
The Cell Transformation Assay: A Historical Assessment of Current Knowledge of Applications in an Integrated Approach to Testing and Assessment for Non-Genotoxic Carcinogens
 , , and
, , and
Abstract
1. Introduction
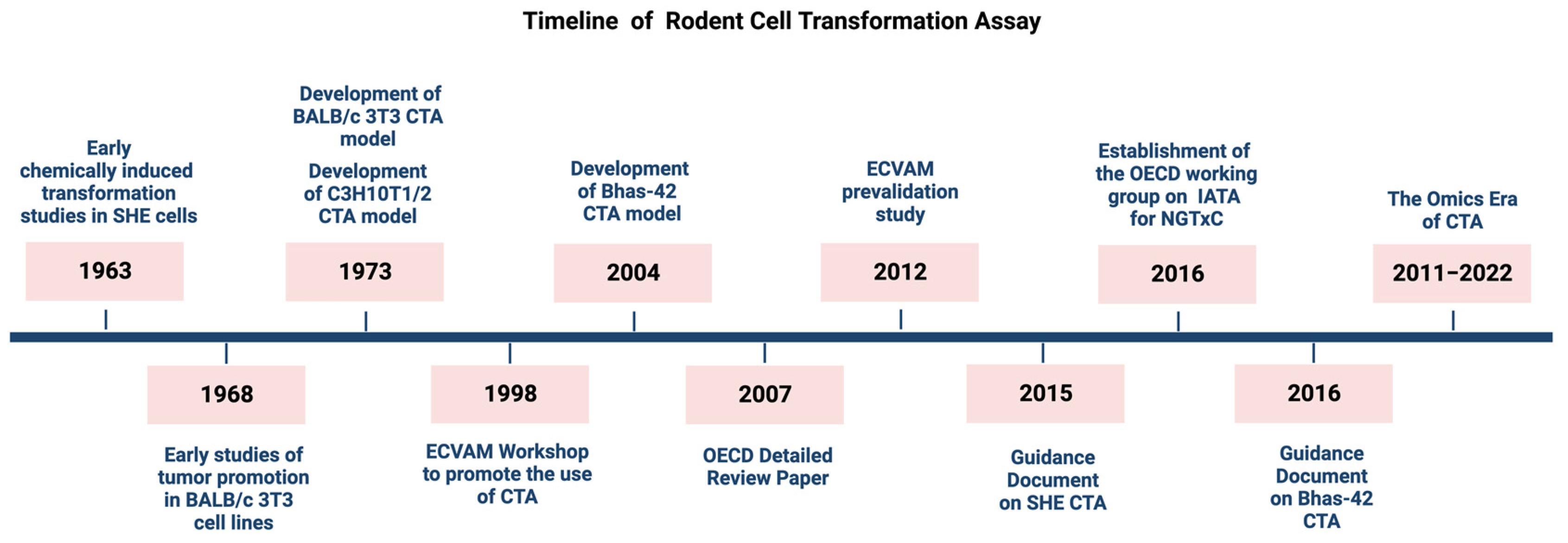
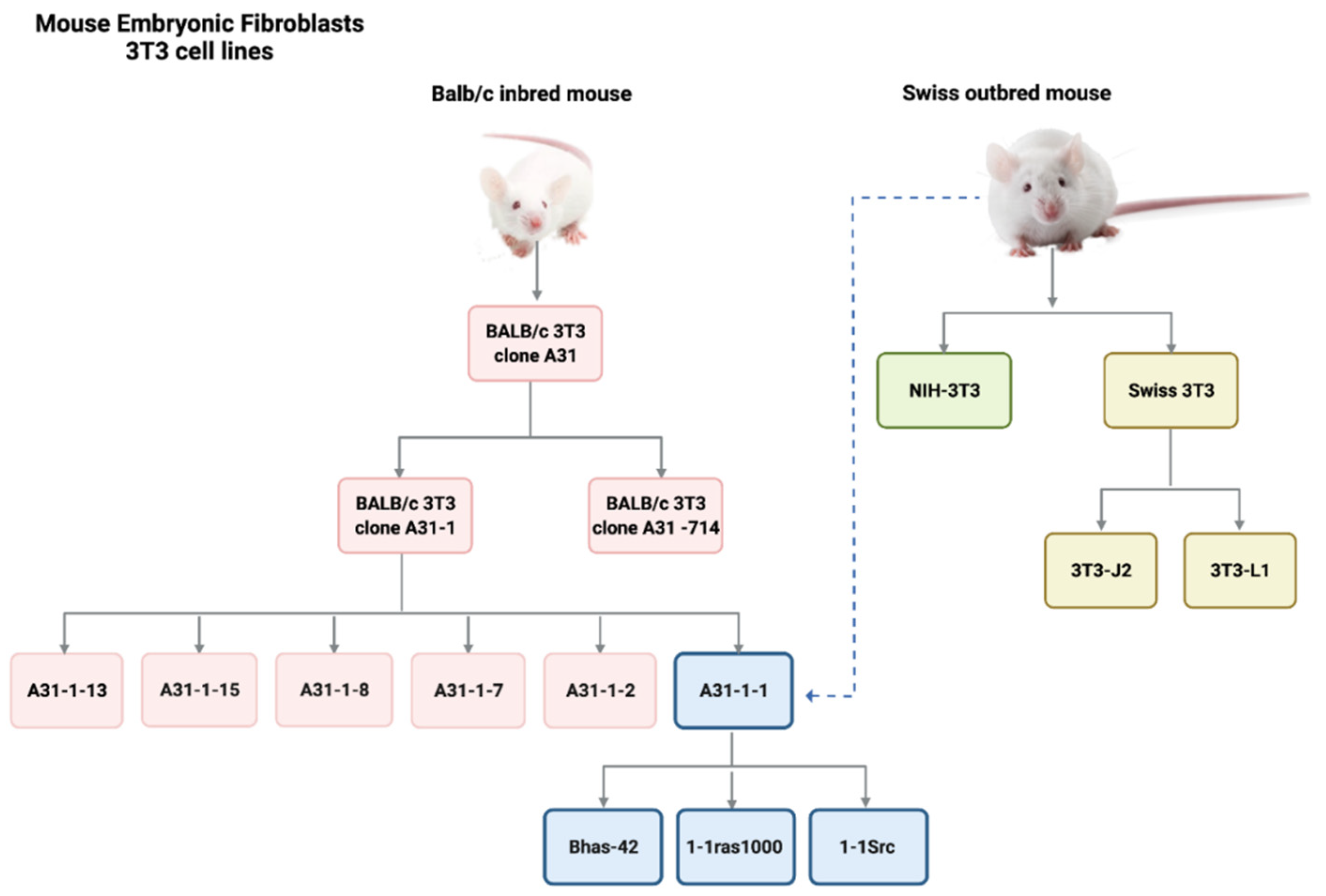
A Historical Perspective on Cell Transformation Assay Models
2. Cellular and Molecular Mechanisms of Cell Transformation and Their Significance for In Vivo Carcinogenesis
2.1. The Role of Fibroblasts in Cancer
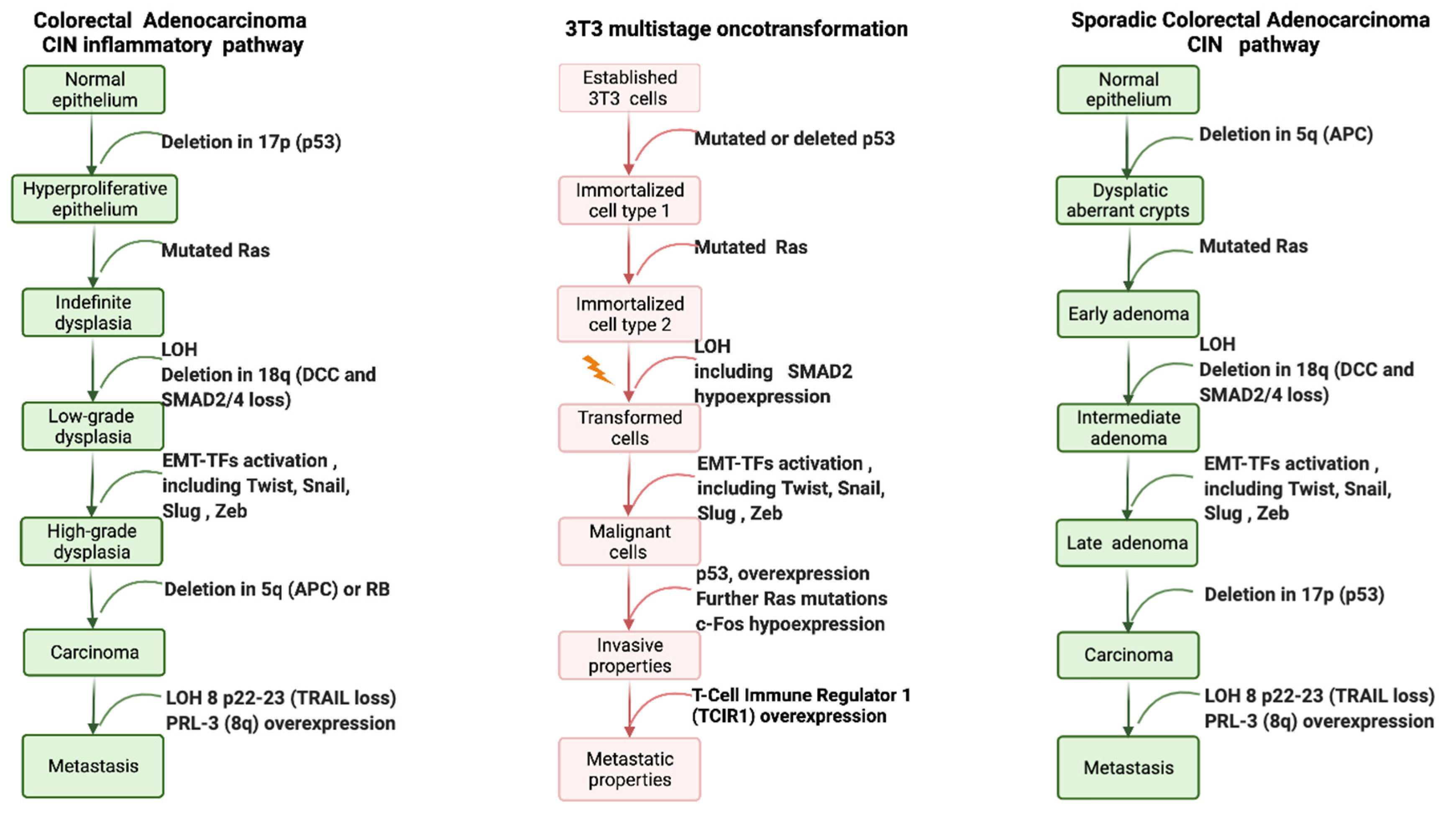
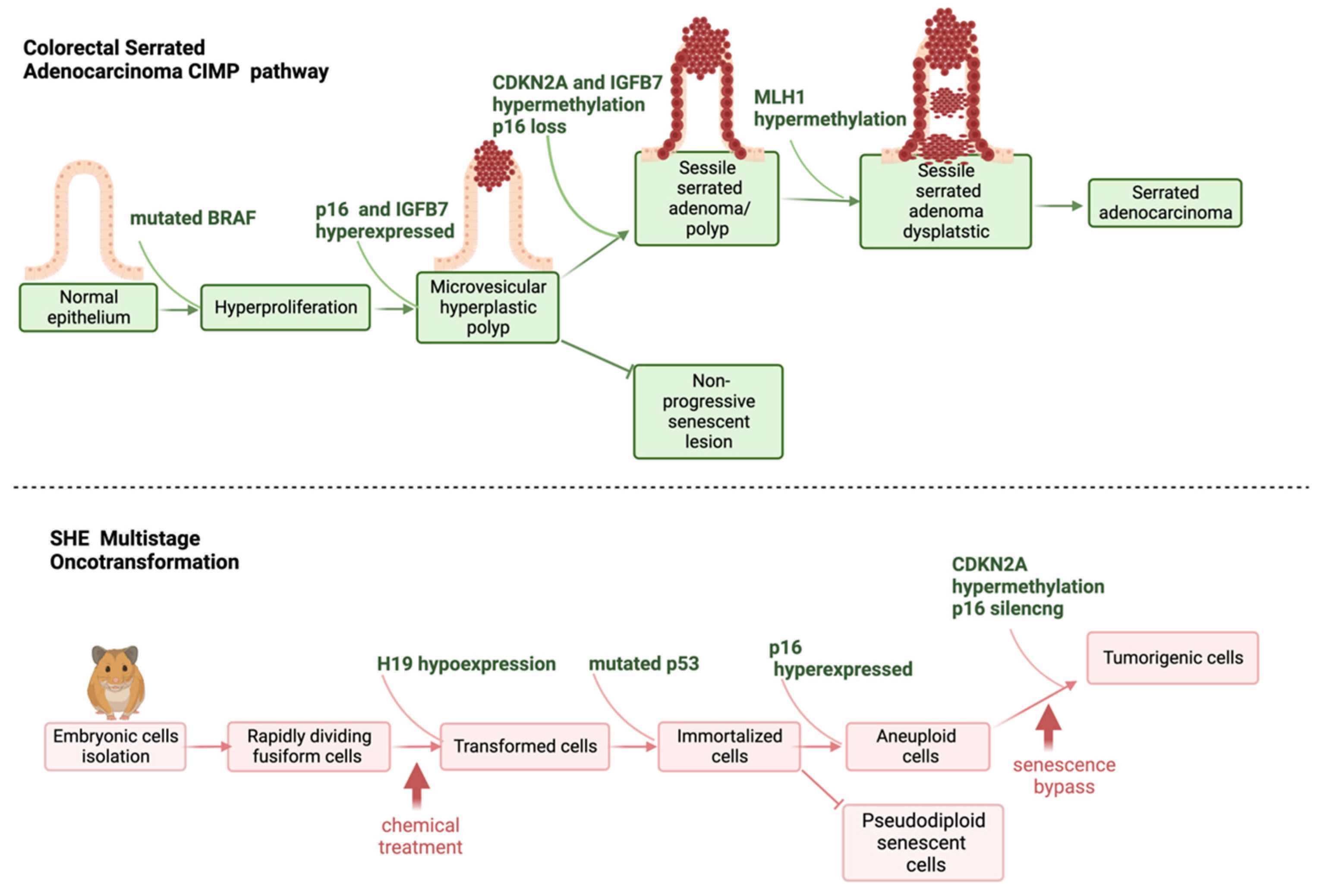
2.2. Multistep Oncotransformation Models
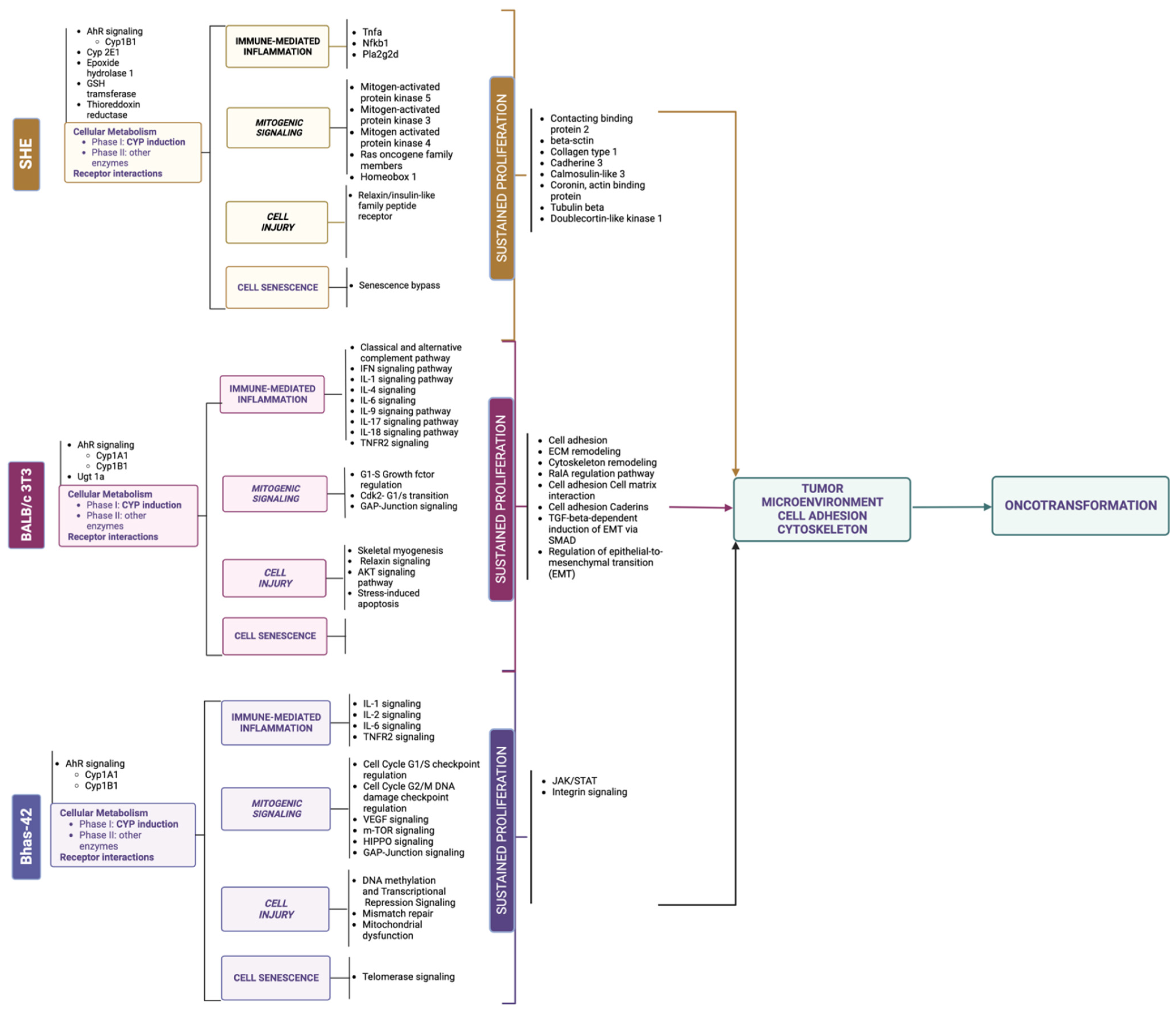
2.3. Molecular Signatures in In Vitro Cell Oncotransformation
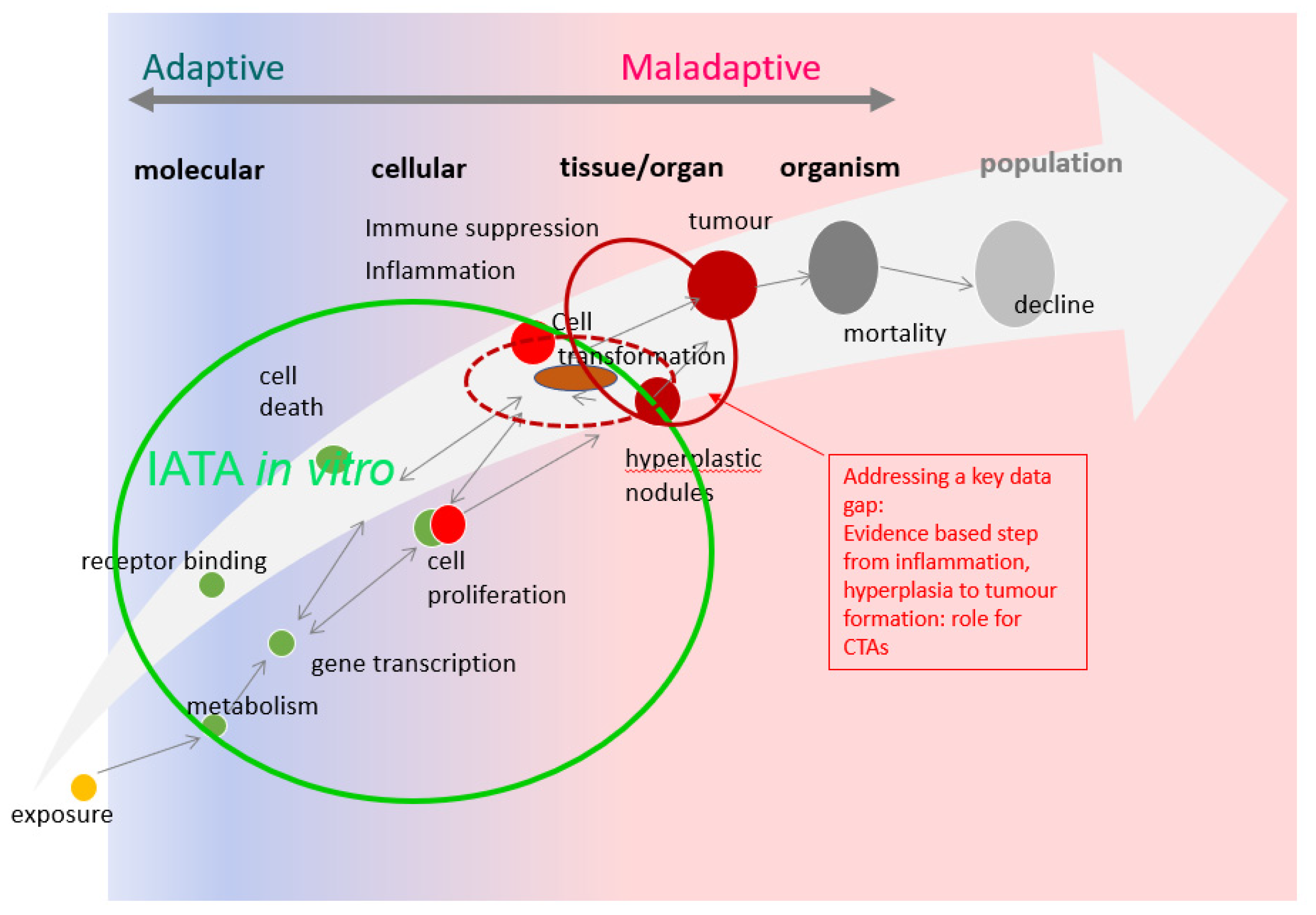
2.4. Current In Vivo and In Vitro CTA Models Depict Different Steps of Carcinogenesis; How Can These Be Appropriately Integrated?
2.4.1. The Mouse Skin Model
2.4.2. Revisiting the Concept of Promotion
2.4.3. The Rat Hepatocarcinogenesis Model
2.4.4. Two-Stage Cell Transformation Assay
2.4.5. Overview of Chemicals Tested in SHE and BALB/c 3T3 Models Recognized to Be Carcinogens versus Non-Genotoxic Carcinogenic Mechanisms
2.4.6. Ability of the SHE CTA to Identify Carcinogens with Non-Genotoxic Properties
2.4.7. The BALB/c 3T3 CTA Performance in Identifying Non-Genotoxic Carcinogenic Chemicals
2.4.8. Bhas 42 CTA Performance in Identifying Non-Genotoxic Carcinogenic Chemicals and Mechanistic Studies
3. Discussion and Next Steps
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barrett, J.C. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ. Health Perspect. 1993, 100, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Colacci, A. The carcinogenesis process. In Oncologia Genetica; Grilli, S., Amadori, D., Eds.; Poletto Editore: Milan, Italy, 2001; pp. 21–39. [Google Scholar]
- Jacobs, M.N.; Colacci, A.; Corvi, R.; Vaccari, M.; Aguila, M.C.; Corvaro, M.; Delrue, N.; Desaulniers, D.; Ertych, N.; Jacobs, A.; et al. Chemical carcinogen safety testing: OECD expert group international consensus on the development of an integrated approach for the testing and assessment of chemical non-genotoxic carcinogens. Arch. Toxicol. 2020, 94, 2899–2923. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Colacci, A.; Louekari, K.; Luijten, M.; Hakkert, B.C.; Paparella, M.; Vasseur, P. International regulatory needs for development ofan IATA for non-genotoxic carcinogenic chemical substances. Altex 2016, 33, 359–392. [Google Scholar] [CrossRef] [PubMed]
- Corvi, R.; Madia, F.; Guyton, K.Z.; Kasper, P.; Rudel, R.; Colacci, A.; Kleinjans, J.; Jennings, P. Moving forward in carcinogenicity assessment: Report of an EURL ECVAM/ESTIV workshop. Toxicol. Vitr. 2017, 45 Pt 3, 278–286. [Google Scholar] [CrossRef]
- Paparella, M.; Colacci, A.; Jacobs, M.N. Uncertainties of testing methods: What do we (want to) know about carcinogenicity? Altex 2017, 34, 235–252. [Google Scholar] [CrossRef]
- Combes, R.; Grindon, C.; Cronin, M.T.; Roberts, D.W.; Garrod, J. Proposed Integrated Decision-tree Testing Strategies for Mutagenicity and Carcinogenicity in Relation to the EU REACH Legislation. Altern. Lab. Anim. 2007, 35, 267–287. [Google Scholar] [CrossRef] [PubMed]
- Corvi, R.; Vanparys, P. International prevalidation study on cell transformation assay. Preface. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 1–2. [Google Scholar] [CrossRef]
