, M.; Dobrzyn, K.; Bogus-Nowakowska, K.; Kaminska, B.; Smolinska, N.; Kaminski, T. Relative abundance of chemerin mRNA transcript and protein in pituitaries of pigs during the estrous cycle and early pregnancy and associations with LH and FSH secretion during the estrous cycle. Anim. Reprod. Sci. 2020, 219, 106532. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, H.; Zeng, B.; Liu, J.; Luo, J.; Chen, T.; Sun, J.; Xi, Q.; Zhang, Y. An Exploration of Non-Coding RNAs in Extracellular Vesicles Delivered by Swine Anterior Pituitary. Front. Genet. 2021, 12, 2326. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, H.; Wang, Y.; Liu, C.; Wang, C.; Guo, J. Transcriptomic analysis of the porcine endometrium during embryo implantation. Genes 2015, 6, 1330–1346. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Bick, J.; Ulbrich, S.E.; Bauersachs, S. Cell type-specific analysis of transcriptome changes in the porcine endometrium on Day 12 of pregnancy. BMC Genom. 2018, 19, 459. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Seo, H.; Choi, Y.; Shim, J.; Kim, H.; Lee, C.K.; Ka, H. Microarray analysis of gene expression in the uterine endometrium during the implantation period in pigs. Asian Australas. J. Anim. Sci. 2012, 25, 1102–1116. [Google Scholar] [CrossRef]
- Kim, J.M.; Park, J.E.; Yoo, I.; Han, J.; Kim, N.; Lim, W.J.; Cho, E.S.; Choi, B.; Choi, S.; Kim, T.H.; et al. Integrated transcriptomes throughout swine oestrous cycle reveal dynamic changes in reproductive tissues interacting networks. Sci. Rep. 2018, 8, 5436. [Google Scholar] [CrossRef] [PubMed]
- Zmijewska, A.; Czelejewska, W.; Waszkiewicz, E.M.; Gajewska, A.; Okrasa, S.; Franczak, A. Transcriptomic analysis of the porcine anterior pituitary gland during the peri-implantation period. Reprod. Domest. Anim. 2020, 55, 1434–1445. [Google Scholar] [CrossRef]
- Zmijewska, A.; Czelejewska, W.; Drzewiecka, E.M.; Franczak, A. Transcriptome profile of the anterior pituitary gland in pigs during maternal recognition of pregnancy. Theriogenology 2022, 116544, 310–321. [Google Scholar] [CrossRef]
- Perry, J.S.; Rowlands, I.W. Early pregnancy in the pig. Reproduction 1962, 4, 175–188. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2019, 48, D682–D688. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Lu, Q.; Ren, S.; Lu, M.; Zhang, Y.; Zhu, D.; Zhang, X.; Li, T. Computational prediction of associations between long non-coding RNAs and proteins. BMC Genom. 2013, 14, 651. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.-X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, Y.; Shen, S.; Lin, L.; Xing, Y. rMATS-DVR: rMATS discovery of differential variants in RNA. Bioinformatics 2017, 33, 2216–2217. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Feng, X.; Tang, Z.; Li, S.C. Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues. Genes 2019, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Carbon, S.; Dietze, H.; Lewis, S.E.; Mungall, C.J.; Munoz-Torres, M.C.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Fey, P.; Thomas, P.D.; et al. Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Hahm, S.; Mizuno, T.M.; Wu, T.J.; Wisor, J.P.; Priest, C.A.; Kozak, C.A.; Boozer, C.N.; Peng, B.; McEvoy, R.C.; Good, P.; et al. Targeted deletion of the Vgf gene indicates that the encoded secretory peptide precursor plays a novel role in the regulation of energy balance. Neuron 1999, 23, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.G.; Wang, Q.; Jia, J.; Chikina, M.; Pincas, H.; Dolios, G.; Sasaki, K.; Wang, R.; Minamino, N.; Salton, S.R.J.; et al. Characterization of Gonadotrope Secretoproteome Identifies Neurosecretory Protein VGF-derived Peptide Suppression of Follicle-stimulating Hormone Gene Expression. J. Biol. Chem. 2016, 291, 21322. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, P.A.; Smiljanic, K.; Maso Prévide, R.; Iben, J.R.; Li, T.; Rokic, M.B.; Sherman, A.; Coon, S.L.; Stojilkovic, S.S. Cell Type- and Sex-Dependent Transcriptome Profiles of Rat Anterior Pituitary Cells. Front. Endocrinol. Lausanne 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, X.; Liu, G.; Fu, J.; Wang, A. Expression of αV and β3 integrin subunits during implantation in pig. Mol. Reprod. Dev. 2007, 74, 1379–1385. [Google Scholar] [CrossRef]
- Daude, N.; Lestage, J.; Reichardt, J.K.V.; Petry, K.G. Expression of Galactose-1-Phosphate Uridyltransferase in the Anterior Pituitary of Rat during the Estrous Cycle. Neuroendocrinology 1996, 64, 42–48. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ERβ: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- He, J.; Wei, C.; Li, Y.; Liu, Y.; Wang, Y.; Pan, J.; Liu, J.; Wu, Y.; Cui, S. Zearalenone and alpha-zearalenol inhibit the synthesis and secretion of pig follicle stimulating hormone via the non-classical estrogen membrane receptor GPR30. Mol. Cell. Endocrinol. 2018, 461, 43–54. [Google Scholar] [CrossRef]
