
Hormone-Related and Drug-Induced Osteoporosis: A Cellular and Molecular Overview



Abstract
1. Introduction
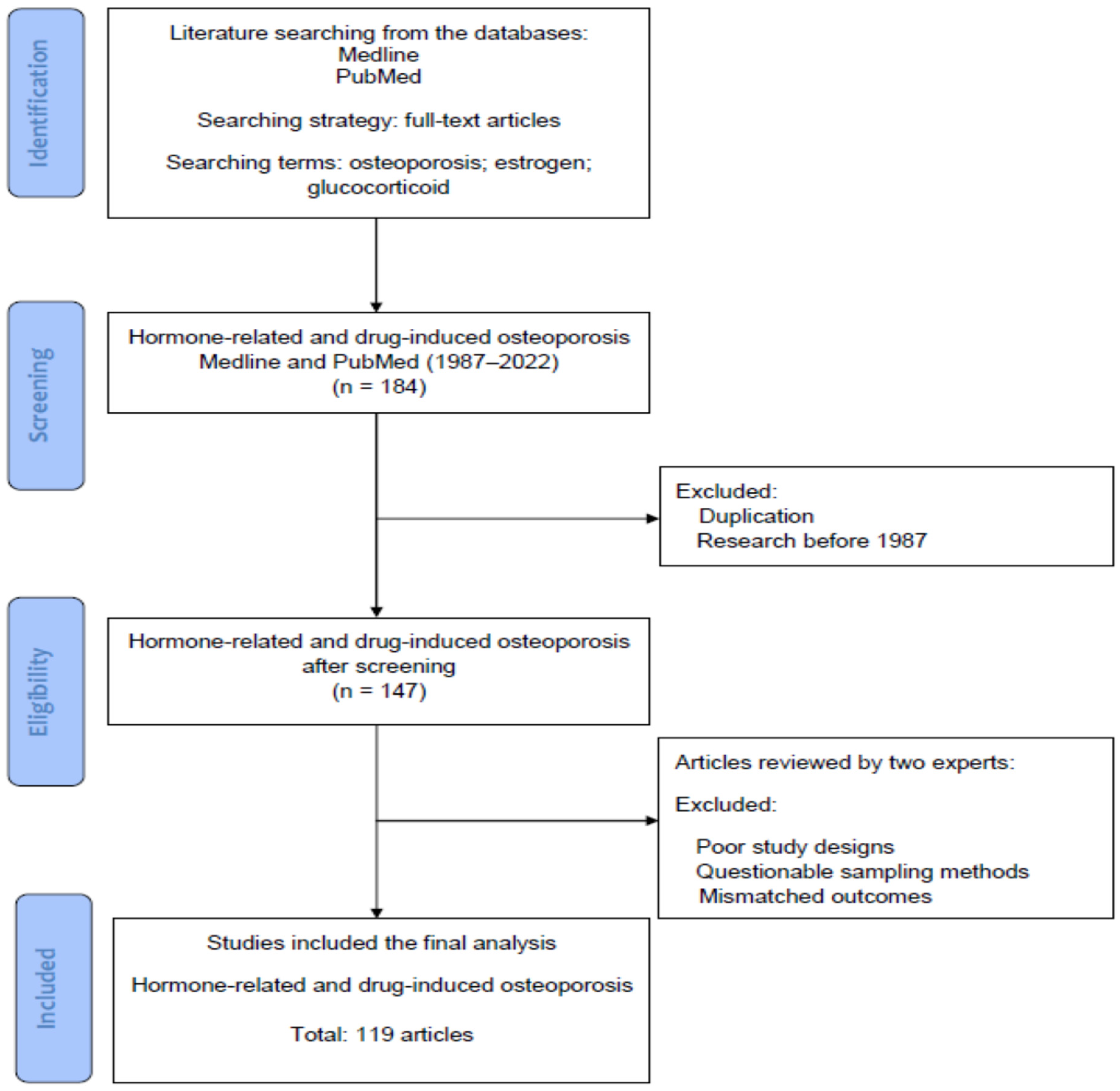
2. Materials and Methods
Searching Terms and Strategies in the Literature
3. Cellular and Molecular Mechanisms of Bone Turnover among Osteoblasts, Osteoclasts and Osteocytes
3.1. Bone Remodeling
3.2. Molecular Mechanisms in Osteoclast and Osteoblast Differentiation
3.3. Hormonal and Environmental Factors
3.4. Coupling between Osteoclasts (OCs) and Osteoblasts (OBs)
3.5. Gene Expression
3.6. Other factors which Influence the Bone Turnover and the Balance of Osteoblassts and Osteoclasts
3.6.1. Secretome Related to Senescence and Bone Loss
3.6.2. Secretome of Osteoblasts Promotes Cell Growth in Osteoblasts
3.6.3. Secretome of MSCs in Ovariectomized Rats Promotes Bone Resorption
3.6.4. Extracellular Vesicles in Bone Homeostasis
3.6.5. The Alteration of Osteocytes Secretory Profile (Secretome and Extracellular Vesicles in Osteocytes) in Response to Mechanical Stress
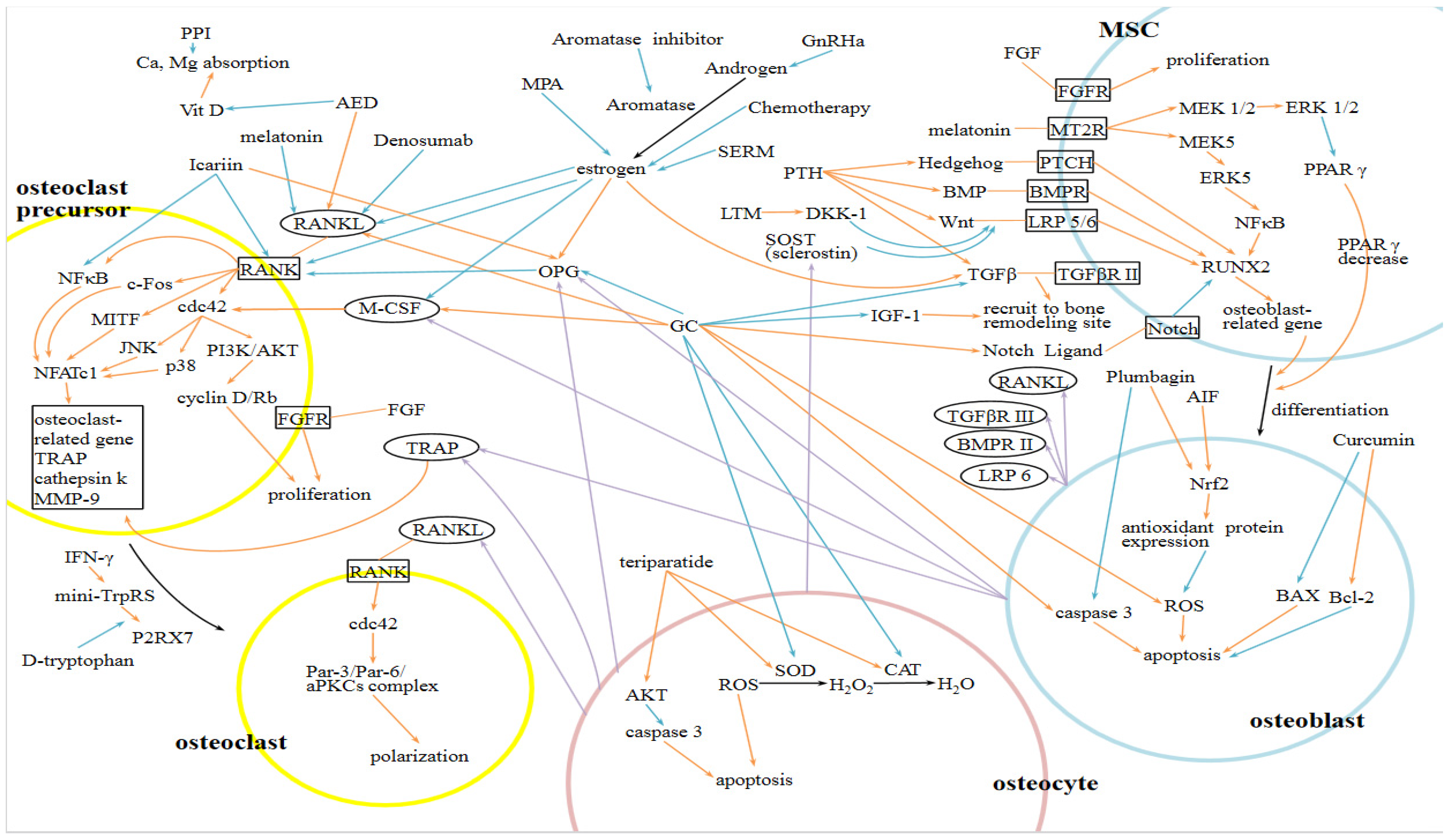
4. Cellular and Molecular Mechanisms of Hormone-Related and Drug-Induced Osteoporosis
4.1. Hormone Related Osteoporosis (HROP)
4.2. Drug Induced Osteoporosis
4.2.1. Glucocorticoids Induced Osteoporosis (GIOP)
4.2.2. Aromatase Inhibitor Associated Osteoporosis
4.2.3. Gonadotropin Releasing Hormone Agonist (GnRHa) Associated Osteoporosis
4.2.4. Chemotherapy Induced Osteoporosis
4.2.5. Selective Estrogen Receptor Modulators (SERMs) Related Osteoporosis
4.2.6. Proton-Pump Inhibitor (PPI) or Diabetes Mellitus (DM) Related Osteoporosis
4.2.7. Anti Convulsion Drug Induced Osteoporosis
4.2.8. Medroxyprogesterone Acetate (MPA) Associated Osteoporosis