- Lilienblum, W.; Dekant, W.; Foth, H.; Gebel, T.; Hengstler, J.G.; Kahl, R.; Kramer, P.-J.; Schweinfurth, H.; Wollin, K.-M. Alternative methods to safety studies in experimental animals: Role in the risk assessment of chemicals under the new European Chemicals Legislation (REACH). Arch. Toxicol. 2008, 82, 211–236. [Google Scholar] [CrossRef]
- Mascolo, M.G.; Perdichizzi, S.; Rotondo, F.; Morandi, E.; Guerrini, A.; Silingardi, P.; Vaccari, M.; Grilli, S.; Colacci, A. BALB/c 3T3 cell transformation assay for the prediction of carcinogenic potential of chemicals and environmental mixtures. Toxicol. Vitr. 2010, 24, 1292–1300. [Google Scholar] [CrossRef]
- Rohrbeck, A.; Salinas, G.; Maaser, K.; Linge, J.; Salovaara, S.; Corvi, R.; Borlak, J. Toxicogenomics Applied to In Vitro Carcinogenicity Testing with Balb/c 3T3 Cells Revealed a Gene Signature Predictive of Chemical Carcinogens. Toxicol. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef]
- Vanparys, P.; Corvi, R.; Aardema, M.J.; Gribaldo, L.; Hayashi, M.; Hoffmann, S.; Schechtman, L. Application of in vitro cell transformation assays in regulatory toxicology for pharmaceuticals, chemicals, food products and cosmetics. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 111–116. [Google Scholar] [CrossRef]
- Vasseur, P.; Lasne, C. OECD Detailed Review Paper (DRP) number 31 on Cell Transformation Assays for Detection of Chemical Carcinogens: Main results and conclusions. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 8–11. [Google Scholar] [CrossRef]
- Groupe, V.; Manaker, R.A. Discrete foci of altered chicken embryo cells associated with Rous sarcoma virus in tissue culture. Virology 1956, 2, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Rubin, H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology 1958, 6, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Dulbecco, R. VIRUS-CELL INTERACTION WITH A TUMOR-PRODUCING VIRUS. Proc. Natl. Acad. Sci. USA 1960, 46, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Sachs, L.; Medina, D. In Vitro Transformation of Normal Cells by Polyoma Virus. Nature 1961, 189, 457–458. [Google Scholar] [CrossRef]
- Berwald, Y.; Sachs, L. In Vitro Cell Transformation with Chemical Carcinogens. Nature 1963, 200, 1182–1184. [Google Scholar] [CrossRef]
- Defendi, V.; Lehman, J.M. Transformation of hamster embryo cellsin vitro by polyoma virus: Morphological, karyological, immunological and transplantation characteristics. J. Cell. Comp. Physiol. 1965, 66, 351–409. [Google Scholar] [CrossRef]
- Ahmadzai, A.A.; Trevisan, J.; Fullwood, N.J.; Carmichael, P.L.; Scott, A.D.; Martin, F.L. The Syrian hamster embryo (SHE) assay (pH 6.7): Mechanisms of cell transformation and application of vibrational spectroscopy to objectively score endpoint alterations. Mutagenesis 2011, 27, 257–266. [Google Scholar] [CrossRef]
- Schechtman, L.M. Rodent cell transformation assays—A brief historical perspective. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 3–7. [Google Scholar] [CrossRef]
- OECD. Guidance document on the in vitro Syrian Hamster Embryo (SHE) cell transformation assay. In Series on Testing and Assessment No. 214; Organization for Economic Co-Operation and Development: Paris, France, 2015. [Google Scholar]