- Shaw, N.D.; Histed, S.N.; Srouji, S.S.; Yang, J.; Lee, H.; Hall, J.E. Estrogen Negative Feedback on Gonadotropin Secretion: Evidence for a Direct Pituitary Effect in Women. J. Clin. Endocrinol. Metab. 2010, 95, 1955. [Google Scholar] [CrossRef]
- O’Neill, P.A.; Davies, M.P.A.; Shaaban, A.M.; Innes, H.; Torevell, A.; Sibson, D.R.; Foster, C.S. Wild-type oestrogen receptor beta (ERβ1) mRNA and protein expression in Tamoxifen-treated post-menopausal breast cancers. Br. J. Cancer 2004, 91, 1694. [Google Scholar] [CrossRef]
- Kato, J.; Hirata, S.; Koh, T.; Yamada-Mouri, N.; Hoshi, K.; Okinaga, S. The multiple untranslated first exons and promoters system of the oestrogen receptor gene in the brain and peripheral tissues of the rat and monkey and the developing rat cerebral cortex. J. Steroid Biochem. Mol. Biol. 1998, 65, 281–293. [Google Scholar] [CrossRef]
- Smith, L.; Coleman, L.J.; Cummings, M.; Satheesha, S.; Shaw, S.O.; Speirs, V.; Hughes, T.A. Expression of oestrogen receptor β isoforms is regulated by transcriptional and post-transcriptional mechanisms. Biochem. J. 2010, 429, 283–290. [Google Scholar] [CrossRef]
- Mitchell, R.; Grieve, G.; Dow, R.; Fink, G. Endogenous GABA Receptor Ligands in Hypophysial Portal Blood. Neuroendocrinology 1983, 37, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.C.; Reymond, M.J.; Arita, J.; Sissom, J.F. Secretion of hypothalamic dopamine into the hypophysial portal vasculature: An overview. Methods Enzymol. 1983, 103, 607–618. [Google Scholar] [CrossRef]
- Caffe, A.R. Architecture of the mammalian pituitary cholinergic system with observations on a putative blood acetylcholine sensor. Histol. Histopathol. 1996, 11, 537–551. [Google Scholar] [PubMed]
- Feldman, S.; Weidenfeld, J. Hypothalamic mechanisms mediating glutamate effects on the hypothalamo-pituitary-adrenocortical axis. J. Neural Transm. 1997, 104, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Zemková, H.; Stojilkovic, S.S. Neurotransmitter receptors as signaling platforms in anterior pituitary cells. Mol. Cell. Endocrinol. 2018, 463, 49–64. [Google Scholar] [CrossRef]
- Anderson, R.A.; Mitchell, R. Effects of β-aminobutyric acid receptor agonists on the secretion of growth hormone, luteinizing hormone, adrenocorticotrophic hormone and thyroid-stimulating hormone from the rat pituitary gland in vitro. J. Endocrinol. 1986, 108, 1–8. [Google Scholar] [CrossRef]
- Zemkova, H.W.; Bjelobaba, I.; Tomic, M.; Zemkova, H.; Stojilkovic, S.S. Molecular, pharmacological and functional properties of GABAA receptors in anterior pituitary cells. J. Physiol. 2008, 586, 3097–3111. [Google Scholar] [CrossRef]
- Lamberts, S.W.J.; Macleod, R.M. Studies on the Mechanism of the GABA-Mediated Inhibition of Prolactin Secretion. Proc. Soc. Exp. Biol. Med. 2016, 158, 10–13. [Google Scholar] [CrossRef]
- Niimi, M.; Sato, M.; Murao, K.; Takahara, J.; Kawanishi, K. Effect of Excitatory Amino Acid Receptor Agonists on Secretion of Growth Hormone as Assessed by the Reverse Hemolytic Plaque Assay. Neuroendocrinology 1994, 60, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Pampillo, M.; Theas, S.; Duvilanski, B.; Seilicovich, A.; Lasaga, M. Effect of ionotropic and metabotropic glutamate agonists and D-aspartate on prolactin release from anterior pituitary cells. Exp. Clin. Endocrinol. Diabetes 2002, 110, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Zanisi, M.; Galbiati, M.; Messi, E.; Martini, L. The Anterior Pituitary Gland as a Possible Site of Action of Kainic Acid. Proc. Soc. Exp. Biol. Med. 2016, 206, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Berger, U.V.; Hediger, M.A. Distribution of the glutamate transporters GLAST and GLT-1 in rat circumventricular organs, meninges, and dorsal root ganglia. J. Comp. Neurol. 2000, 421, 385–399. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Vanoni, C.; Bossi, M.; Massari, S.; Basudev, H.; Longhi, R.; Pietrini, G. The GLT-1 and GLAST Glutamate Transporters Are Expressed on Morphologically Distinct Astrocytes and Regulated by Neuronal Activity in Primary Hippocampal Cocultures. J. Neurochem. 2000, 75, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, P.A.; Sherman, A.; Stojilkovic, S.S. Common and diverse elements of ion channels and receptors underlying electrical activity in endocrine pituitary cells. Mol. Cell. Endocrinol. 2018, 463, 23–36. [Google Scholar] [CrossRef]
- Ashworth, R.; Hinkle, P.M. Thyrotropin-releasing hormone-induced intracellular calcium responses in individual rat lactotrophs and thyrotrophs. Endocrinology 1996, 137, 5205–5212. [Google Scholar] [CrossRef]