5. Medication for Treating Osteoporosis
5.1. Bisphosphonates
5.2. Parathyroid Hormone (PTH)
5.3. Potential Treatment Target in the Future
6. Discussion and Conclusions
- Get adequate calcium and vitamin D: Adequate calcium and vitamin D intake is essential for maintaining bone health. Calcium-rich foods include dairy products, leafy green vegetables, and fortified foods, while vitamin D can be obtained mainly by sunlight, fatty fish, and egg yolks. The absorption of calcium is influenced by not only the concentration of calcium, but also the bioavailability of calcium. Several non-dairy foods contain factors, such as oxalic acid and phytic acid, that negatively affect the absorption of calcium by forming salts with low solubility, leading to a low absorption of calcium. Similarly, phytic acid in leafy green vegetables substantially reduces the absorption of calcium. In dairy products, calcium phosphates remain sparingly soluble salts and the absorption is further facilitated by casein [120]. Naturally calcium-rich waters are also considerable sources of dietary calcium due to the high bioavailability of calcium similar to dairy products [121].
- Engage in weight-bearing exercises: Weight-bearing exercises, such as walking, running, and strength training, can help maintain bone density and reduce the risk of osteoporosis/fragility fracture risk.
- Avoid smoking and excessive alcohol consumption: Both smoking and excessive alcohol consumption can contribute to bone loss and increase the risk of osteoporosis.
- Consider bone density testing: Bone density testing, such as a dual-energy x-ray absorptiometry (DXA) scan, can help detect osteoporosis early and guide treatment decisions.