- Kakunaga, T. A quantitative system for assay of malignant transformation by chemical carcinogens using a clone derived from BALB/3T3. Int. J. Cancer 1973, 12, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Schechtman, L.M. BALB/c 3T3 cell transformation: Protocols, problems and improvements. IARC Sci. Publ. 1985, 67, 165–184. [Google Scholar] [PubMed]
- Adatia, R.; Albini, A.; Colacci, A.; De Giovanni, C.; Nanni, P.; Giunciuglio, D.; Parodi, S.; Grilli, S.; Perocco, P. Induction of chemotactic and invasive phenotype in BALB/c 3T3 cells by 1,2-dibromoethane transformation. Invasion Metastasis 1993, 13, 234–243. [Google Scholar] [PubMed]
- Melchiori, A.; Colacci, A.; Lollini, P.L.; De Giovanni, C.; Carlone, S.; Grilli, S.; Parodi, S.; Albini, A. Induction of invasive and experimental metastasis potential in BALB/c 3T3 cells by benzo(a)pyrene transformation. Invasion Metastasis 1992, 12, 1–11. [Google Scholar]
- Colacci, A.; Albini, A.; Melchiori, A.; Nanni, P.; Nicoletti, G.; Noonan, D.; Parodi, S.; Grilli, S. Induction of a malignant phenotype in BALB/c 3T3 cells by 1,1,2,2-tetrachloroethane. Int. J. Oncol. 1993, 2, 937–945. [Google Scholar]
- Jacobs, M. In vitro metabolism and bioavailability tests for endocrine active substances: What is needed next for regulatory purposes? Altex 2013, 30, 331–351. [Google Scholar] [CrossRef]
- Colacci, A.; Mascolo, M.G.; Perdichizzi, S.; Quercioli, D.; Gazzilli, A.; Rotondo, F.; Morandi, E.; Guerrini, A.; Silingardi, P.; Grilli, S.; et al. Different sensitivity of BALB/c 3T3 cell clones in the response to carcinogens. Toxicol. Vitr. 2011, 25, 1183–1190. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Kakunaga, T.; Crow, J.D. Cell Variants Showing Differential Susceptibility to Ultraviolet Light—Induced Transformation. Science 1980, 209, 505–507. [Google Scholar] [CrossRef]
- Uchio-Yamada, K.; Kasai, F.; Ozawa, M.; Kohara, A. Incorrect strain information for mouse cell lines: Sequential influence of misidentification on sublines. Vitr. Cell. Dev. Biol. Anim. 2016, 53, 225–230. [Google Scholar] [CrossRef]
- Sasaki, K.; Mizusawa, H.; Ishidate, M.; Tanaka, N. Establishment of a Highly Reproducible Transformation Assay of a Ras-Transfected Balb 3T3 Clone by Treatment with Promoters. In Antimutagenesis and Anticarcinogenesis Mechanisms II; Springer: Berlin/Heidelberg, Germany, 1990; pp. 411–416. [Google Scholar] [CrossRef]
- Tatsuka, M.; Ota, T.; Yamagishi, N.; Kashihara, Y.; Wada, M.; Matsuda, N.; Mitsui, H.; Seiki, M.; Odashima, S. Different metastatic potentials of ras- and src-transformed BALB/c 3T3 A31 variant cells. Mol. Carcinog. 1996, 15, 300–308. [Google Scholar] [CrossRef]
- Kuroki, T.; Miyashita, S.Y.; Yuasa, Y. Development of a 3T3-like line from an embryo culture of an inbred strain of Syrian golden hamster. Cancer Res. 1975, 35, 1819–1825. [Google Scholar]
- Reznikoff, C.A.; Bertram, J.S.; Brankow, D.W.; Heidelberger, C. Quantitative and qualitative studies of chemical transformation of cloned C3H mouse embryo cells sensitive to postconfluence inhibition of cell division. Cancer Res. 1973, 33, 3239–3249. [Google Scholar] [PubMed]
- OECD. Detailed review paper on cell transformation assays for detection of chemical carcinogens series on testing and assessment. In Series on Testing and Assessment No.31; Organisation for Economic Co-Operation and Development, Environment Directorate: Paris, France, 2007. [Google Scholar]