- Zemková, H.; Vaneěcček, J. Differences in Gonadotropin-Releasing Hormone-Induced Calcium Signaling between Melatonin-Sensitive and Melatonin-Insensitive Neonatal Rat Gonadotrophs. Endocrinology 2000, 141, 1017–1026. [Google Scholar] [CrossRef]
- Kanasaki, H.; Oride, A.; Hara, T.; Mijiddorj, T.; Sukhbaatar, U.; Kyo, S. Interactions between Two Different G Protein-Coupled Receptors in Reproductive Hormone-Producing Cells: The Role of PACAP and Its Receptor PAC1R. Int. J. Mol. Sci. 2016, 17, 1635. [Google Scholar] [CrossRef]
- Stojilkovic, S.S.; Prévide, R.M.; Sherman, A.S.; Fletcher, P.A. Pituitary corticotroph identity and receptor-mediated signaling: A transcriptomics perspective. Curr. Opin. Endocr. Metab. Res. 2022, 25, 100364. [Google Scholar] [CrossRef]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 51, 3792. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ray, A.; Barpanda, A.; Dash, A.; Gupta, I.; Nissa, M.U.; Zhu, H.; Shah, A.; Duttagupta, S.P.; Goel, A.; et al. Evaluation of autoantibody signatures in pituitary adenoma patients using human proteome arrays. Proteom. Clin. Appl. 2022, 16, 2100111. [Google Scholar] [CrossRef]
- DeAlmeida, V.I.; Mayo, K.E. The growth hormone-releasing hormone receptor. Vitam. Horm. 2001, 63, 233–276. [Google Scholar] [CrossRef]
- Zemkova, H.; Tomić, M.; Kucka, M.; Aguilera, G.; Stojilkovic, S.S. Spontaneous and CRH-Induced Excitability and Calcium Signaling in Mice Corticotrophs Involves Sodium, Calcium, and Cation-Conducting Channels. Endocrinology 2016, 157, 1576–1589. [Google Scholar] [CrossRef] [PubMed]
- Vezzosi, D.; Bertherat, J. Phosphodiesterases in endocrine physiology and disease. Eur. J. Endocrinol. 2011, 165, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Corvol, J.C.; Studler, J.M.; Schonn, J.S.; Girault, J.A.; Hervé, D. Galpha(olf) is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J. Neurochem. 2001, 76, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. G-proteins as transducers in transmembrane signalling. Prog. Biophys. Mol. Biol. 2003, 83, 101–130. [Google Scholar] [CrossRef]
- Tahir, S.A.; Gao, J.; Miura, Y.; Blando, J.; Tidwell, R.S.S.; Zhao, H.; Subudhi, S.K.; Tawbi, H.; Keung, E.; Wargo, J.; et al. Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc. Natl. Acad. Sci. USA 2019, 116, 22246–22251. [Google Scholar] [CrossRef]
- Ben-Jonathan, N.; Hnasko, R. Dopamine as a Prolactin (PRL) Inhibitor. Endocr. Rev. 2001, 22, 724–763. [Google Scholar] [CrossRef]
- Roof, A.K.; Gutierrez-Hartmann, A. Consider the context: Ras/ERK and PI3K/AKT/mTOR signaling outcomes are pituitary cell type-specific. Mol. Cell. Endocrinol. 2018, 463, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Ontsuka, K.; Kotobuki, Y.; Shiraishi, H.; Serada, S.; Ohta, S.; Tanemura, A.; Yang, L.; Fujimoto, M.; Arima, K.; Suzuki, S.; et al. Periostin, a matricellular protein, accelerates cutaneous wound repair by activating dermal fibroblasts. Exp. Dermatol. 2012, 21, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, K.; Arima, K.; Ohta, S.; Suzuki, S.; Inamitsu, M.; Yamamoto, K. Periostin in Allergic Inflammation. Allergol. Int. 2014, 63, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Yelamanchi, S.D.; Tyagi, A.; Mohanty, V.; Dutta, P.; Korbonits, M.; Chavan, S.; Advani, J.; Madugundu, A.K.; Dey, G.; Datta, K.K.; et al. Proteomic Analysis of the Human Anterior Pituitary Gland. OMICS 2018, 22, 759. [Google Scholar] [CrossRef] [PubMed]
- Labrèche, C. The Role of Periostin in ErbB2-Driven Mammary Tumorigenesis and Its Gene Regulation in ErbB2+ Cancer Cells. Ph.D. Thesis, University of Ottawa, Ottawa, ON, Canada, 2021. [Google Scholar]
- Ben-Shlomo, A.; Melmed, S. Hypothalamic Regulation of Anterior Pituitary Function. In The Pituitary; Elsevier: Amsterdam, The Netherlands, 2011; pp. 21–45. ISBN 9780123809261. [Google Scholar]
- Coss, D. Regulation of reproduction via tight control of gonadotropin hormone levels. Mol. Cell. Endocrinol. 2018, 463, 116–130. [Google Scholar] [CrossRef]
- Mulvaney, J.M.; Roberson, M.S. Divergent Signaling Pathways Requiring Discrete Calcium Signals Mediate Concurrent Activation of Two Mitogen-activated Protein Kinases by Gonadotropin-releasing Hormone. J. Biol. Chem. 2000, 275, 14182–14189. [Google Scholar] [CrossRef]
- Wei, W.; Jiao, Y.; Postlethwaite, A.; Stuart, J.M.; Wang, Y.; Sun, D.; Gu, W. Dual-specificity phosphatases 2: Surprising positive effect at the molecular level and a potential biomarker of diseases. Genes Immun. 2013, 14, 1–6. [Google Scholar] [CrossRef]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 Are Essential for Constitutive and Inducible Phosphorylation of Eukaryotic Initiation Factor 4E but Not for Cell Growth or Development. Mol. Cell. Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef]