- Discuss with a healthcare provider: Healthcare providers can provide guidance on lifestyle modifications, supplements, and medications that can help prevent or treat osteoporosis. In addition, individuals who are at an increased risk for osteoporosis, such as postmenopausal women and individuals with a family history of the disease, should take extra precautions to maintain bone health.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parfitt, A.M. Bone Remodeling and Bone Loss: Understanding The Pathophysiology of Osteoporosis. Clin. Obstet. Gynecol. 1987, 30, 789–811. [Google Scholar] [CrossRef] [PubMed]
- Chapurlat, R.; Bui, M.; Sornay-Rendu, E.; Zebaze, R.; Delmas, P.D.; Liew, D.; Lespessailles, E.; Seeman, E. Deterioration of Cortical and Trabecular Microstructure Identifies Women With Osteopenia or Normal Bone Mineral Density at Imminent and Long-Term Risk for Fragility Fracture: A Prospective Study. J. Bone Miner. Res. 2019, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Sioka, C.; Fotopoulos, A.; Georgiou, A.; Xourgia, X.; Papadopoulos, A.; Kalef-Ezra, J.A. Age at menarche, age at menopause and duration of fertility as risk factors for osteoporosis. Climacteric 2010, 13, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Svejme, O.; Ahlborg, H.; Nilsson, J.-Å.; Karlsson, M. Early menopause and risk of osteoporosis, fracture and mortality: A 34-year prospective observational study in 390 women: Early menopause and osteoporosis, fracture and mortality. BJOG Int. J. Obstet. Gynaecol. 2012, 119, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Akdeniz, N.; Akpolat, V.; Kale, A.; Erdemoğlu, M.; Kuyumcuoglu, U.; Celik, Y. Risk factors for postmenopausal osteoporosis: Anthropometric measurements, age, age at menopause and the time elapsed after menopause onset. Gynecol. Endocrinol. 2009, 25, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Johnell, O.; Kanis, J.A. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos. Int. 2006, 17, 1726–1733. [Google Scholar] [CrossRef]
- Baccaro, L.F.; Conde, D.M.; Costa-Paiva, L.; Pinto-Neto, A.M. The epidemiology and management of postmenopausal osteoporosis: A viewpoint from Brazil. Clin. Interv. Aging 2015, 10, 583–591. [Google Scholar] [CrossRef]
- Stein, E.; Shane, E. Secondary osteoporosis. Endocrinol. Metab. Clin. North Am. 2003, 32, 115–134. [Google Scholar] [CrossRef]
- Panday, K.; Gona, A.; Humphrey, M.B. Medication-induced osteoporosis: Screening and treatment strategies. Ther. Adv. Musculoskelet. Dis. 2014, 6, 185–202. [Google Scholar] [CrossRef]
- Goh, M.; Nguyen, H.H.; Khan, N.N.; Milat, F.; Boyle, J.A.; Vincent, A.J. Identifying and addressing osteoporosis knowledge gaps in women with premature ovarian insufficiency and early menopause: A mixed-methods study. Clin. Endocrinol. 2019, 91, 498–507. [Google Scholar] [CrossRef]
- Ayub, N.; Faraj, M.; Ghatan, S.; Reijers, J.A.A.; Napoli, N.; Oei, L. The Treatment Gap in Osteoporosis. J. Clin. Med. 2021, 10, 3002. [Google Scholar] [CrossRef]
- Tay, C.L.; Ng, W.L.; Beh, H.C.; Lim, W.C.; Hussin, N. Screening and management of osteoporosis: A survey of knowledge, attitude and practice among primary care physicians in Malaysia. Arch. Osteoporos. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.; Canalis, E. Management of endocrine disease: Secondary osteoporosis: Pathophysiology and management. Eur. J. Endocrinol. 2015, 173, R131–R151. [Google Scholar] [CrossRef]
- Thakur, P.; Kuriakose, C.; Cherian, K.E.; Asha, H.S.; Kapoor, N.; Paul, T.V. Knowledge gap regarding osteoporosis among medical professionals in Southern India. J. Evaluation Clin. Pract. 2019, 26, 272–280. [Google Scholar] [CrossRef]
- Tella, S.H.; Gallagher, J.C. Bazedoxifene + conjugated estrogens in HT for the prevention of osteoporosis and treatment of vasomotor symptoms associated with the menopause. Expert Opin. Pharmacother. 2013, 14, 2407–2420. [Google Scholar] [CrossRef]
- Pennisi, P.; Trombetti, A.; Rizzoli, R. Glucocorticoid-induced Osteoporosis and Its Treatment. Clin. Orthop. Relat. Res. 2006, 443, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Barton, D.L.; Loprinzi, C.L. Chemotherapy-Induced Ovarian Failure. Drug Saf. 2005, 28, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, S.; Çelik, Ç.; Görkemli, H.; Kıyıcı, A.; Kaya, B.; Kiyici, A. Compared effects of surgical and natural menopause on climacteric symptoms, osteoporosis, and metabolic syndrome. Int. J. Gynecol. Obstet. 2009, 106, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Ofer, L.; Dean, M.N.; Zaslansky, P.; Kult, S.; Shwartz, Y.; Zaretsky, J.; Griess-Fishheimer, S.; Monsonego-Ornan, E.; Zelzer, E.; Shahar, R. A novel nonosteocytic regulatory mechanism of bone modeling. PLoS Biol. 2019, 17, e3000140. [Google Scholar] [CrossRef]
- Khosla, S.; Westendorf, J.J.; Mödder, U.I. Concise Review: Insights from Normal Bone Remodeling and Stem Cell-Based Therapies for Bone Repair. Stem Cells 2010, 28, 2124–2128. [Google Scholar] [CrossRef]
- Dempster, D.W.; Zhou, H.; Ruff, V.A.; Melby, T.E.; Alam, J.; Taylor, K.A. Longitudinal Effects of Teriparatide or Zoledronic Acid on Bone Modeling- and Remodeling-Based Formation in the SHOTZ Study: Longitudinal effects of Tptd and Zol on bone formation. J. Bone Miner. Res. 2018, 33, 627–633. [Google Scholar] [CrossRef]
- Yi, S.-J.; Lee, H.; Lee, J.; Lee, K.; Kim, J.; Kim, Y.; Park, J.-I.; Kim, K. Bone Remodeling: Histone Modifications as Fate Determinants of Bone Cell Differentiation. Int. J. Mol. Sci. 2019, 20, 3147. [Google Scholar] [CrossRef]
- Biros, E.; Malabu, U.H.; Vangaveti, V.N.; Birosova, E.; Moran, C.S. The IFN-γ/miniTrpRS signaling axis: An insight into the pathophysiology of osteoporosis and therapeutic potential. Cytokine Growth Factor Rev. 2022, 64, 7–11. [Google Scholar] [CrossRef]