- Sasaki, K.; Mizusawa, H.; Ishidate, M. Isolation and characterization of ras-transfected BALB/3T3 clone showing morphological transformation by 12-O-tetradecanoyl-phorbol-13-acetate. Jpn. J. Cancer Res. 1988, 79, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, K.; Sasaki, K.; Asada, S.; Tanaka, N.; Umeda, M. An assay method for the prediction of tumor promoting potential of chemicals by the use of Bhas 42 cells. Mutat. Res. Toxicol. Environ. Mutagen. 2004, 557, 191–202. [Google Scholar] [CrossRef]
- Ohmori, K.; Umeda, M.; Tanaka, N.; Takagi, H.; Yoshimura, I.; Sasaki, K.; Asada, S.; Sakai, A.; Araki, H.; Asakura, M.; et al. An Inter-laboratory Collaborative Study by the Non-Genotoxic Carcinogen Study Group in Japan, on a Cell Transformation Assay for Tumour Promoters Using Bhas 42 cells. Altern. Lab. Anim. 2005, 33, 619–639. [Google Scholar] [CrossRef]
- Sasaki, K.; Umeda, M.; Sakai, A.; Yamazaki, S.; Tanaka, N. Transformation Assay in Bhas 42 Cells: A Model Using Initiated Cells to Study Mechanisms of Carcinogenesis and Predict Carcinogenic Potential of Chemicals. J. Environ. Sci. Health Part C 2015, 33, 1–35. [Google Scholar] [CrossRef]
- Arai, S.; Sakai, A.; Hayashi, K.; Sasaki, K.; Muramatsu, D.; Endou, N.; Umeda, M.; Tanaka, N. A High-Throughput Cell Transformation Assay Applicable to Automation for Detecting Potential Chemical Carcinogens Using Bhas 42 Cells. Altern. Anim. Test. Exp. 2013, 18, 1–18. [Google Scholar] [CrossRef]
- Corvi, R.; Aardema, M.J.; Gribaldo, L.; Hayashi, M.; Hoffmann, S.; Schechtman, L.; Vanparys, P. ECVAM prevalidation study on in vitro cell transformation assays: General outline and conclusions of the study. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 12–19. [Google Scholar] [CrossRef]
- LeBoeuf, R.; Kerckaert, G.; Aardema, M.; Gibson, D.; Brauninger, R.; Isfort, R. The pH 6.7 Syrian hamster embryo cell transformation assay for assessing the carcinogenic potential of chemicals. Mutat. Res. Mol. Mech. Mutagen. 1996, 356, 85–127. [Google Scholar] [CrossRef]
- Matthews, E.J.; Spalding, J.W.; Tennant, R.W. Transformation of BALB/c-3T3 cells: IV. Rank-ordered potency of 24 chemical responses detected in a sensitive new assay procedure. Environ. Health Perspect. 1993, 101 (Suppl. 2), 319–345. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, M.; Argnani, A.; Horn, W.; Silingardi, P.; Giungi, M.; Mascolo, M.G.; Bartoli, S.; Grilli, S.; Colacci, A. Effects of the protease inhibitor antipain on cell malignant transformation. Anticancer Res. 1999, 19, 589–596. [Google Scholar] [PubMed]
- Schechtman, L.M.; Kiss, E.; McCarvill, J.; Nims, R.; Kouri, R.E.; Lubet, R.A. A method for the amplification of chemically induced transformation in C3H/10T1/2 clone 8 cells: Its use as a potential screening assay. J. Natl. Cancer Inst. 1987, 79, 487–498. [Google Scholar] [PubMed]
- ESAC. ECVAM Scientific Advisory Committee Working Group Peer Review Consensus Report on an ECVAM-Coordinated Prevalidation Study Concerning Three Protocols of the Cell Transformation Assay (CTA) for In Vitro Carcinogenicity Testing; European Commission Joint Research Center: Ispra, Italy; Institute for Health and Consumer Protection: Ispra, Italy; European Union Reference Laboratory for Alternatives to Animal Testing (EURL ECVAM): Ispra, Italy, 2011; Available online: europa.eu (accessed on 21 January 2023).