- Kilka, S.; Erdmann, F.; Migdoll, A.; Fischer, G.; Weiwad, M. The proline-rich N-terminal sequence of calcineurin Abeta determines substrate binding. Biochemistry 2009, 48, 1900–1910. [Google Scholar] [CrossRef]
- Brinegar, A.E.; Xia, Z.; Loehr, J.A.; Li, W.; Rodney, G.G.; Cooper, T.A. Extensive alternative splicing transitions during postnatal skeletal muscle development are required for calcium handling functions. Elife 2017, 6, e27192. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta Rev. Cancer 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.G.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D.; Curran, T.; Verma, I.M. c-fos protein can induce cellular transformation: A novel mechanism of activation of a cellular oncogene. Cell 1984, 36, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Boutillier, A.L.; Sassone-Corsi, P.; Loeffler, J.P. The Protooncogene c-fos Is Induced by Corticotropin-Releasing Factor and Stimulates Proopiomelanocortin Gene Transcription in Pituitary Cells. Mol. Endocrinol. 1991, 5, 1301–1310. [Google Scholar] [CrossRef]
- Chung, H.O.; Kato, T.; Kato, Y. Molecular cloning of c-jun and c-fos cDNAs from porcine anterior pituitary and their involvement in gonadotropin-releasing hormone stimulation. Mol. Cell. Endocrinol. 1996, 119, 75–82. [Google Scholar] [CrossRef]
- Allen, D.L.; Mitchner, N.A.; Uveges, T.E.; Nephew, K.P.; Khan, S.; Ben-Jonathan, N. Cell-Specific Induction of c-fos Expression in the Pituitary Gland by Estrogen. Endocrinology 1997, 138, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Van Haasteren, G.; Li, S.; Ryser, S.; Schlegel, W. Essential contribution of intron sequences to Ca2+-dependent activation of c-fos transcription in pituitary cells. Neuroendocrinology 2000, 72, 368–378. [Google Scholar] [CrossRef]
- Elkeles, A.; Juven-Gershon, T.; Israeli, D.; Wilder, S.; Zalcenstein, A.; Oren, M. The c-fos Proto-Oncogene Is a Target for Transactivation by the p53 Tumor Suppressor. Mol. Cell. Biol. 1999, 19, 2594. [Google Scholar] [CrossRef]
- Listerman, I.; Sapra, A.K.; Neugebauer, K.M. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat. Struct. Mol. Biol. 2006, 13, 815–822. [Google Scholar] [CrossRef]
- Vargas, D.Y.; Shah, K.; Batish, M.; Levandoski, M.; Sinha, S.; Marras, S.A.E.; Schedl, P.; Tyagi, S. Single-Molecule Imaging of Transcriptionally Coupled and Uncoupled Splicing. Cell 2011, 147, 1054–1065. [Google Scholar] [CrossRef]
- Przygrodzka, E.; Witek, K.J.; Kaczmarek, M.M.; Andronowska, A.; Ziecik, A.J. Expression of factors associated with apoptosis in the porcine corpus luteum throughout the luteal phase of the estrous cycle and early pregnancy: Their possible involvement in acquisition of luteolytic sensitivity. Theriogenology 2015, 83, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Ohmichi, M.; Hirota, K.; Koike, K.; Kurachi, H.; Ohtsuka, S.; Matsuzaki, N.; Yamaguchi, M.; Miyake, A.; Tanizawa, O. Binding sites for interleukin-6 in the anterior pituitary gland. Neuroendocrinology 1992, 55, 199–203. [Google Scholar] [CrossRef]
- Gautron, L.; Lafon, P.; Tramu, G.; Layé, S. In vivo activation of the interleukin-6 receptor/gp130 signaling pathway in pituitary corticotropes of lipopolysaccharide-treated rats. Neuroendocrinology 2003, 77, 32–43. [Google Scholar] [CrossRef]
- Schwartz, J. Intercellular communication in the anterior pituitary. Endocr. Rev. 2000, 21, 488–513. [Google Scholar] [CrossRef] [PubMed]
- Spangelo, B.L.; Judd, A.M.; Isakson, P.C.; Macleod, R.M. Interleukin-6 stimulates anterior pituitary hormone release in vitro. Endocrinology 1989, 125, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Matsuzaki, N.; Hirota, K.; Miyake, A.; Tanizawa, O. Interleukin 6 possibly induced by interleukin 1β in the pituitary gland stimulates the release of gonadotropins and prolactin. Acta Endocrinol. Copenh. 1990, 122, 201–205. [Google Scholar] [CrossRef]
- Lyson, K.; McCann, S.M. The Effect of Interleukin-6 on Pituitary Hormone Release in vivo and in vitro. Neuroendocrinology 1991, 54, 262–266. [Google Scholar] [CrossRef]
- Fukata, J.; Usui, T.; Naitoh, Y.; Nakai, Y.; Imura, H. Effects of recombinant human interleukin-1α, -1β, 2 and 6 on ACTH synthesis and release in the mouse pituitary tumour cell line AtT-20. J. Endocrinol. 1989, 122, 33–39. [Google Scholar] [CrossRef]
- Callahan, T.A.; Piekut, D.T. Differential Fos expression induced by IL-1β and IL-6 in rat hypothalamus and pituitary gland. J. Neuroimmunol. 1997, 73, 207–211. [Google Scholar] [CrossRef]