- Ito, Y.; Teitelbaum, S.L.; Zou, W.; Zheng, Y.; Johnson, J.F.; Chappel, J.; Ross, F.P.; Zhao, H. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J. Clin. Investig. 2010, 120, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.L.; Cao, X. Bone marrow mesenchymal stem cells and TGF-β signaling in bone remodeling. J. Clin. Investig. 2014, 124, 466–472. [Google Scholar] [CrossRef]
- Xiong, J.; O’Brien, C.A. Osteocyte RANKL: New insights into the control of bone remodeling. J. Bone Miner. Res. 2012, 27, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.C.; Sai, A. Molecular biology of bone remodeling: Implications for new therapeutic targets for osteoporosis. Maturitas 2010, 65, 301–307. [Google Scholar] [CrossRef]
- Remer, T.; Boye, K.R.; Hartmann, M.; Neu, C.M.; Schoenau, E.; Manz, F.; A Wudy, S. Adrenarche and Bone Modeling and Remodeling at the Proximal Radius: Weak Androgens Make Stronger Cortical Bone in Healthy Children. J. Bone Miner. Res. 2003, 18, 1539–1546. [Google Scholar] [CrossRef]
- Canalis, E. Mechanisms of glucocorticoid-induced osteoporosis. Curr. Opin. Rheumatol. 2003, 15, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Usategui-Martín, R.; Rigual, R.; Ruiz-Mambrilla, M.; Fernández-Gómez, J.-M.; Dueñas, A.; Pérez-Castrillón, J.L. Molecular Mechanisms Involved in Hypoxia-Induced Alterations in Bone Remodeling. Int. J. Mol. Sci. 2022, 23, 3233. [Google Scholar] [CrossRef]
- Maria, S.; Samsonraj, R.M.; Munmun, F.; Glas, J.; Silvestros, M.; Kotlarczyk, M.P.; Rylands, R.; Dudakovic, A.; van Wijnen, A.J.; Enderby, L.T.; et al. Biological effects of melatonin on osteoblast/osteoclast cocultures, bone, and quality of life: Implications of a role for MT2 melatonin receptors, MEK1/2, and MEK5 in melatonin-mediated osteoblastogenesis. J. Pineal Res. 2017, 64, e12465. [Google Scholar] [CrossRef]
- Itkin, T.; Kaufmann, K.B.; Gur-Cohen, S.; Ludin, A.; Lapidot, T. Fibroblast growth factor signaling promotes physiological bone remodeling and stem cell self-renewal. Curr. Opin. Hematol. 2013, 20, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Licini, C.; Vitale-Brovarone, C.; Mattioli-Belmonte, M. Collagen and non-collagenous proteins molecular crosstalk in the pathophysiology of osteoporosis. Cytokine Growth Factor Rev. 2019, 49, 59–69. [Google Scholar] [CrossRef]
- Masaoutis, C.; Theocharis, S. The Role of Exosomes in Bone Remodeling: Implications for Bone Physiology and Disease. Dis. Markers 2019, 2019, 9417914. [Google Scholar] [CrossRef] [PubMed]
- Oton-Gonzalez, L.; Mazziotta, C.; Iaquinta, M.R.; Mazzoni, E.; Nocini, R.; Trevisiol, L.; D’Agostino, A.; Tognon, M.; Rotondo, J.C.; Martini, F. Genetics and Epigenetics of Bone Remodeling and Metabolic Bone Diseases. Int. J. Mol. Sci. 2022, 23, 1500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Feng, P.; Mo, G.; Li, D.; Li, Y.; Mo, L.; Yang, Z.; Liang, D. Icariin influences adipogenic differentiation of stem cells affected by osteoblast-osteoclast co-culture and clinical research adipogenic. Biomed. Pharmacother. 2017, 88, 436–442. [Google Scholar] [CrossRef]
- Chandra, A.; Rajawat, J. Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics. Int. J. Mol. Sci. 2021, 22, 3553. [Google Scholar] [CrossRef] [PubMed]
- Romanello, M.; Piatkowska, E.; Antoniali, G.; Cesaratto, L.; Vascotto, C.; Iozzo, R.V.; Delneri, D.; Brancia, F.L. Osteoblastic cell secretome: A novel role for progranulin during risedronate treatment. Bone 2014, 58, 81–91. [Google Scholar] [CrossRef]
- Rao, V.V.; Wechsler, M.E.; Cravens, E.; Wojda, S.J.; Caldwell, A.S.; Kirkpatrick, B.E.; Donahue, S.W.; Anseth, K.S. Granular PEG hydrogels mediate osteoporotic MSC clustering via N-cadherin influencing the pro-resorptive bias of their secretory profile. Acta Biomater. 2022, 145, 77–87. [Google Scholar] [CrossRef]
- Huang, X.; Lan, Y.; Shen, J.; Chen, Z.; Xie, Z. Extracellular Vesicles in Bone Homeostasis: Emerging Mediators of Osteoimmune Interactions and Promising Therapeutic Targets. Int. J. Biol. Sci. 2022, 18, 4088–4100. [Google Scholar] [CrossRef]
- Eichholz, K.F.; Woods, I.; Riffault, M.; Johnson, G.P.; Corrigan, M.; Lowry, M.C.; Shen, N.; Labour, M.-N.; Wynne, K.; O’Driscoll, L.; et al. Human bone marrow stem/stromal cell osteogenesis is regulated via mechanically activated osteocyte-derived extracellular vesicles. Stem Cells Transl. Med. 2020, 9, 1431–1447. [Google Scholar] [CrossRef]
- Gurban, C.V.; Balaş, M.O.; Vlad, M.M.; Caraba, A.E.; Jianu, A.M.; Bernad, E.S.; Borza, C.; Bănicioiu-Covei, S.; Motoc, A.G.M. Bone turnover markers in postmenopausal osteoporosis and their correlation with bone mineral density and menopause duration. Romanian J. Morphol. Embryol. 2019, 60, 1127–1135. [Google Scholar]
- Huang, S.; Zhu, X.; Xiao, D.; Zhuang, J.; Liang, G.; Liang, C.; Zheng, X.; Ke, Y.; Chang, Y. LncRNA SNHG1 was down-regulated after menopause and participates in postmenopausal osteoporosis. Biosci. Rep. 2019, 39, BSR20190445. [Google Scholar] [CrossRef]
