
Impaired Integrated Stress Response and Mitochondrial Integrity Modulate Genotoxic Stress Impact and Lower the Threshold for Immune Signalling

 , , and
, , and 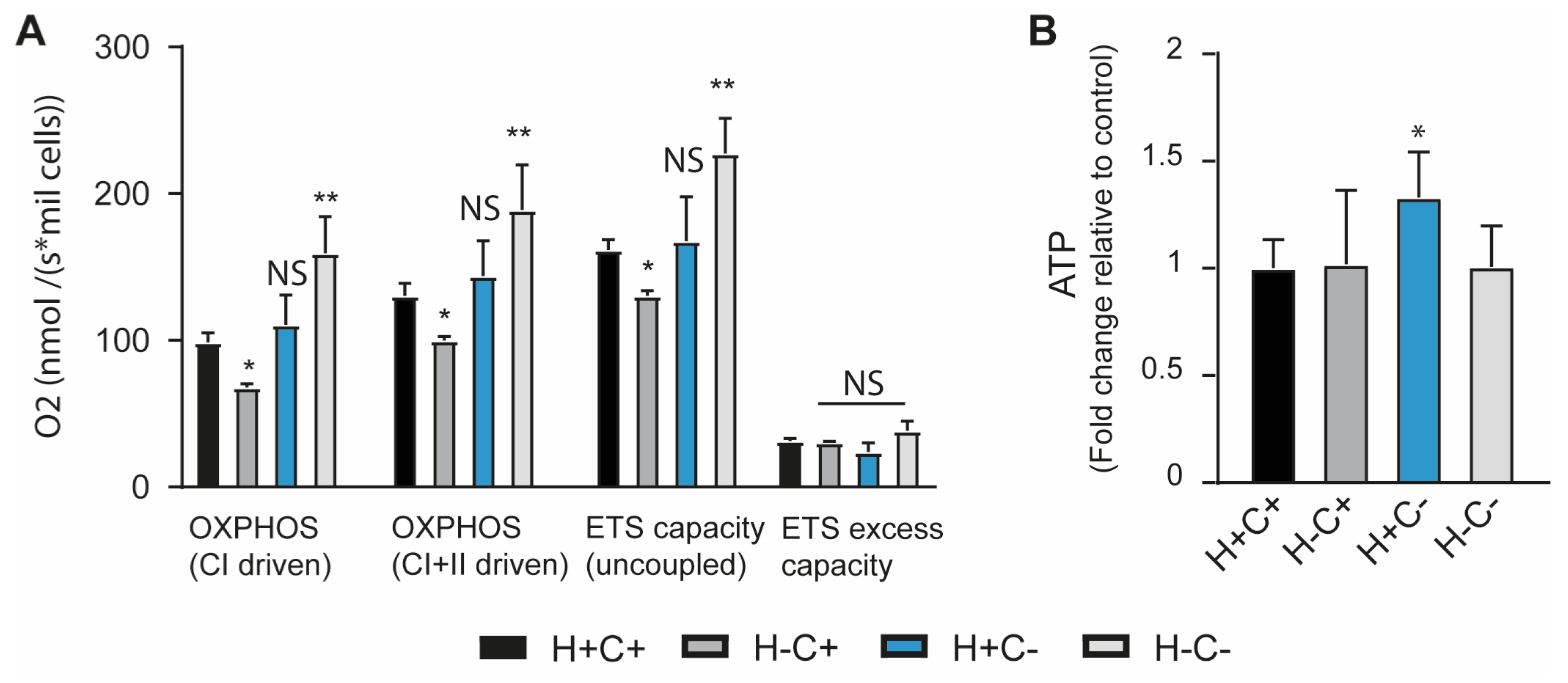{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Mitochondrial Homeostasis Is Influenced by Mitochondrial Quality Control and ISR Integrity
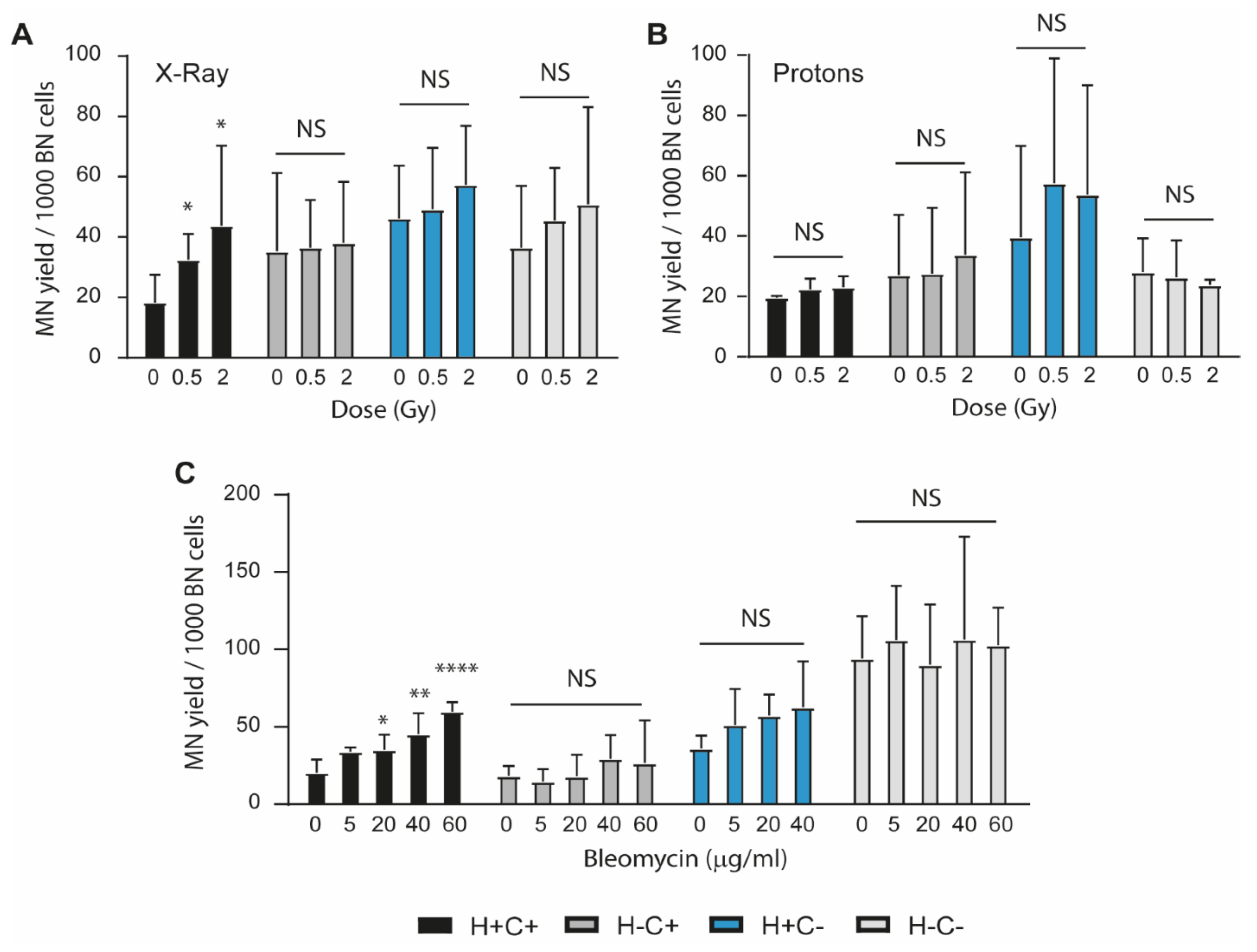
2.2. Sensitivity to Genotoxic Stressors
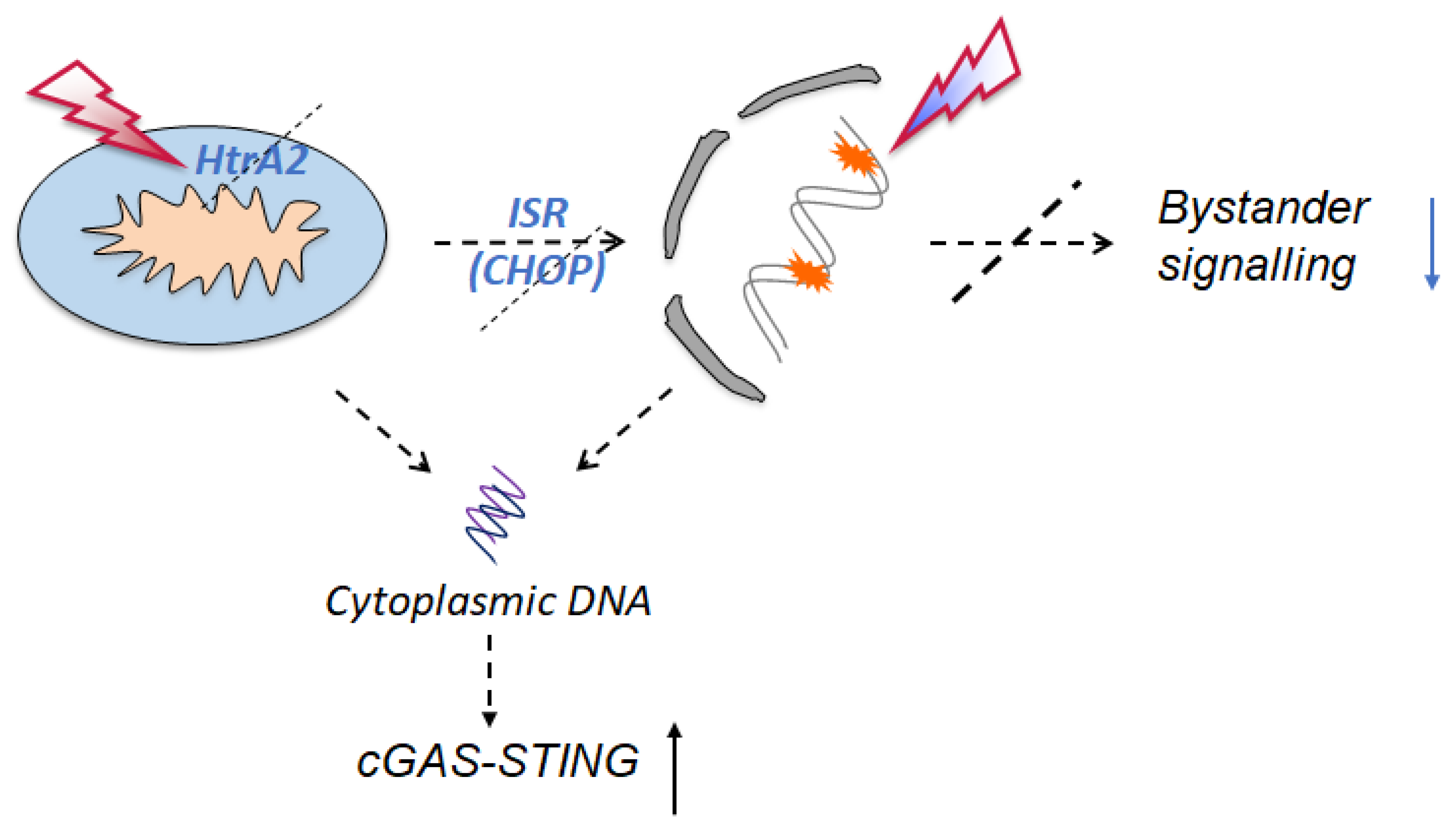
2.3. Signalling Mechanisms Revealed by RNA Sequencing
3. Materials and Methods
3.1. Cell Culture
3.2. Genotoxic Treatment
3.3. Bystander Effect
3.4. Cell Viability
3.5. ATP Level Measurement
3.6. Protein Level Measurement (Bradford Assay)
3.7. Gene Expression Analysis
3.8. High Resolution Respirometry
3.9. Micronuclei (MN) Analysis
3.10. Statistical Analyses
3.11. RNA Sequencing Analysis
3.12. Differential Expression Analysis and Pathway Enrichment Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopez-Otin, C.; Blasco, M.A.; Partrige, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Savu, D.I.; Moisoi, N. Mitochondria-Nucleus communication in neurodegenerative Disease. Who talks first, who talks louder? BBA Bioenerg. 2022, 1863, 148588. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-lazikani, B.; Pearl, F.M.G. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samal, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, K.; Hong, M. Radiation-Induced Bystander Effect and Cytoplasmic Irradiation Studies with Microbeams. Biology 2022, 11, 945. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Kaspar, S.; Oertlin, C.; Szczepanovska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 2021, 7, eabf0971. [Google Scholar] [CrossRef]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Liv, G.P.; et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef]