- Arzt, E. gp130 cytokine signaling in the pituitary gland: A paradigm for cytokine–neuro-endocrine pathways. J. Clin. Investig. 2001, 108, 1729–1733. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Melmed, S. Critical role for STAT3 in murine pituitary adrenocorticotropin hormone leukemia inhibitory factor signaling. J. Biol. Chem. 1999, 274, 10723–10730. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Zatelli, M.C.; Melmed, S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J. Clin. Investig. 2000, 106, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Glover-Cutter, K.; Kim, S.; Espinosa, J.; Bentley, D.L. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat. Struct. Mol. Biol. 2008, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Aregger, M.; Cowling, V.H. Human cap methyltransferase (RNMT) N-terminal non-catalytic domain mediates recruitment to transcription initiation sites. Biochem. J. 2013, 455, 67. [Google Scholar] [CrossRef]
- Martínez-Montes, A.M.; Fernández, A.; Pérez-Montarelo, D.; Alves, E.; Benítez, R.M.; Nuñez, Y.; Óvilo, C.; Ibañez-Escriche, N.; Folch, J.M.; Fernández, A.I. Using RNA-Seq SNP data to reveal potential causal mutations related to pig production traits and RNA editing. Anim. Genet. 2017, 48, 151–165. [Google Scholar] [CrossRef]
- Zhou, Z.; Licklider, L.J.; Gygi, S.P.; Reed, R. Comprehensive proteomic analysis of the human spliceosome. Nature 2002, 419, 182–185. [Google Scholar] [CrossRef]
- Chen, Y.I.G.; Moore, R.E.; Ge, H.Y.; Young, M.K.; Lee, T.D.; Stevens, S.W. Proteomic analysis of in vivo-assembled pre-mRNA splicing complexes expands the catalog of participating factors. Nucleic Acids Res. 2007, 35, 3928–3944. [Google Scholar] [CrossRef]
- Berg, M.G.; Singh, L.N.; Younis, I.; Liu, Q.; Pinto, A.M.; Kaida, D.; Zhang, Z.; Cho, S.; Sherrill-Mix, S.; Wan, L.; et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell 2012, 150, 53–64. [Google Scholar] [CrossRef]
- Nguyen, T.H.D.; Galej, W.P.; Bai, X.C.; Savva, C.G.; Newman, A.J.; Scheres, S.H.W.; Nagai, K. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 2015, 523, 47–52. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.; Dienstbier, M.; Cowley, S.A.; Vazquez, P.; Drozdz, M.; Taylor, S.; James, W.S.; Murphy, S. Differentially expressed, variant U1 snRNAs regulate gene expression in human cells. Genome Res. 2013, 23, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Zeng, Y.; Yang, R.; Xu, H.; Chen, Z.; Zhong, J.; Xie, H.; Xu, Y.; Zeng, X. U6 is not a suitable endogenous control for the quantification of circulating microRNAs. Biochem. Biophys. Res. Commun. 2014, 454, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Ma, N.; Xu, Y.; Jiang, L.; Yang, J.; Wang, C.; Jiao, Y.; Gao, X. Differential distribution of U6 (RNU6-1) expression in human carcinoma tissues demonstrates the requirement for caution in the internal control gene selection for microRNA quantification. Int. J. Mol. Med. 2015, 36, 1400–1408. [Google Scholar] [CrossRef]
- Yin, Y.; Lu, J.Y.; Zhang, X.; Shao, W.; Xu, Y.; Li, P.; Hong, Y.; Cui, L.; Shan, G.; Tian, B.; et al. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature 2020, 580, 147–150. [Google Scholar] [CrossRef]
- Li, S.; Zhang, S.; Huang, M.; Hu, H.; Xie, Y. U1RNP/lncRNA/Transcription Cycle Axis Promotes Tumorigenesis of Hepatocellular Carcinoma. Diagnostics 2022, 12, 1133. [Google Scholar] [CrossRef] [PubMed]
- Goldammer, G.; Neumann, A.; Strauch, M.; Müller-McNicoll, M.; Heyd, F.; Preußner, M. Characterization of cis-acting elements that control oscillating alternative splicing. RNA Biol. 2018, 15, 1081–1092. [Google Scholar] [CrossRef]
- Dasgupta, T.; Ladd, A.N. The importance of CELF control: Molecular and biological roles of the CUG-BP, Elav-like family of RNA binding proteins. Wiley Interdiscip. Rev. RNA 2012, 3, 104. [Google Scholar] [CrossRef]
- Wagnon, J.L.; Briese, M.; Sun, W.; Mahaffey, C.L.; Curk, T.; Rot, G.; Ule, J.; Frankel, W.N. CELF4 Regulates Translation and Local Abundance of a Vast Set of mRNAs, Including Genes Associated with Regulation of Synaptic Function. PLoS Genet. 2012, 8, e1003067. [Google Scholar] [CrossRef]
- Yang, Y.; Mahaffey, C.L.; Bérubé, N.; Maddatu, T.P.; Cox, G.A.; Frankel, W.N. Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genet. 2007, 3, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Kühn, U.; Gündel, M.; Knoth, A.; Kerwitz, Y.; Rüdel, S.; Wahle, E. Poly(A) tail length is controlled by the nuclear Poly(A)-binding protein regulating the interaction between Poly(A) polymerase and the cleavage and polyadenylation specificity factor. J. Biol. Chem. 2009, 284, 22803–22814. [Google Scholar] [CrossRef]
- De Klerk, E.; Venema, A.; Anvar, S.Y.; Goeman, J.J.; Hu, O.H.; Trollet, C.; Dickson, G.; Den Dunnen, J.T.; Van Der Maarel, S.M.; Raz, V.; et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 2012, 40, 9089–9101. [Google Scholar] [CrossRef]