- Abrahamsen, B.; Vestergaard, P.; Rud, B.; Bärenholdt, O.; Jensen, J.-E.B.; Nielsen, S.P.; Mosekilde, L.; Brixen, K. Ten-Year Absolute Risk of Osteoporotic Fractures According to BMD T Score at Menopause: The Danish Osteoporosis Prevention Study. J. Bone Miner. Res. 2006, 21, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Boschitsch, E.P.; Durchschlag, E.; Dimai, H.P. Age-related prevalence of osteoporosis and fragility fractures: Real-world data from an Austrian Menopause and Osteoporosis Clinic. Climacteric 2017, 20, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Fistarol, M.; Rezende, C.R.; Campos, A.L.F.; Kakehasi, A.M.; Geber, S. Time since menopause, but not age, is associated with increased risk of osteoporosis. Climacteric 2019, 22, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Oelzner, P.; Schwabe, A.; Lehmann, G.; Eidner, T.; Franke, S.; Wolf, G.; Hein, G. Significance of risk factors for osteoporosis is dependent on gender and menopause in rheumatoid arthritis. Rheumatol. Int. 2008, 28, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, J.; Sheng, Z.; Wu, X.; Liao, E. Relationship between body composition and age, menopause and its effects on bone mineral density at segmental regions in Central Southern Chinese postmenopausal elderly women with and without osteoporosis. Arch. Gerontol. Geriatr. 2010, 53, e192–e197. [Google Scholar] [CrossRef]
- Parker, S.E.; Troisi, R.; Wise, L.A.; Palmer, J.R.; Titus-Ernstoff, L.; Strohsnitter, W.C.; Hatch, E.E. Menarche, Menopause, Years of Menstruation, and the Incidence of Osteoporosis: The Influence of Prenatal Exposure to Diethylstilbestrol. J. Clin. Endocrinol. Metab. 2014, 99, 594–601. [Google Scholar] [CrossRef]
- Qiu, C.; Chen, H.; Wen, J.; Zhu, P.; Lin, F.; Huang, B.; Wu, P.; Lin, Q.; Lin, Y.; Rao, H.; et al. Associations Between Age at Menarche and Menopause With Cardiovascular Disease, Diabetes, and Osteoporosis in Chinese Women. J. Clin. Endocrinol. Metab. 2013, 98, 1612–1621. [Google Scholar] [CrossRef]
- Yoldemir, T.; Erenus, M.; Durmusoglu, F. The impact of serum FSH and estradiol on postmenopausal osteoporosis related to time since menopause. Gynecol. Endocrinol. 2012, 28, 884–888. [Google Scholar] [CrossRef]
- Zhao, L.; Cui, B.; Liu, J.-M.; Zhang, M.-J.; Zhao, H.-Y.; Sun, L.-H.; Tao, B.; Zhang, L.-Z.; Ning, G. Interactions of osteoporosis candidate genes for age at menarche, age at natural menopause, and maximal height in Han Chinese women. Menopause 2011, 18, 1018–1025. [Google Scholar] [CrossRef]
- Özkaya, E.; Cakir, E.; Okuyan, E.; Çakir, C.; Üstün, G.; Küçüközkan, T. Comparison of the effects of surgical and natural menopause on carotid intima media thickness, osteoporosis, and homocysteine levels. Menopause 2011, 18, 73–76. [Google Scholar] [CrossRef]
- Chen, G.; Chen, L.; Wen, J.; Yao, J.; Li, L.; Lin, L.; Tang, K.; Huang, H.; Liang, J.; Lin, W.; et al. Associations Between Sleep Duration, Daytime Nap Duration, and Osteoporosis Vary by Sex, Menopause, and Sleep Quality. J. Clin. Endocrinol. Metab. 2014, 99, 2869–2877. [Google Scholar] [CrossRef]
- Management of osteoporosis in postmenopausal women. Menopause 2010, 17, 25–54. [CrossRef] [PubMed]
- Liu, H.; Huang, H.; Li, B.; Wu, D.; Wang, F.; Zheng, X.; Chen, Q.; Wu, B.; Fan, X. Olive oil in the prevention and treatment of osteoporosis after artificial menopause. Clin. Interv. Aging 2014, 9, 2087–2095. [Google Scholar] [CrossRef]
- Ring, M. Women’s Health. Prim. Care Clin. Off. Pract. 2017, 44, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, N.; Sánchez-Borrego, R.; Villero, J.; Baró, F.; Calaf, J.; Cancelo, M.J.; Coronado, P.; Estévez, A.; Fernández-Moya, J.M.; González, S.; et al. 2013 Up-date of the consensus statement of the Spanish Menopause Society on postmenopausal osteoporosis. Maturitas 2013, 76, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Compston, J. How to manage osteoporosis after the menopause. Best Pract. Res. Clin. Rheumatol. 2005, 19, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- De Quadros, V.P.; Tobar, N.; Viana, L.R.; Dos Santos, R.W.; Kiyataka, P.H.M.; Gomes-Marcondes, M.C.C. The 17β-oestradiol treatment minimizes the adverse effects of protein restriction on bone parameters in ovariectomized Wistar rats. Bone Jt. Res. 2019, 8, 573–581. [Google Scholar] [CrossRef]
- Placios, S.; Ríos, A.M. Bazedoxifene/conjugated estrogens combination for the treatment of the vasomotor symptoms associated with menopause and for prevention of osteoporosis in postmenopausal women. Drugs Today 2015, 51, 107. [Google Scholar] [CrossRef]
- Umland, E.M.; Karel, L.; Santoro, N. Bazedoxifene and Conjugated Equine Estrogen: A Combination Product for the Management of Vasomotor Symptoms and Osteoporosis Prevention Associated with Menopause. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2016, 36, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.; Angioli, R.; Coleman, R.L.; Glasspool, R.; Plotti, F.; Simoncini, T.; Terranova, C. European Menopause and Andropause Society (EMAS) and International Gynecologic Cancer Society (IGCS) position statement on managing the menopause after gynecological cancer: Focus on menopausal symptoms and osteoporosis. Maturitas 2020, 134, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Baldi, S.; Becorpi, A. Prevention, diagnosis and treatment of osteoporosis following menopause induced due to oncological disease. Bone Abstr. 2009, 6, 261–263. [Google Scholar]
- Andersen, C.Y.; Kristensen, S.G. Novel use of the ovarian follicular pool to postpone menopause and delay osteoporosis. Reprod. Biomed. Online 2015, 31, 128–131. [Google Scholar] [CrossRef]
- Sciannamblo, M.; Russo, G.; Cuccato, D.; Chiumello, G.; Mora, S. Reduced Bone Mineral Density and Increased Bone Metabolism Rate in Young Adult Patients with 21-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab. 2006, 91, 4453–4458. [Google Scholar] [CrossRef]
- Ishida, Y.; Heersche, J.N.M. Glucocorticoid-Induced Osteoporosis: Both In Vivo and In Vitro Concentrations of Glucocorticoids Higher Than Physiological Levels Attenuate Osteoblast Differentiation. J. Bone Miner. Res. 1998, 13, 1822–1826. [Google Scholar] [CrossRef]