- Kang, S.; Louboutin, J.P.; Datta, P.; Landel, C.P.; Martinez, D.; Zervos, A.S.; Strayer, D.S.; Alnemri, T.F.; Alnemri, E.S. Loss of HtrA2/Omi activity in non-neuronal tissues of adult mice causes premature aging. Cell Death Differ. 2013, 20, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain specific transcriptional stress response. Cell Death Differ. 2009, 16, 449464. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Bose, R.; Bose, K. Unraveling the Dichotomy of Enigmatic Serine Protease HtrA2. Front. Mol. Biosci. 2022, 9, 824846. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, S.; Zhou, L.; Seyhan, A.A.; Hernandez Borrero, L.; Zhang, Y.; El-Deiry, W.S. Targeting the Integrated Stress Response in Cancer Therapy. Front. Pharmacol. 2021, 12, 747837. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting proten synthesis and oxidation in the stresses endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Das, S.; Joshi, M.B.; Parashiva, G.K.; Rao, S.B.S. Stimulation of cytoprotective autophagy and components of mitochondrial biogenesis/proteostasis in response to ionising radiation as a credible pro-survival strategy. Free. Radic. Biol. Med. 2020, 152, 715–727. [Google Scholar] [CrossRef]
- Temelie, M.; Savu, D.I.; Moisoi, N. Intracellular and intercellular signalling mechanisms following DNA damage are modulated by PINK1. Oxidative Med. Cell. Longev. 2018, 2018, 1391387. [Google Scholar] [CrossRef]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar] [PubMed]