- Jenal, M.; Elkon, R.; Loayza-Puch, F.; Van Haaften, G.; Kühn, U.; Menzies, F.M.; Vrielink, J.A.F.O.; Bos, A.J.; Drost, J.; Rooijers, K.; et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012, 149, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, D.; Pal, G.; Beaulieu, Y.B.; Chabot, B.; Bachand, F. Regulated Intron Retention and Nuclear Pre-mRNA Decay Contribute to PABPN1 Autoregulation. Mol. Cell. Biol. 2015, 35, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, X.; Gygi, S.P.; Harper, J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007, 447, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Day, R. Proopiomelanocortin. In Encyclopedia of Neuroscience; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1139–1141. [Google Scholar]
- Hermus, A.R.M.M.; Sweep, C.G.J. Cytokines and the hypothalamic-pituitary-adrenal axis. J. Steroid Biochem. Mol. Biol. 1990, 37, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.G.; Weatherbee, J.; Sharp, B.M. A Central Mechanism Is Involved in the Secretion of ACTH in Response to IL-6 in Rats: Comparison to and Interaction with IL-1β. Neuroendocrinology 1992, 56, 516–525. [Google Scholar] [CrossRef]
- Späth-Schwalbe, E.; Born, J.; Schrezenmeier, H.; Bornstein, S.R.; Stromeyer, P.; Drechsler, S.; Fehm, H.L.; Porzsolt, F. Interleukin-6 stimulates the hypothalamus-pituitary-adrenocortical axis in man. J. Clin. Endocrinol. Metab. 1994, 79, 1212–1214. [Google Scholar] [CrossRef]
- Brunton, P.J.; Russell, J.A.; Douglas, A.J. Adaptive Responses of the Maternal Hypothalamic-Pituitary-Adrenal Axis during Pregnancy and Lactation. J. Neuroendocrinol. 2008, 20, 764–776. [Google Scholar] [CrossRef]
- Nepomnaschy, P.A.; Welch, K.B.; McConnell, D.S.; Low, B.S.; Strassmann, B.I.; England, B.G. Cortisol levels and very early pregnancy loss in humans. Proc. Natl. Acad. Sci. USA 2006, 103, 3938–3942. [Google Scholar] [CrossRef]
- Parker, V.J.; Douglas, A.J. Stress in early pregnancy: Maternal neuro-endocrine-immune responses and effects. J. Reprod. Immunol. 2010, 85, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Leaflet, A.S.R. Reproductive Biology of Pigs. Anim. Ind. Rep. 2009, AS655, ASL R2443. [Google Scholar] [CrossRef]
- Ka, H.; Seo, H.; Choi, Y.; Yoo, I.; Han, J. Endometrial response to conceptus-derived estrogen and interleukin-1β at the time of implantation in pigs. J. Anim. Sci. Biotechnol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Akins, E.L.; Morrissette, M.C. Gross ovarian changes during estrous cycle of swine. Am. J. Vet. Res. 1968, 29, 1953–1957. [Google Scholar]
- Anderson, L.L. Growth, protein content and distribution of early pig embryos. Anat. Rec. 1978, 190, 143–153. [Google Scholar] [CrossRef]
- Shen, Y.; Mao, H.; Huang, M.; Chen, L.; Chen, J.; Cai, Z.; Wang, Y.; Xu, N. Long Noncoding RNA and mRNA Expression Profiles in the Thyroid Gland of Two Phenotypically Extreme Pig Breeds Using Ribo-Zero RNA Sequencing. Genes 2016, 7, 34. [Google Scholar] [CrossRef]
- Paukszto, L.; Mikolajczyk, A.; Jastrzebski, J.P.; Majewska, M.; Dobrzyn, K.; Kiezun, M.; Smolinska, N.; Kaminski, T. Transcriptome, Spliceosome and Editome Expression Patterns of the Porcine Endometrium in Response to a Single Subclinical Dose of Salmonella Enteritidis Lipopolysaccharide. Int. J. Mol. Sci. 2020, 21, 4217. [Google Scholar] [CrossRef] [PubMed]
- Makowczenko, K.G.; Jastrzebski, J.P.; Paukszto, L.; Dobrzyn, K.; Kiezun, M.; Smolinska, N.; Kaminski, T. Chemerin Impact on Alternative mRNA Transcription in the Porcine Luteal Cells. Cells 2022, 11, 715. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 24 July 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Jakobi, T.; Uvarovskii, A.; Dieterich, C. Detect Module—Circtools Documentation. Available online: https://docs.circ.tools/en/latest/Detect.html (accessed on 15 September 2020).
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Pertea, G. prepDE.py. Available online: https://github.com/gpertea/stringtie/blob/master/prepDE.py (accessed on 11 September 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Love, M.I.; Anders, S.; Huber, W. Analyzing RNA-seq Data with DESeq2. Available online: http://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#countmat (accessed on 6 November 2018).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.; Schiffer, L.; Re, A.; Azhar, R.; Basunia, A.; Rodriguez, C.; Chan, T.; Chapman, P.; Davis, S.R.; Gomez-Cabrero, D.; et al. Software for the Integration of Multiomics Experiments in Bioconductor. Cancer Res. 2017, 77, e39–e42. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tseng, Y.-T.; Xing, Y. rmats2sashimiplot. Available online: https://github.com/Xinglab/rmats2sashimiplot (accessed on 17 November 2022).