- Sasaki, K.; Bohnenberger, S.; Hayashi, K.; Kunkelmann, T.; Muramatsu, D.; Phrakonkham, P.; Poth, A.; Sakai, A.; Salovaara, S.; Tanaka, N.; et al. Recommended protocol for the BALB/c 3T3 cell transformation assay. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 30–35. [Google Scholar] [CrossRef]
- Maire, M.-A.; Rast, C.; Vasseur, P. Photo catalogue for the classification of cell colonies in the Syrian hamster embryo (SHE) cell transformation assay at pH 7.0. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 744, 97–110. [Google Scholar] [CrossRef]
- Callegaro, G.; Stefanini, F.M.; Colacci, A.; Vaccari, M.; Urani, C. An improved classification of foci for carcinogenicity testing by statistical descriptors. Toxicol. Vitr. 2015, 29, 1839–1850. [Google Scholar] [CrossRef]
- Callegaro, G.; Malkoc, K.; Corvi, R.; Urani, C.; Stefanini, F.M. A comprehensive statistical classifier of foci in the cell transformation assay for carcinogenicity testing. Toxicol. Vitr. 2017, 45 Pt 3, 351–358. [Google Scholar] [CrossRef]
- Masumoto, M.; Fukuda, I.; Furihata, S.; Arai, T.; Kageyama, T.; Ohmori, K.; Shirakawa, S.; Fukuda, J. Deep neural network for the determination of transformed foci in Bhas 42 cell transformation assay. Sci. Rep. 2021, 11, 23344. [Google Scholar] [CrossRef]
- OECD. Guidance document on the in vitro Bhas 42 cell transformation assay. In Series on Testing and Assessment No.31; Organization for Economic Co-Operation and Development: Paris, France, 2017. [Google Scholar]
- Newbold, R.F.; Overell, R.W.; Connell, J.R. Induction of immortality is an early event in malignant transformation of mammalian cells by carcinogens. Nature 1982, 299, 633–635. [Google Scholar] [CrossRef]
- Stoker, M.G.P.; Shearer, M.; O’Neill, C. Growth Inhibition of Polyoma-Transformed Cells by Contact with Static Normal Fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Vaccari, M.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Barcellos-Hoff, M.H.; Brown, D.G.; Chapellier, M.; Christopher, J.; Curran, C.S.; et al. The effect of environmental chemicals on the tumor microenvironment. Carcinogenesis 2015, 36, S160–S183. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; DePinho, R.A. Cellular senescence: Mitotic clock or culture shock? Cell 2000, 102, 407–410. [Google Scholar] [CrossRef]
- Pickles, J.C.; Pant, K.; Mcginty, L.A.; Yasaei, H.; Roberts, T.; Scott, A.D.; Newbold, R.F. A mechanistic evaluation of the Syrian hamster embryo cell transformation assay (pH 6.7) and molecular events leading to senescence bypass in SHE cells. Mutat. Res. Toxicol. Environ. Mutagen. 2016, 802, 50–58. [Google Scholar] [CrossRef]
- Goldstein, I.; Marcel, V.; Olivier, M.; Oren, M.; Rotter, V.; Hainaut, P. Understanding wild-type and mutant p53 activities in human cancer: New landmarks on the way to targeted therapies. Cancer Gene Ther. 2011, 18, 2–11. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Varghese, R.T.; Kinney, N.A.; Garner, H.R. Estimating the number of genetic mutations (hits) required for carcinogenesis based on the distribution of somatic mutations. PLoS Comput. Biol. 2019, 15, e1006881. [Google Scholar] [CrossRef]
- Yamagishi, H.; Kuroda, H.; Imai, Y.; Hiraishi, H. Molecular pathogenesis of sporadic colorectal cancers. Chin. J. Cancer 2016, 35, 4. [Google Scholar] [CrossRef]
- Worthley, D.L.; Leggett, B.A. Colorectal cancer: Molecular features and clinical opportunities. Clin. Biochem. Rev. 2010, 31, 31–38. [Google Scholar]
- Mascolo, M.G.; Perdichizzi, S.; Vaccari, M.; Rotondo, F.; Zanzi, C.; Grilli, S.; Paparella, M.; Jacobs, M.N.; Colacci, A. The transformics assay: First steps for the development of an integrated approach to investigate the malignant cell transformation in vitro. Carcinogenesis 2018, 39, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Pillo, G.; Mascolo, M.G.; Zanzi, C.; Rotondo, F.; Serra, S.; Bortone, F.; Grilli, S.; Vaccari, M.; Jacobs, M.N.; Colacci, A. Mechanistic Interrogation of Cell Transformation In Vitro: The Transformics Assay as an Exemplar of Oncotransformation. Int. J. Mol. Sci. 2022, 23, 7603. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, K.; Kamei, A.; Watanabe, Y.; Abe, K. Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells. Int. J. Mol. Sci. 2022, 23, 3216. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.M.; Campbell, B.J. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trends Mol. Med. 2002, 8, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Balmain, A.; Harris, C.C. Carcinogenesis in mouse and human cells: Parallels and paradoxes. Carcinogenesis 2000, 21, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Goodlad, R.A. Mouse Models for Gastrointestinal Cancer. In The Cancer Handbook, 2nd ed.; Alison, M.R., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Yamada, Y.; Mori, H. Multistep carcinogenesis of the colon in ApcMin/+ mouse. Cancer Sci. 2007, 98, 6–10. [Google Scholar] [CrossRef]
- Desaulniers, D.; Vasseur, P.; Jacobs, A.; Aguila, M.C.; Ertych, N.; Jacobs, M.N. Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications. Int. J. Mol. Sci. 2021, 22, 10969. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.-D. Regulatory Mechanisms of Tumor Suppressor P16INK4A and Their Relevance to Cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 576, 22–38. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef]
- Mojarad, E.N.; Kuppen, P.J.K.; Aghdaei, H.A.; Zali, M.R. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Nguyen, H.T.; Duong, H. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef]
- Zhao, R.; Choi, B.Y.; Lee, M.-H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16 INK4a) in Cancer. Ebiomedicine 2016, 8, 30–39. [Google Scholar] [CrossRef]
- Harvey, D.M.; Levine, A.J. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 1991, 5, 2375–2385. [Google Scholar] [CrossRef]
- Jacobsen, K.; Groth, A.; Willumsen, B.M. Ras-inducible immortalized fibroblasts: Focus formation without cell cycle deregulation. Oncogene 2002, 21, 3058–3067. [Google Scholar] [CrossRef]
- Nakazawa, H.; Aguelon, A.-M.; Yamasaki, H. Relationship between chemically induced ha-ras mutation and transformation of BALB/c 3T3 cells: Evidence for chemical-specific activation and cell type-specific recruitment of oncogene in transformation. Mol. Carcinog. 1990, 3, 202–209. [Google Scholar] [CrossRef]
- Miller, M.S.; Miller, L.D. RAS Mutations and Oncogenesis: Not all RAS Mutations are Created Equally. Front. Genet. 2012, 2, 100. [Google Scholar] [CrossRef]
- Landkocz, Y.; Poupin, P.; Atienzar, F.; Vasseur, P. Transcriptomic effects of di-(2-ethylhexyl)-phthalate in Syrian hamster embryo cells: An important role of early cytoskeleton disturbances in carcinogenesis? BMC Genom. 2011, 12, 524. [Google Scholar] [CrossRef]
- Mostowy, S.; Shenoy, A.R. The cytoskeleton in cell-autonomous immunity: Structural determinants of host defence. Nat. Rev. Immunol. 2015, 15, 559–573. [Google Scholar] [CrossRef]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Kavasi, R.-M.; Voudouri, K.; Berdiaki, A.; Spyridaki, I.; Tsatsakis, A.; Nikitovic, D. Role of the extracellular matrix in cancer-associated epithelial to mesenchymal transition phenomenon. Dev. Dyn. 2018, 247, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, M.; Silingardi, P.; Argnani, A.; Horn, W.; Giungi, M.; Mascolo, M.G.; Grilli, S.; Colacci, A. In vitro effects of fenretinide on cell-matrix interactions. Anticancer Res. 2000, 20, 3059–3066. [Google Scholar] [PubMed]