- Dong, C.; Tu, W.; He, M.; Fu, J.; Kobayashi, A.; Konishi, T.; Shao, C. Role of endoplasmic reticulum and mitochondrion in proton microbeam radiation-induced bystander effect. Radiat. Res. 2020, 193, 63–72. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular function of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, H.; Ouyang, X.; Dong, Y.; Sarapultsev, A.; Luo, S.; Hu, D. Multifaceted functions of STING in human health and disease: From molecular mechanism to targeted strategy. Signal Transduct. Ther. 2022, 7, 394. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fisher, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Cannon, T.; Voisin, A.; Pentinen, A.-M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.-J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease—Like symptims in Pink1−/− mice. Nature 2020, 571, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Tigano, M.; Vargas, D.C.; Tremblay-Belzile, S.; Fu, Y.; Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 2021, 591, 477–481. [Google Scholar] [CrossRef]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Zhang, J.; Yang, M.; Zhu, M.; Yin, Y.; Fan, X.; Fei, Y. The paradoxical role of radiation-induced cGAS-STING signalling network in tumour immunity. Immunology 2022, 168, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zeng, Y.; Peng, Y.; Liu, J.; Wu, F. Cancer immunotherapy strategies that target the cGAS-STING pathway. Front. Immunol. 2022, 13, 996663. [Google Scholar] [CrossRef]