- Broad Institute. Picard Tools. Available online: https://broadinstitute.github.io/picard/ (accessed on 13 July 2022).
- Wang, X.; Lu, P.; Luo, Z. GMATo: A novel tool for the identification and analysis of microsatellites in large genomes. Bioinformation 2013, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Funkhouser, S.A.; Steibel, J.P.; Bates, R.O.; Raney, N.E.; Schenk, D.; Ernst, C.W. Evidence for transcriptome-wide RNA editing among Sus scrofa PRE-1 SINE elements. BMC Genom. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Nitkiewicz, A.; Smolinska, N.; Przala, J.; Kaminski, T. Expression of orexin receptors 1 (OX1R) and 2 (OX2R) in the porcine ovary during the oestrous cycle. Regul. Pept. 2010, 165, 186–190. [Google Scholar] [CrossRef]
- Orzechowska, K.; Kopij, G.; Paukszto, L.; Dobrzyn, K.; Kiezun, M.; Jastrzebski, J.; Kaminski, T.; Smolinska, N. Chemerin effect on transcriptome of the porcine endometrium during implantation determined by RNA-sequencing. Biol. Reprod. 2022, 107, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Condition | Mid-Luteal Phase (Days 10–12) | Early Pregnancy (Days 15–16) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Samples | 1_ML | 2_ML | 3_ML | 4_ML | 5_ML | 1_EP | 2_EP | 3_EP | 4_EP | 5_EP |
| Raw reads | 72.432 | 72.180 | 66.126 | 69.288 | 72.645 | 72.387 | 72.401 | 72.615 | 72.714 | 72.007 |
| Processed reads | 58.689 | 57.916 | 52.628 | 51.839 | 57.544 | 53.791 | 57.401 | 55.282 | 63.397 | 52.511 |
| Mapped reads | 56.773 | 55.304 | 50.692 | 50.814 | 55.763 | 53.031 | 56.484 | 52.697 | 61.209 | 51.657 |
| Uniquely mapped reads | 52.615 | 51.013 | 47.023 | 47.889 | 52.567 | 51.455 | 53.689 | 49.443 | 58.120 | 48.469 |
| % of uniquely mapped reads | 89.65% | 88.08% | 89.35% | 92.38% | 91.35% | 95.66% | 93.53% | 89.44% | 91.68% | 92.30% |
| Multi-mapped reads | 4.158 | 4.291 | 3.668 | 2.924 | 3.196 | 1.575 | 2.795 | 3.253 | 3.089 | 3.188 |
| % of bases mapped to CDS | 26.06% | 24.76% | 24.02% | 18.71% | 23.57% | 18.94% | 19.03% | 22.94% | 18.72% | 19.80% |
| % of bases mapped to UTR | 14.40% | 13.14% | 13.37% | 10.63% | 13.12% | 12.68% | 11.44% | 12.65% | 11.28% | 11.07% |
| % of bases mapped to introns | 33.71% | 36.19% | 37.59% | 44.86% | 37.73% | 42.27% | 46.44% | 39.69% | 46.73% | 44.30% |
| % of bases mapped to intergenic | 25.83% | 25.92% | 25.02% | 25.80% | 25.58% | 26.11% | 23.09% | 24.72% | 23.27% | 24.83% |
| Pathway Name | Pathway ID | Input (Background) Number | FDR | Figure |
|---|---|---|---|---|
| Axon guidance | ssc04360 | 30 (155) | 9.66 × 10−7 | — |
| Focal adhesion | ssc04510 | 30 (155) | 9.66 × 10−7 | — |
| Phosphatidylinositol signaling system | ssc04070 | 22 (89) | 1.48 × 10−6 | Figure S2 |
| Tight junction | ssc04530 | 28 (143) | 1.55 × 10−6 | — |
| cAMP signaling pathway | ssc04024 | 30 (175) | 5.56 × 10−6 | Figure S3 |
| Protein processing in endoplasmic reticulum | ssc04141 | 26 (138) | 6.76 × 10−6 | Figure S4 |
| Spliceosome | ssc03040 | 23 (113) | 9.36 × 10−6 | Figure S5 |
| mTOR signaling pathway | ssc04150 | 25 (133) | 1.02 × 10−5 | — |
| MAPK signaling pathway | ssc04010 | 34 (233) | 1.67 × 10−5 | Figure S6 |
| RNA transport | ssc03013 | 24 (129) | 1.67 × 10−5 | Figure S7 |
| Thyroid hormone signaling pathway | ssc04919 | 22 (112) | 2.03 × 10−5 | — |
| Cholinergic synapse | ssc04725 | 18 (77) | 2.03 × 10−5 | Figure S8 |
| Phospholipase D signaling pathway | ssc04072 | 24 (132) | 2.03 × 10−5 | Figure S9 |
| Endocytosis | ssc04144 | 31 (206) | 2.24 × 10−5 | — |
| Regulation of actin cytoskeleton | ssc04810 | 28 (179) | 3.22 × 10−5 | — |
| Relaxin signaling pathway | ssc04926 | 21 (109) | 3.33 × 10−5 | — |
| PI3K-Akt signaling pathway | ssc04151 | 36 (271) | 3.61 × 10−5 | Figure S10 |
| Glycerophospholipid metabolism | ssc00564 | 17 (74) | 3.61 × 10−5 | — |
| Glutamatergic synapse | ssc04724 | 18 (83) | 3.64 × 10−5 | Figure S11 |
| Rap1 signaling pathway | ssc04015 | 27 (174) | 4.59 × 10−5 | — |
| Inositol phosphate metabolism | ssc00562 | 16 (68) | 4.85 × 10−5 | Figure S12 |
| Dopaminergic synapse | ssc04728 | 19 (97) | 6.46 × 10−5 | Figure S13 |
| GABAergic synapse | ssc04727 | 15 (65) | 1.03 × 10−4 | Figure S14 |
| GnRH signaling pathway | ssc04912 | 16 (75) | 1.18 × 10−4 | Figure S15 |
| Insulin resistance | ssc04931 | 18 (96) | 1.50 × 10−4 | — |
| Ras signaling pathway | ssc04014 | 27 (191) | 1.50 × 10−4 | — |
| Autophagy—animal | ssc04140 | 20 (116) | 1.50 × 10−4 | — |
| Prolactin signaling pathway | ssc04917 | 14 (60) | 1.50 × 10−4 | — |
| Purine metabolism | ssc00230 | 19 (107) | 1.58 × 10−4 | — |
| Circadian entrainment | ssc04713 | 14 (62) | 1.87 × 10−4 | — |
| HIF-1 signaling pathway | ssc04066 | 17 (91) | 2.15 × 10−4 | — |