- Adami, G.; Saag, K.G. Glucocorticoid-induced osteoporosis update. Curr. Opin. Rheumatol. 2019, 31, 388–393. [Google Scholar] [CrossRef]
- Faienza, M.F.; Brunetti, G.; Colucci, S.; Piacente, L.; Ciccarelli, M.; Giordani, L.; Del Vecchio, G.C.; D’Amore, M.; Albanese, L.; Cavallo, L.; et al. Osteoclastogenesis in Children with 21-Hydroxylase Deficiency on Long-Term Glucocorticoid Therapy: The Role of Receptor Activator of Nuclear Factor-κB Ligand/Osteoprotegerin Imbalance. J. Clin. Endocrinol. Metab. 2009, 94, 2269–2276. [Google Scholar] [CrossRef]
- Boling, E.P. Secondary osteoporosis: Underlying disease and the risk for glucocorticoid-induced osteoporosis. Clin. Ther. 2004, 26, 1–14. [Google Scholar] [CrossRef]
- Adler, R.A. Glucocorticoid-Induced Osteoporosis: Management Challenges in Older Patients. J. Clin. Densitom. 2019, 22, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Adinoff, A. Prevention of Glucocorticoid-Induced Osteoporosis: Experience in a Managed Care Setting. Pediatrics 2002, 110, 462–463. [Google Scholar] [CrossRef]
- Berris, K.K.; Repp, A.L.; Kleerekoper, M. Glucocorticoid-induced osteoporosis. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Mellibovsky, L.; Prieto-Alhambra, D.; Mellibovsky, F.; Güerri-Fernández, R.; Nogués, X.; Randall, C.; Hansma, P.K.; Díez-Perez, A. Bone Tissue Properties Measurement by Reference Point Indentation in Glucocorticoid-Induced Osteoporosis. J. Bone Miner. Res. 2015, 30, 1651–1656. [Google Scholar] [CrossRef]
- Woolf, A.D. An update on glucocorticoid-induced osteoporosis. Curr. Opin. HIV AIDS 2007, 6, 544–549. [Google Scholar] [CrossRef]
- Kilbreath, S.; Refshauge, K.M.; Beith, J.; Ward, L.; Sawkins, K.; Paterson, R.; Clifton-Bligh, P.; Sambrook, P.N.; Simpson, J.M.; Nery, L. Prevention of osteoporosis as a consequence of aromatase inhibitor therapy in postmenopausal women with early breast cancer: Rationale and design of a randomized controlled trial. Contemp. Clin. Trials 2011, 32, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Hadji, P. Aromatase inhibitor-associated bone loss in breast cancer patients is distinct from postmenopausal osteoporosis. Crit. Rev. Oncol. 2009, 69, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, T.; Maita, S.; Tsuchiya, N.; Ma, Z.; Narita, S.; Horikawa, Y.; Yamamoto, S.; Yonese, J.; Fukui, I.; Takahashi, S.; et al. Relationship Between Bone Mineral Density and Androgen-deprivation Therapy in Japanese Prostate Cancer Patients. Urology 2010, 75, 1131–1137. [Google Scholar] [CrossRef]
- Miyaji, Y.; Saika, T.; Yamamoto, Y.; Kusaka, N.; Arata, R.; Ebara, S.; Nasu, Y.; Tsushima, T.; Kumon, H. Effects of gonadotropin-releasing hormone agonists on bone metabolism markers and bone mineral density in patients with prostate cancer. Urology 2004, 64, 128–131. [Google Scholar] [CrossRef]
- Shahinian, V.B.; Kuo, Y.-F.; Freeman, J.L.; Goodwin, J.S. Risk of Fracture after Androgen Deprivation for Prostate Cancer. New Engl. J. Med. 2005, 352, 154–164. [Google Scholar] [CrossRef]
- Khriguian, J.; Tsui, J.M.G.; Vaughan, R.; Kucharczyk, M.J.; Nabid, A.; Bettahar, R.; Vincent, L.; Martin, A.-G.; Jolicoeur, M.; Yassa, M.; et al. The Clinical Significance of Bone Mineral Density Changes Following Long-Term Androgen Deprivation Therapy in Localized Prostate Cancer Patients. J. Urol. 2021, 205, 1648–1654. [Google Scholar] [CrossRef]
- A Moyad, M. Complementary therapies for reducing the risk of osteoporosis in patients receiving luteinizing hormone-releasing hormone treatment/orchiectomy for prostate cancer: A review and assessment of the need for more research. Urology 2002, 59, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bui, K.T.; Willson, M.L.; Goel, S.; Beith, J.; Goodwin, A. Ovarian suppression for adjuvant treatment of hormone receptor-positive early breast cancer. Cochrane Database Syst. Rev. 2020, 3, CD013538. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.L.; Loveridge, N.; Lindsay, P.C.; Lunt, M.; Garrahan, N.; Compston, J.E.; Reeve, J. Cortical Remodeling Following Suppression of Endogenous Estrogen with Analogs of Gonadotrophin Releasing Hormone. J. Bone Miner. Res. 1997, 12, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Surrey, E.S. Gonadotropin-releasing hormone agonist and add-back therapy: What do the data show? Curr. Opin. Obstet. Gynecol. 2010, 22, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H. Selective estrogen receptor modulators (SERMS): Keys to understanding their function. Menopause 2020, 27, 1171–1176. [Google Scholar] [CrossRef]
- Chou, Y.-S.; Jiang, H.-J.; Chen, C.-H.; Ho, P.-S.; Lee, T.-C. Proton pump inhibitor use and risk of hip fracture in patients with type 2 diabetes. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Tung, Y.-C.; Chai, C.-Y.; Lu, Y.-Y.; Su, Y.-F.; Tsai, T.-H.; Kuo, K.-L.; Lin, C.-L. Increased Risk of Osteoporosis in Patients With Peptic Ulcer Disease: A Nationwide Population-Based Study. Medicine 2016, 95, e3309. [Google Scholar] [CrossRef]
- Targownik, L.E.; Lix, L.M.; Metge, C.J.; Prior, H.J.; Leung, S.; Leslie, W.D. Use of proton pump inhibitors and risk of osteoporosis-related fractures. Can. Med. Assoc. J. 2008, 179, 319–326. [Google Scholar] [CrossRef]
- Andersen, B.N.; Johansen, P.B.; Abrahamsen, B. Proton pump inhibitors and osteoporosis. Curr. Opin. Rheumatol. 2016, 28, 420–425. [Google Scholar] [CrossRef]