- Zucker, B.; Luthi-Carter, R.; Kama, J.A.; Dunah, A.W.; Stern, E.A.; Fox, J.H.; Standaert, D.G.; Young, A.B.; Augood, S.J. Transcriptional dysregulation in striatal projection- and interneurons in a mouse model of Huntington’s disease: Neuronal selectivity and potential neuroprotective role of HAP1. Huma Mol. Genet. 2005, 14, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 5, 1084–1104. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temelie, M.; Talpur, R.; Dominguez-Prieto, M.; Dantas Silva, A.; Cenusa, C.; Craciun, L.; Savu, D.I.; Moisoi, N. Impaired Integrated Stress Response and Mitochondrial Integrity Modulate Genotoxic Stress Impact and Lower the Threshold for Immune Signalling. Int. J. Mol. Sci. 2023, 24, 5891. https://doi.org/10.3390/ijms24065891
Temelie M, Talpur R, Dominguez-Prieto M, Dantas Silva A, Cenusa C, Craciun L, Savu DI, Moisoi N. Impaired Integrated Stress Response and Mitochondrial Integrity Modulate Genotoxic Stress Impact and Lower the Threshold for Immune Signalling. International Journal of Molecular Sciences. 2023; 24(6):5891. https://doi.org/10.3390/ijms24065891
Chicago/Turabian StyleTemelie, Mihaela, Rubab Talpur, Marta Dominguez-Prieto, Ayanda Dantas Silva, Constantin Cenusa, Liviu Craciun, Diana Iulia Savu, and Nicoleta Moisoi. 2023. "Impaired Integrated Stress Response and Mitochondrial Integrity Modulate Genotoxic Stress Impact and Lower the Threshold for Immune Signalling" International Journal of Molecular Sciences 24, no. 6: 5891. https://doi.org/10.3390/ijms24065891
APA StyleTemelie, M., Talpur, R., Dominguez-Prieto, M., Dantas Silva, A., Cenusa, C., Craciun, L., Savu, D. I., & Moisoi, N. (2023). Impaired Integrated Stress Response and Mitochondrial Integrity Modulate Genotoxic Stress Impact and Lower the Threshold for Immune Signalling. International Journal of Molecular Sciences, 24(6), 5891. https://doi.org/10.3390/ijms24065891