- Maire, M.; Rast, C.; Vasseur, P. Di-(2-ethylhexyl) phthalate (DEHP) increases Bcl-2/Bax ratio and modifies c-myc expression in Syrian hamster embryo (SHE) cells. Toxicol. Lett. 2005, 158, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Berenblum, I.; Shubik, P. The Role of Croton Oil Applications, Associated with a Single Painting of a Carcinogen, in Tumour Induction of the Mouse’s Skin. Br. J. Cancer 1947, 1, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Ewing, M.W.; Conti, C.J.; Kruszewski, F.H.; Slaga, T.J.; DiGiovanni, J. Tumor progression in Sencar mouse skin as a function of initiator dose and promoter dose, duration, and type. Cancer Res. 1988, 48 Pt 1, 7048–7054. [Google Scholar]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef]
- Hennings, H.; Shores, R.; Wenk, M.L.; Spangler, E.F.; Tarone, R.; Yuspa, S.H. Malignant conversion of mouse skin tumours is increased by tumour initiators and unaffected by tumour promoters. Nature 1983, 304, 67–69. [Google Scholar] [CrossRef]
- Yuspa, S.H. Mechanisms of initiation and promotion in mouse epidermis. IARC Sci. Publ. 1984, 56, 191–204. [Google Scholar]
- Lazar, A.D.; Dinescu, S.; Costache, M. Deciphering the Molecular Landscape of Cutaneous Squamous Cell Carcinoma for Better Diagnosis and Treatment. J. Clin. Med. 2020, 9, 2228. [Google Scholar] [CrossRef]
- Lopez, A.T.; Liu, L.; Geskin, L. Molecular Mechanisms and Biomarkers of Skin Photocarcinogenesis. In Human Skin Cancers: Pathways, Mechanisms, Targets and Treatments; Blumenberg, M., Ed.; IntechOpen: London, UK, 2017; pp. 175–200. [Google Scholar] [CrossRef]
- Dotto, G.P.; Rustgi, A.K. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef]
- Berenblum, I.; Shubik, P. A new, quantitative, approach to the study of the stages of chemical carcinogenesis in the mouse’s skin. Br. J. Cancer 1947, 1, 383. [Google Scholar] [CrossRef]
- Goel, G.; Makkar, H.P.; Francis, G.; Becker, K. Phorbol esters: Structure, biological activity, and toxicity in animals. Int. J. Toxicol. 2007, 26, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Mérida, I.; Torres-Ayuso, P.; Ávila-Flores, A.; Arranz-Nicolás, J.; Andrada, E.; Tello-Lafoz, M.; Liébana, R.; Arcos, R. Diacylglycerol kinases in cancer. Adv. Biol. Regul. 2017, 63, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Mérida, I.; Arranz-Nicolás, J.; Rodríguez-Rodríguez, C.; Ávila-Flores, A. Diacylglycerol kinase control of protein kinase C. Biochem. J. 2019, 476, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Ragnoli, B.; Malerba, M. Potential role of diacylglycerol kinases in immune-mediated diseases. Clin. Sci. 2020, 134, 1637–1658. [Google Scholar] [CrossRef]
- Strawn, L.; Babb, A.; Testerink, C.; Kooijman, E.E. The Physical Chemistry of the Enigmatic Phospholipid Diacylglycerol Pyrophosphate. Front. Plant Sci. 2012, 3, 40. [Google Scholar] [CrossRef]
- Peraino, C.; Michael Fry, R.; Staffeldt, E. Reduction and enhancement by phenobarbital of hepatocarcinogenesis induced in the rat by 2-acetylaminofluorene. Cancer Res. 1971, 31, 1506–1512. [Google Scholar]
- Scherer, E. Neoplastic progression in expreimental hepatocarcinogenesis. Biochim. Biophys. Acta Rev. Cancer 1984, 738, 219–236. [Google Scholar] [CrossRef]
- Dragan, Y.P.; Pitot, H.C. The role of the stages of initiation and promotion in phenotypic diversity during hepatocarcinogenesis in the rat. Carcinogenesis 1992, 13, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Dragan, Y.P.; Sargent, L.; Xu, Y.D.; Xu, Y.-H.; Pitot, H.C. The Initiation-Promotion-Progression Model of Rat Hepatocarcinogenesis. Proc. Soc. Exp. Biol. Med. 1993, 202, 16–24. [Google Scholar] [CrossRef]
- Pitot, H.C.; Dragan, Y.P.; Teeguarden, J.; Hsia, S.; Campbell, H. Quantitation of Multistage Carcinogenesis in Rat Liver. Toxicol. Pathol. 1996, 24, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Pitot, H.; Dragan, Y.; Sargent, L.; Xu, Y. Biochemical markers associated with the stages of promotion and progression during hepatocarcinogenesis in the rat. Environ. Health Perspect. 1991, 93, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Römer, M.; Eichner, J.; Metzger, U.; Templin, M.F.; Plummer, S.; Ellinger-Ziegelbauer, H.; Zell, A. Cross-Platform Toxicogenomics for the Prediction of Non-Genotoxic Hepatocarcinogenesis in Rat. PLoS ONE 2014, 9, e97640. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.G. Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol. Res. 2018, 7, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Chouroulinkov, I.; Lasne, C. Chemical carcinogenesis in tissue culture: Criteria and transformation tests. IARC Sci. Publ. 1976, 13, 207–227. [Google Scholar]
- Chouroulinkov, I.; Lasne, C. Two-stage (initiation-promotion) carcinogenesis in vivo and in vitro. Bull. Du Cancer 1978, 65, 255–264. [Google Scholar]
- Colacci, A.; Perocco, P.; Bartoli, S.; Da Via, C.; Silingardi, P.; Vaccari, M.; Grilli, S. Initiating activity of 1,1,2,2-tetrachloroethane in two-stage BALB/c 3T3 cell transformation. Cancer Lett. 1992, 64, 145–153. [Google Scholar] [CrossRef]
- Colacci, A.; Perocco, P.; Vaccari, M.; Da Vià, C.; Silingardi, P.; Manzini, E.; Horn, W.; Bartoli, S.; Grilli, S. 1,2-Dibromoethane as an Initiating Agent for Cell Transformation. Jpn. J. Cancer Res. 1995, 86, 168–173. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Umeda, M. Improvement in the efficiency of the in vitro transformation assay method using BALB/3T3 A31–1–1 cells. Carcinogenesis 1995, 16, 1887–1894. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasaki, K.; Asada, S.; Tsuchiya, T.; Hayashi, M.; Yoshimura, I.; Tanaka, N.; Umeda, M. Technical Modification of the Balb/c 3T3 Cell Transformation Assay: The use of Serum-reduced Medium to Optimise the Practicability of the Protocol. Altern. Lab. Anim. 2008, 36, 653–665. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Umeda, M.; Nishiyama, H.; Yoshimura, I.; Ajimi, S.; Asakura, M.; Baba, H.; Dewa, Y.; Ebe, Y.; Fushiwaki, Y.; et al. An Interlaboratory Validation Study of the Improved Transformation Assay Employing Balb/c 3T3 Cells: Results of a Collaborative Study on the Two-stage Cell Transformation Assay by the Non-genotoxic Carcinogen Study Group. Altern. Lab. Anim. 1999, 27, 685–702. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, T.; Umeda, M.; Tanaka, N.; Sakai, A.; Nishiyama, H.; Yoshimura, I.; Ajimi, S.; Asada, S.; Asakura, M.; Baba, H.; et al. Application of the Improved BALB/c 3T3 Cell Transformation Assay to the Examination of the Initiating and Promoting Activities of Chemicals: The Second Inter-laboratory Collaborative Study by the Non-genotoxic Carcinogen Study Group of Japan. Altern. Lab. Anim. 2010, 38, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.; Fujiki, H. Promotion of BALB/3T3 Cell Transformation by the Okadaic Acid Class of Tumor Promoters, Okadaic Acid and Dinophysistoxin-1. Jpn. J. Cancer Res. 1991, 82, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Valdiglesias, V.; Prego-Faraldo, M.V.; Pásaro, E.; Méndez, J.; Laffon, B. Okadaic Acid: More than a Diarrheic Toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. Molecular and Cellular Mechanism of Okadaic Acid (OKA)-Induced Neurotoxicity: A Novel Tool for Alzheimer’s Disease Therapeutic Application. Mol. Neurobiol. 2014, 50, 852–865. [Google Scholar] [CrossRef]
- Sakai, A.; Sasaki, K.; Muramatsu, D.; Arai, S.; Endou, N.; Kuroda, S.; Hayashi, K.; Lim, Y.-m.; Yamazaki, S.; Umeda, M. A Bhas 42 cell transformation assay on 98 chemicals: The characteristics and performance for the prediction of chemical carcinogenicity. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2010, 702, 100–122. [Google Scholar] [CrossRef]