- E Targownik, L.; Goertzen, A.L.; Luo, Y.; Leslie, W. Long-Term Proton Pump Inhibitor Use Is Not Associated With Changes in Bone Strength and Structure. Am. J. Gastroenterol. 2017, 112, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Parveen, B.; Tiwari, A.K.; Jain, M.; Pal, S.; Chattopadhyay, N.; Tripathi, M.; Vohora, D. The anti-epileptic drugs valproate, carbamazepine and levetiracetam cause bone loss and modulate Wnt inhibitors in normal and ovariectomised rats. Bone 2018, 113, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Ensrud, K.; Walczak, T.S.; Blackwell, T.; Ensrud, E.R.; Bowman, P.J.; Stone, K.L. Antiepileptic drug use increases rates of bone loss in older women: A prospective study. Neurology 2004, 62, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.S.; Hill, K.D.; O’Brien, T.J.; Gorelik, A.; Habib, N.; Wark, J.D. Falls and fractures in patients chronically treated with antiepileptic drugs. Neurology 2012, 79, 145–151. [Google Scholar] [CrossRef]
- Ensrud, K.E.; Walczak, T.S.; Blackwell, T.L.; Ensrud, E.R.; Barrett-Connor, E.; Orwoll, E.S. For the Osteoporotic Fractures in Men (MrOS) Study Research Group Antiepileptic drug use and rates of hip bone loss in older men: A prospective study. Neurology 2008, 71, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Scholes, D.; LaCroix, A.Z.; Ichikawa, L.E.; Barlow, W.E.; Ott, S.M. Injectable Hormone Contraception and Bone Density: Results from a Prospective Study. Epidemiology 2002, 13, 581–587. [Google Scholar] [CrossRef]
- Cromer, B.A. Bone mineral density in adolescent and young adult women on injectable or oral contraception. Curr. Opin. Obstet. Gynecol. 2003, 15, 353–357. [Google Scholar] [CrossRef]
- Lo, S.S.T.; Fan, S.Y.S. Bone loss associated with long-term use of depot medroxyprogesterone acetate. Hong Kong Med. J. 2005, 11, 491–495. [Google Scholar] [PubMed]
- Sambrook, P.N.; Kotowicz, M.; Nash, P.; Styles, C.B.; Naganathan, V.; Henderson-Briffa, K.N.; A Eisman, J.; Nicholson, G.C.; Nicholson, G. Prevention and Treatment of Glucocorticoid-Induced Osteoporosis: A Comparison of Calcitriol, Vitamin D Plus Calcium, and Alendronate Plus Calcium. J. Bone Miner. Res. 2003, 18, 919–924. [Google Scholar] [CrossRef]
- Wang, Y.-K.; Zhang, Y.-M.; Qin, S.-Q.; Wang, X.; Ma, T.; Guo, J.-B.; Zhu, C.; Luo, Z.-J. Effects of alendronate for treatment of glucocorticoid-induced osteoporosis: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e12691. [Google Scholar] [CrossRef]
- Kan, S.-L.; Yuan, Z.-F.; Li, Y.; Ai, J.; Xu, H.; Sun, J.-C.; Feng, S.-Q. Alendronate prevents glucocorticoid-induced osteoporosis in patients with rheumatic diseases: A meta-analysis. Medicine 2016, 95, e3990. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, E.; Ito, S.; Takai, C.; Kobayashi, D.; Nomura, Y.; Otani, H.; Abe, A.; Ishikawa, H.; Murasawa, A.; Narita, I.; et al. The Efficacy of Minodronate in the Treatment of Glucocorticoid-induced Osteoporosis. Intern. Med. 2018, 57, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Beest, F.J.P.-V.; Goettsch, W.G.; Erkens, J.A.; Herings, R.M. Determinants of persistence with bisphosphonates: A study in women with postmenopausal osteoporosis. Clin. Ther. 2006, 28, 236–242. [Google Scholar] [CrossRef]
- Reid, D.M.; Devogelaer, J.-P.; Saag, K.; Roux, C.; Lau, C.-S.; Reginster, J.-Y.; Papanastasiou, P.; Ferreira, A.; Hartl, F.; Fashola, T.; et al. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): A multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009, 373, 1253–1263. [Google Scholar] [CrossRef]
- Campbell, S.C.; Bhoopalam, N.; Moritz, T.E.; Pandya, M.; Iyer, P.; VanVeldhuizen, P.; Ellis, N.K.; Thottapurathu, L.; Garewal, H.; Warren, S.R.; et al. The Use of Zoledronic Acid in Men Receiving Androgen Deprivation Therapy for Prostate Cancer With Severe Osteopenia or Osteoporosis. Urology 2010, 75, 1138–1143. [Google Scholar] [CrossRef]
- Lane, N.E.; Sanchez, S.; Modin, G.W.; Genant, H.K.; Pierini, E.; Arnaud, C.D. Bone Mass Continues to Increase at the Hip After Parathyroid Hormone Treatment Is Discontinued in Glucocorticoid-Induced Osteoporosis: Results of a Randomized Controlled Clinical Trial. J. Bone Miner. Res. 2010, 15, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Han, C.; Tian, P.; Li, P.-F.; Ma, X.-L. Role of Teriparatide in Glucocorticoid-induced Osteoporosis through Regulating Cellular Reactive Oxygen Species: T eriparatide IN G lucocorticoid -I nduced O steoporosis. Orthop. Surg. 2018, 10, 152–159. [Google Scholar] [CrossRef]
- Quattrocchi, E.; Kourlas, H. Teriparatide: A review. Clin. Ther. 2004, 26, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Glüer, C.; Marin, F.; Ringe, J.D.; Hawkins, F.; Möricke, R.; Papaioannu, N.; Farahmand, P.; Minisola, S.; Martínez, G.; Nolla, J.M.; et al. Comparative effects of teriparatide and risedronate in glucocorticoid-induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J. Bone Miner. Res. 2013, 28, 1355–1368. [Google Scholar] [CrossRef]
- Hsu, E.; Nanes, M. Advances in treatment of glucocorticoid-induced osteoporosis. Curr. Opin. Endocrinol. Diabetes 2017, 24, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.E.; Wilson, H.A.; Zapalowski, C.; Fink, H.A.; Minisola, S.; Adler, R.A. Uncertainties in the prevention and treatment of glucocorticoid-induced osteoporosis. J. Bone Miner. Res. 2011, 26, 1989–1996. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Pang, Q.; Tao, D. Alpinumisoflavone protects against glucocorticoid-induced osteoporosis through suppressing the apoptosis of osteoblastic and osteocytic cells. Biomed. Pharmacother. 2017, 96, 993–999. [Google Scholar] [CrossRef]
- Chen, Z.; Xue, J.; Shen, T.; Ba, G.; Yu, D.; Fu, Q. Curcumin alleviates glucocorticoid-induced osteoporosis by protecting osteoblasts from apoptosis in vivo and in vitro. Clin. Exp. Pharmacol. Physiol. 2016, 43, 268–276. [Google Scholar] [CrossRef]