| mRNA surveillance pathway | ssc03015 | 14 (65) | 2.61 × 10−4 | Figure S16 |
| Neurotrophin signaling pathway | ssc04722 | 18 (103) | 2.61 × 10−4 | — |
| Inflammatory mediator regulation of TRP channels | ssc04750 | 16 (84) | 2.74 × 10−4 | — |
| Ubiquitin-mediated proteolysis | ssc04120 | 19 (114) | 2.76 × 10−4 | Figure S17 |
| cGMP-PKG signaling pathway | ssc04022 | 21 (136) | 2.90 × 10−4 | Figure S18 |
| Retrograde endocannabinoid signaling | ssc04723 | 19 (115) | 2.90 × 10−4 | — |
| Fc gamma R-mediated phagocytosis | ssc04666 | 15 (76) | 2.99 × 10−4 | — |
| Mitophagy—animal | ssc04137 | 13 (58) | 2.99 × 10−4 | — |
| Chemokine signaling pathway | ssc04062 | 22 (150) | 3.51 × 10−4 | — |
| ErbB signaling pathway | ssc04012 | 14 (70) | 4.23 × 10−4 | — |
| Estrogen signaling pathway | ssc04915 | 18 (111) | 4.82 × 10−4 | Figure S19 |
| Oxytocin signaling pathway | ssc04921 | 19 (124) | 5.91 × 10−4 | — |
| ECM–receptor interaction | ssc04512 | 13 (64) | 6.07 × 10−4 | — |
| Sphingolipid signaling pathway | ssc04071 | 16 (94) | 6.46 × 10−4 | — |
| Amino sugar and nucleotide sugar metabolism | ssc00520 | 11 (48) | 7.51 × 10−4 | — |
| Apelin signaling pathway | ssc04371 | 18 (117) | 7.71 × 10−4 | — |
| Aminoacyl–tRNA biosynthesis | ssc00970 | 11 (49) | 8.21 × 10−4 | — |
| Fatty acid metabolism | ssc01212 | 11 (49) | 8.21 × 10−4 | — |
| Adherens junction | ssc04520 | 12 (59) | 9.29 × 10−4 | — |
| Gene Symbol | Type | Primers Sequences | Product Length | Reference |
|---|---|---|---|---|
| CACNA1D | DEG | F: CATCGCATCACTGCTGCTTC | 185 bp | [The present study] |
| R: TCACAGCGTTCCAGTCTTCC | ||||
| DUSP2 | DEG | F: CATCCCTGTGGAGGACAACC | 187 bp | [The present study] |
| R: GGCCTCATCTAGACGCACTC | ||||
| GRIK2 | DEG | F: TGGGAATGACCGGTTTGAGG | 372 bp | [The present study] |
| R: AGCACACAACTGACACCCAA | ||||
| ISYNA1 | DEG | F: TACATCCCGGAGTTCATCGC | 201 bp | [The present study] |
| R: AGCAGTGTCATTGAGGCCAG | ||||
| NELFCD | DEG | F: ACACCTCTGACTTCGTGCAG | 119 bp | [The present study] |
| R: CGGGCAAAACCCACCTATGA | ||||
| POMC | DEG | F: AAAGTAACTTGCTGGCGTGC | 363 bp | [The present study] |
| R: CGTTGGGATACACCTTCACCG | ||||
| VGF | DEG | F: TGAAATCGCCCAGGTTGCC | 164 bp | [The present study] |
| R: AACATCCTTTGGCCCGATCA | ||||
| E*T00000074581 | DEL | F: TTTTCCCAAAGGCAGGAGCA | 414 bp | [The present study] |
| R: TGATCTGTTTCGGCAGGCTT | ||||
| E*T00000076568 | DEL | F: CAAGGCGGTCGTTAGGATCA | 344 bp | [The present study] |
| R: CACTAATGAAGGCGCTGCAC | ||||
| MSTRG.14404.1 | DEL | F: GTGACATGTGTGGGACGGTA | 110 bp | [The present study] |
| R: CACGTCTTCCTGACAGCCTC | ||||
| MSTRG.18944.1 | DEL | F: AGGAGTTCAAGGCCAACAGG | 160 bp | [The present study] |
| R: GTCCACGTACACCCCCTTTC | ||||
| MSTRG.20172.2 | DEL | F: TATCCTGCACCGAGCAATGG | 119 bp | [The present study] |
| R: CAACCCAGACCATCCCATCC | ||||
| MSTRG.27546.1 | DEL | F: GCTTTGTGTGGCCTGGACTA | 333 bp | [The present study] |
| R: TCCACATAGGCACAGAGGAGA | ||||
| MSTRG.32275.1 | DEL | F: AGACAGTAAGCACACAGCGG | 164 bp | [The present study] |
| R: TGTGGAGTTGGATCATGGCG | ||||
| CELF4 | DAS | F: TACCATCTGCCCCAGGAGTT | 78 or 157 bp | [The present study] |
| R: GTTGTCGAAGCTCACGAAGC | ||||
| POSTN | DAS | F: TCGACTAGTGGTGGCGAAAC | 81 or 165 bp | [The present study] |
| R: GGTGGCTTGTATCTTCCTCACA | ||||
| GAPDH | HKG | F: CCTTCATTGACCTCCACTACATGG | 183 bp | [182] |
| R: CCACAACATACGTAGCACCAGCATC | ||||
| PPIA | HKG | F: GCACTGGTGGCAAGTCCAT | 71 bp | [183] |
| R: AGGACCCGTATGCTTCAGGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makowczenko, K.G.; Jastrzebski, J.P.; Kiezun, M.; Paukszto, L.; Dobrzyn, K.; Smolinska, N.; Kaminski, T. Adaptation of the Porcine Pituitary Transcriptome, Spliceosome and Editome during Early Pregnancy. Int. J. Mol. Sci. 2023, 24, 5946. https://doi.org/10.3390/ijms24065946
Makowczenko KG, Jastrzebski JP, Kiezun M, Paukszto L, Dobrzyn K, Smolinska N, Kaminski T. Adaptation of the Porcine Pituitary Transcriptome, Spliceosome and Editome during Early Pregnancy. International Journal of Molecular Sciences. 2023; 24(6):5946. https://doi.org/10.3390/ijms24065946
Chicago/Turabian StyleMakowczenko, Karol G., Jan P. Jastrzebski, Marta Kiezun, Lukasz Paukszto, Kamil Dobrzyn, Nina Smolinska, and Tadeusz Kaminski. 2023. "Adaptation of the Porcine Pituitary Transcriptome, Spliceosome and Editome during Early Pregnancy" International Journal of Molecular Sciences 24, no. 6: 5946. https://doi.org/10.3390/ijms24065946
APA StyleMakowczenko, K. G., Jastrzebski, J. P., Kiezun, M., Paukszto, L., Dobrzyn, K., Smolinska, N., & Kaminski, T. (2023). Adaptation of the Porcine Pituitary Transcriptome, Spliceosome and Editome during Early Pregnancy. International Journal of Molecular Sciences, 24(6), 5946. https://doi.org/10.3390/ijms24065946