- Kirkland, D.; Zeiger, E.; Madia, F.; Gooderham, N.; Kasper, P.; Lynch, A.; Morita, T.; Ouedraogo, G.; Morte, J.M.P.; Pfuhler, S.; et al. Can in vitro mammalian cell genotoxicity test results be used to complement positive results in the Ames test and help predict carcinogenic or in vivo genotoxic activity? I. Reports of individual databases presented at an EURL ECVAM Workshop. Mutat. Res. Toxicol. Environ. Mutagen. 2014, 775–776, 55–68. [Google Scholar] [CrossRef]
- Kirkland, D.; Kasper, P.; Martus, H.-J.; Müller, L.; van Benthem, J.; Madia, F.; Corvi, R. Updated recommended lists of genotoxic and non-genotoxic chemicals for assessment of the performance of new or improved genotoxicity tests. Mutat. Res. Toxicol. Environ. Mutagen. 2016, 795, 7–30. [Google Scholar] [CrossRef]
- Madia, F.; Kirkland, D.; Morita, T.; White, P.; Asturiol, D.; Corvi, R. EURL ECVAM genotoxicity and carcinogenicity database of substances eliciting negative results in the Ames test: Construction of the database. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 854, 503199. [Google Scholar] [CrossRef]
- IARC. IARC Monographs Preamble; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- NTP. Report on Carcinogens, Fifteenth Edition. In Report on Carcinogens, National Toxicology Program Department of Health and Human Services; Public Health Service: Durham, NC, USA, 2021. [Google Scholar]
- Das, A.M.; Gogia, A.; Janardhanan, R.; Babu-Rajendran, R.; Das, B.C. Environmental Contamination and Chronic Exposure to Endocrine-Disrupting Phthalates: An Overlooked and Emerging Determinant for Hormone-Sensitive Cancers. J. Indian Inst. Sci. 2022, 102, 731–742. [Google Scholar] [CrossRef]
- Rusyn, I.; Peters, J.M.; Cunningham, M.L. Effects of DEHP in the liver: Modes of action and species-specific differences. Crit. Rev. Toxicol. 2006, 36, 459. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Chen, H.-S.; Long, C.-Y.; Tsai, C.-F.; Hsieh, T.-H.; Hsu, C.; Tsai, E.-M. Possible mechanism of phthalates-induced tumorigenesis. Kaohsiung J. Med. Sci. 2012, 28 (Suppl. 7), S22–S27. [Google Scholar] [CrossRef] [PubMed]
- NTP. Toxicology and Carcinogenesis Studies of Diethanolamine (CAS No. 111-42-2) ln F344/N Rats and B6C3F1 Mice (Dermal Studies)—TR 478; NTP: Durham, NC, USA, 1999; pp. 1–212. [Google Scholar]
- Leung, H.-W.; Kamendulis, L.M.; Stott, W.T. Review of the carcinogenic activity of diethanolamine and evidence of choline deficiency as a plausible mode of action. Regul. Toxicol. Pharmacol. 2005, 43, 260–271. [Google Scholar] [CrossRef]
- NTP. Bioassay of N,N’-Diethylthiourea for Possible Carcinogenesis/CAS No. 105-55-5)—NCI-CG-TR-149; U.S. Department of Health, Education and Welfare: Washington, DC, USA; National Cancer Institute: Bethesda, MD, USA; National Institute of Health: Bethesda, MD, USA, 1979. [Google Scholar]
- OECD. OECD Report of the Workshop on a Framework for the Development and Use of Integrated Approaches to Testing and Assessment. In OECD Series on Testing and Assessment; OECD Publishing: Paris, France, 2017. [Google Scholar] [CrossRef]
- Ament, Z.; Waterman, C.L.; West, J.A.; Waterfield, C.; Currie, R.A.; Wright, J.; Griffin, J.L. A Metabolomics Investigation of Non-genotoxic Carcinogenicity in the Rat. J. Proteome Res. 2013, 12, 5775–5790. [Google Scholar] [CrossRef] [PubMed]
- NTP. Technical Report on the Toxicology and Carcinogenesis Studies of Ethylbenzene (CAS No. 100-41-4) in F344/N Rats and B6C3F1 Mice (Inhalation Studies)—NTP TR 466; U.S. Department of Health and Human Services, Public Health Service: Washington, DC, USA; National Institutes of Health: Durham, NC, USA, 1999. [Google Scholar]
- Huff, J.; Chan, P.; Melnick, R. Clarifying carcinogenicity of ethylbenzene. Regul. Toxicol. Pharmacol. 2010, 58, 167–169. [Google Scholar] [CrossRef]
- Stott, W.T.; Johnson, K.A.; Bahnemann, R.; Day, S.J.; McGuirk, R.J. Evaluation of Potential Modes of Action of Inhaled Ethylbenzene in Rats and Mice. Toxicol. Sci. 2003, 71, 53–66. [Google Scholar] [CrossRef]
- Saghir, S.A.; Zhang, F.; Rick, D.L.; Kan, L.; Bus, J.S.; Bartels, M.J. In vitro metabolism and covalent binding of ethylbenzene to microsomal protein as a possible mechanism of ethylbenzene-induced mouse lung tumorigenesis. Regul. Toxicol. Pharmacol. 2010, 57, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Salim, Y.M.; EL-Gendy, I.R.; Nassae, A.M. Toxicological effects of methyl eugenol and fenitrothion on hematological, hepatic, renal, and oxidative stress-related biochemical characteristics of white albino rats. Interdiscip Toxicol. 2020, 13, 13–20. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Militão, G.C.G.; De Morais, M.C.; De Sousa, D.P. The Dual Antioxidant/Prooxidant Effect of Eugenol and Its Action in Cancer Development and Treatment. Nutrients 2017, 9, 1367. [Google Scholar] [CrossRef]
- Hardt, A.; Stippel, D.; Odenthal, M.; Hölscher, A.H.; Dienes, H.-P.; Drebber, U. Development of Hepatocellular Carcinoma Associated with Anabolic Androgenic Steroid Abuse in a Young Bodybuilder: A Case Report. Case Rep. Pathol. 2012, 2012, 195607. [Google Scholar] [CrossRef]
- Salerno, M.; Cascio, O.; Bertozzi, G.; Sessa, F.; Messina, A.; Monda, V.; Cipolloni, L.; Biondi, A.; Daniele, A.; Pomara, C. Anabolic androgenic steroids and carcinogenicity focusing on Leydig cell: A literature review. Oncotarget 2018, 9, 19415–19426. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.L.; Martin, E.J.; Brinton, L.A.; Hoover, R.N. Rauwolfia use and breast cancer: A case-control study. J. Natl. Cancer Inst. 1986, 76, 817–822. [Google Scholar] [PubMed]
- Grossman, E.; Messerli, F.H.; Boyko, V.; Goldbourt, U. Is there an association between hypertension and cancer mortality? Am. J. Med. 2002, 112, 479–486. [Google Scholar] [CrossRef]
- Tischler, A.; McClain, R.M.; Childers, H.; Downing, J. Neurogenic signals regulate chromaffin cell proliferation and mediate the mitogenic effect of reserpine in the adult rat adrenal medulla. Lab. Investig. 1991, 65, 374–376. [Google Scholar]
- Clevenger, C.V.; Furth, P.A.; Hankinson, S.E.; Schuler, L.A. The Role of Prolactin in Mammary Carcinoma. Endocr. Rev. 2003, 24, 1–27. [Google Scholar] [CrossRef] [PubMed]
- NTP. Technical Report on the Toxicity Studies of Wy-14,643 (CAS No. 50892-23-4) Administered in Feed to Sprague-Dawley Rats, B6C3F 1 Mice, and Syrian Hamsters—NTP TR 62; U.S. Department of Health and Human Services, Public Health Service: Washington, DC, USA; National Institutes of Health: Durham, NC, USA, 2007. [Google Scholar]
- Reddy, J.K.; Rao, M.S.; Azarnoff, D.L.; Sell, S. Mitogenic and carcinogenic effects of a hypolipidemic peroxisome proliferator, [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14, 643), in rat and mouse liver. Cancer Res. 1979, 39, 152–161. [Google Scholar] [PubMed]
- Marsman, D.S.; Popp, J.A. Biological potential of basophilic hepatocellular foci and hepatic adenoma induced by the peroxisome proliferator, Wy-14,643. Carcinogenesis 1994, 15, 111–117. [Google Scholar] [CrossRef]
- Hayashi, F.; Tamura, H.; Yamada, J.; Kasai, H.; Suga, T. Characteristics of the hepatocarcinogenesis caused by dehydroepiandrosterone, a peroxisome proliferator, in male F-344 rats. Carcinogenesis 1994, 15, 2215–2219. [Google Scholar] [CrossRef]
- Lake, B.G.; Evans, J.G.; Cunninghame, M.E.; Price, R.J. Comparison of the hepatic effects of nafenopin and WY-14,643 on peroxisome proliferation and cell replication in the rat and Syrian hamster. Environ. Health Perspect. 1993, 101 (Suppl. S5), 241–247. [Google Scholar] [CrossRef]