- Sato, A.Y.; Cregor, M.; Delgado-Calle, J.; Condon, K.W.; Allen, M.R.; Peacock, M.; I Plotkin, L.; Bellido, T. Protection From Glucocorticoid-Induced Osteoporosis by Anti-Catabolic Signaling in the Absence of Sost/Sclerostin: Anti-catabolic signaling protects sost −/− mice from Gc-induced osteoporosis. J. Bone Miner. Res. 2016, 31, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, D.; Yang, J.-Y.; Yan, T.-B. Plumbagin protects against glucocorticoid-induced osteoporosis through Nrf-2 pathway. Cell Stress Chaperones 2015, 20, 621–629. [Google Scholar] [CrossRef]
- Lekva, T.; Bollerslev, J.; Kristo, C.; Olstad, O.K.; Ueland, T.; Jemtland, R. The Glucocorticoid-Induced Leucine Zipper Gene (GILZ) Expression Decreases after Successful Treatment of Patients with Endogenous Cushing’s Syndrome and May Play a Role in Glucocorticoid-Induced Osteoporosis. J. Clin. Endocrinol. Metab. 2010, 95, 246–255. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, W.; Zhu, D.; Li, Z.; Wang, Z.; Li, J.; Mei, X.; Xu, W.; Cheng, K.; Zhong, B. Nanoparticles functionalized with stem cell secretome and CXCR4-overexpressing endothelial membrane for targeted osteoporosis therapy. J. Nanobiotechnology 2022, 20, 35. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; He, S.-H.; Xie, P.-Y.; Li, W.; Zhang, X.-X.; Li, T.-F.; Li, D.-F. Recent Progresses in the Treatment of Osteoporosis. Front. Pharmacol. 2021, 12, 717065. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, F.; Muhamad, I.I.; Nazari, Z.; Mobini, P.; Khatir, N.M. Investigation of acyclovir-loaded, acrylamide-based hydrogels for potential use as vaginal ring. Mater. Today Commun. 2018, 16, 274–280. [Google Scholar] [CrossRef]
- Shkembi, B.; Huppertz, T. Calcium Absorption from Food Products: Food Matrix Effects. Nutrients 2021, 14, 180. [Google Scholar] [CrossRef]
- Vannucci, L.; Fossi, C.; Quattrini, S.; Guasti, L.; Pampaloni, B.; Gronchi, G.; Giusti, F.; Romagnoli, C.; Cianferotti, L.; Marcucci, G.; et al. Calcium Intake in Bone Health: A Focus on Calcium-Rich Mineral Waters. Nutrients 2018, 10, 1930. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Medications | Mechanisms to Induce Osteoporosis | Pathways/Related Mediators |
|---|---|---|
| Glucocorticoids (GCs) | promote the differentiation, proliferation, and activation of osteoclasts; suppress the differentiation of osteoblasts; induce the apoptosis of osteoblasts and osteocytes | RANKL, M-CSF, OPG, DKK-1, Notch pathway, TGFβ, IGF-1, SOD, CAT, caspase 3 |
| Aromatase Inhibitors (AIs) | reduce the production of estrogen, leading to uncoupling of bone resorption and formation, increased differentiation of osteoclasts and decreased differentiation of osteoblasts | OPG, RANKL, M-CSF, TGFβ, IL-1, TNF |
| GnRH agonists | ||
| Chemotherapy | ||
| Serotonin selective reuptake inhibitors (SSRIs) | ||
| Medroxyprogesterone acetate (MPA) | ||
| Proton pump inhibitors (PPIs) | decrease intestinal calcium absorption | |
| Antiepileptic drugs (AEDs) | decrease intestinal calcium absorption; increase proliferation of osteoclasts | RANKL, DKK-1 |
| Bisphosphonates | 2000: Fracture risk reduction with alendronate in postmenopausal women 2000: Randomized trial of the effects of risedronate on vertebral fractures in postmenopausal women 2007: Effects of continuing or stopping alendronate after 5 years of treatment in postmenopausal women 2007: Once yearly zoledronic acid for treatment of osteoporosis in postmenopausal women |
| Estrogen | 2002: Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results from the Women’s Health Initiative Randomized Controlled Trial |
| RANKL inhibitor | 2009: Denosumab for prevention of fractures in postmenopausal women with osteoporosis 2017: 10 years of denosumab treatment in postmenopausal women with osteoporosis |
| SERM | 2008: Efficacy of Bazedoxifene in Reducing New Vertebral Fracture Risk in Postmenopausal Women with Osteoporosis: Results from a 3-Year, Randomized, Placebo-, and Active-Controlled Clinical Trial |
| PTH analogues | 2001: Effect of parathyroid hormone on fractures and bone mineral density in postmenopausal women with osteoporosis 2016: Effect of Abaloparatide vs. Placebo on new vertebral fracture in postmenopausal women with osteoporosis |
| Sclerostine inhibitor | 2018: The foundation effect of Building bone with 1 year of Romosozumab leads to continued lower fracture risk after transition to Denosumab in postmenopausal women |
| Cathepsin K inhibitor | 2014: The Effect of the Cathepsin K Inhibitor ONO-5334 on Trabecular and Cortical Bone in Postmenopausal Osteoporosis: the OCEAN Study 2016: Continuous Treatment with Odanacatib for up to 8 Years in Postmenopausal Women with Low Bone mineral Density: a Phase 2 Study |
| Advantages | Disadvantages | |
|---|---|---|
| Bisphosphonates |
|
|
| PTH analogs |
|
|
| RANKL inhibitor |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-T.; Chen, L.-R.; Chen, K.-H. Hormone-Related and Drug-Induced Osteoporosis: A Cellular and Molecular Overview. Int. J. Mol. Sci. 2023, 24, 5814. https://doi.org/10.3390/ijms24065814
Wang L-T, Chen L-R, Chen K-H. Hormone-Related and Drug-Induced Osteoporosis: A Cellular and Molecular Overview. International Journal of Molecular Sciences. 2023; 24(6):5814. https://doi.org/10.3390/ijms24065814
Chicago/Turabian StyleWang, Li-Ting, Li-Ru Chen, and Kuo-Hu Chen. 2023. "Hormone-Related and Drug-Induced Osteoporosis: A Cellular and Molecular Overview" International Journal of Molecular Sciences 24, no. 6: 5814. https://doi.org/10.3390/ijms24065814
APA StyleWang, L.-T., Chen, L.-R., & Chen, K.-H. (2023). Hormone-Related and Drug-Induced Osteoporosis: A Cellular and Molecular Overview. International Journal of Molecular Sciences, 24(6), 5814. https://doi.org/10.3390/ijms24065814