- Torrey, C.E.; Campbell, J.A.; Hoivik, D.J.; Miller, R.T.; Allen, J.S.; Mann, P.C.; Selinger, K.; Rickert, D.; Savina, P.M.; Santostefano, M.J. Evaluation of the carcinogenic potential of clofibrate in the p53+/− mouse. Int. J. Toxicol. 2005, 24, 289–299. [Google Scholar] [CrossRef]
- Gad, S.C.; Burton, E.; Chengelis, C.P.; Levin, S.; Piper, C.E.; Oshiro, Y.; Semler, D.E. Promotional activities of the non-genotoxic carcinogen bemitradine (SC-33643). J. Appl. Toxicol. 1992, 12, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Heuvel, J.P.V.; Rusyn, I. Genomic Profiling in Nuclear Receptor-Mediated Toxicity. Toxicol. Pathol. 2007, 35, 474–494. [Google Scholar] [CrossRef]
- NTP. Toxicology and Carcinogenesis Studies of Butyl Benzylphthalate (CAS No. 85-68-7) in F344/N Rats (Feed Studies)-TR-458; U.S. Department of Health and Human Services, Public Health Service: Washington, DC, USA; National Institutes of Health: Durham, NC, USA, 1997. [Google Scholar]
- Kohno, H.; Suzuki, R.; Sugie, S.; Tsuda, H.; Tanaka, T. Lack of modifying effects of 4-tert-octylphenol and benzyl butyl phthalate on 3, 2′-dimethyl-4-aminobiphenyl-induced prostate carcinogenesis in rats. Cancer Sci. 2004, 95, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Aschengrau, A.; Coogan, P.F.; Quinn, M.M.; Cashins, L.J. Occupational exposure to estrogenic chemicals and the occurrence of breast cancer: An exploratory analysis. Am. J. Ind. Med. 1998, 34, 6–14. [Google Scholar] [CrossRef]
- López-Carrillo, L.; Hernández-Ramírez, R.U.; Calafat, A.M.; Torres-Sánchez, L.; Galván-Portillo, M.; Needham, L.L.; Ruiz-Ramos, R.; Cebrián, M.E. Exposure to Phthalates and Breast Cancer Risk in Northern Mexico. Environ. Health Perspect. 2010, 118, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Babich, M.A.; Baetcke, K.P.; Cook, J.C.; Corton, J.C.; David, R.M.; DeLuca, J.G.; Lai, D.Y.; McKee, R.H.; Peters, J.; et al. PPARα Agonist-Induced Rodent Tumors: Modes of Action and Human Relevance. Crit. Rev. Toxicol. 2003, 33, 655–780. [Google Scholar] [CrossRef]
- Mankidy, R.; Wiseman, S.; Ma, H.; Giesy, J.P. Biological impact of phthalates. Toxicol. Lett. 2012, 217, 50–58. [Google Scholar] [CrossRef]
- Moody, D.E.; Reddy, J.K. The hepatic effects of hypolipidemic drugs (clofibrate, nafenopin, tibric acid, and Wy-14,643) on hepatic peroxisomes and peroxisome-associated enzymes. Am. J. Pathol. 1978, 90, 435–446. [Google Scholar]
- Reddy, J.K.; Qureshi, S.A. Tumorigenicity of the hypolipidaemic peroxisome proliferator ethyl-α-p-chlorophenoxyisobutyrate (clofibrate) in rats. Br. J. Cancer 1979, 40, 476–482. [Google Scholar] [CrossRef]
- Reddy, J.K.; Lalwani, N.D.; Reddy, M.K.; Qureshi, S.A. Excessive accumulation of autofluorescent lipofuscin in the liver during hepatocarcinogenesis by methyl clofenapate and other hypolipidemic peroxisome proliferators. Cancer Res. 1982, 42, 259–266. [Google Scholar]
- Tucker, M.J.; Orton, T. Comparative Toxicology of Hypolipidaemic Fibrates; Taylor & Francis: Bristol, PA, USA, 1995. [Google Scholar]
- Iversen, O.H. Carcinogenesis Studies with Benzoyl Peroxide (Panoxyl Gel 5%). J. Investig. Dermatol. 1986, 86, 442–448. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free. Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Gundimeda, U.; Anderson, W.B.; Colburn, N.H.; Slaga, T.J. Tumor Promoter Benzoyl Peroxide Induces Sulfhydryl Oxidation in Protein Kinase C: Its Reversibility Is Related to the Cellular Resistance to Peroxide-Induced Cytotoxicity. Arch. Biochem. Biophys. 1999, 363, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Blair, A.; Gibson, R.; Everett, G.D.; Cantor, K.P.; Schuman, L.M.; Burmeister, L.F.; Van Lier, S.F.; Dick, F. Pesticide exposures and other agricultural risk factors for leukemia among men in Iowa and Minnesota. Cancer Res. 1990, 50, 6585–6591. [Google Scholar] [PubMed]
- Chen, H.-S.; Chiang, P.-H.; Wang, Y.-C.; Kao, M.-C.; Shieh, T.-H.; Tsai, C.-F.; Tsai, E.-M. Benzyl butyl phthalate induces necrosis by AhR mediation of CYP1B1 expression in human granulosa cells. Reprod. Toxicol. 2012, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.N.; Marczylo, E.L.; Guerrero-Bosagna, C.; Rüegg, J. Marked for Life: Epigenetic Effects of Endocrine Disrupting Chemicals. Annu. Rev. Environ. Resour. 2017, 42, 105–160. [Google Scholar] [CrossRef]
- Innes, J.; Ulland, B.; Valerio, M.G.; Petrucelli, L.; Fishbein, L.; Hart, E.; Pallotta, A.; Bates, R.; Falk, H.; Gart, J. Bioassay of pesticides and industrial chemicals for tumorigenicity in mice: A preliminary note. J. Natl. Cancer Inst. 1969, 42, 1101–1114. [Google Scholar]
- Fitzgerald, D.J.; Piccoli, C.; Yamasaki, H. Detection of non-genotoxic carcinogens in the BALB/c 3T3 cell transformation/mutation assay system. Mutagenesis 1989, 4, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Naus, C.C. Role of connexin genes in growth control. Carcinogenesis 1996, 17, 1199–1214. [Google Scholar] [CrossRef]
- Llop, S.; Ballester, F.; Vizcaino, E.; Murcia, M.; Lopez-Espinosa, M.-J.; Rebagliato, M.; Vioque, J.; Marco, A.; Grimalt, J.O. Concentrations and determinants of organochlorine levels among pregnant women in Eastern Spain. Sci. Total. Environ. 2010, 408, 5758–5767. [Google Scholar] [CrossRef]
- Glynn, A.; Aune, M.; Darnerud, P.O.; Cnattingius, S.; Bjerselius, R.; Becker, W.; Lignell, S. Determinants of serum concentrations of organochlorine compounds in Swedish pregnant women: A cross-sectional study. Environ. Health 2007, 6, 2. [Google Scholar] [CrossRef]
- Gaben, A.-M.; Mester, J. Balbc mouse 3T3 fibroblasts expressing human estrogen receptor: Effect of estradiol on cell growth. Biochem. Biophys. Res. Commun. 1991, 176, 1473–1481. [Google Scholar] [CrossRef]
- Matthews, E.J.; Spalding, J.W.; Tennant, R.W. Transformation of BALB/c-3T3 cells: V. Transformation responses of 168 chemicals compared with mutagenicity in Salmonella and carcinogenicity in rodent bioassays. Environ. Health Perspect. 1993, 101 (Suppl. S2), 347–482. [Google Scholar]
- IARC. Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide. In IARC Monographs on the Evaluation of Carcinogenic Risk to Humans; International Agency for Research on Cancer: Lyon, France, 1999; Volume 71, pp. 391–395. [Google Scholar]
- NTP. NTP Toxicology and Carcinogenesis Studies of Methyleugenol (CAS NO. 93-15-2) in F344/N Rats and B6C3F1 Mice (Gavage Studies); NTP: Morrisville, NC, USA, 2000; pp. 1–412. [Google Scholar]
- Pavlatos, A.M.; Fultz, O.; Monberg, M.J.; Vootkur, A. Review of oxymetholone: A 17α-alkylated anabolic-androgenic steroid. Clin. Ther. 2001, 23, 789–801. [Google Scholar] [CrossRef]
- Supasyndh, O.; Satirapoj, B.; Aramwit, P.; Viroonudomphol, D.; Chaiprasert, A.; Thanachatwej, V.; Vanichakarn, S.; Kopple, J.D. Effect of Oral Anabolic Steroid on Muscle Strength and Muscle Growth in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2013, 8, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Kicman, A.T. Pharmacology of anabolic steroids. Br. J. Pharmacol. 2008, 154, 502–521. [Google Scholar] [CrossRef] [PubMed]
- Albano, G.; Amico, F.; Cocimano, G.; Liberto, A.; Maglietta, F.; Esposito, M.; Rosi, G.; Di Nunno, N.; Salerno, M.; Montana, A. Adverse Effects of Anabolic-Androgenic Steroids: A Literature Review. Healthcare 2021, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Fujino, Y.; Ku, Y.; Suzuki, Y.; Ajiki, T.; Hasegawa, Y.; Kuroda, Y. Ampullary carcinoma developing after androgenic steroid therapy for aplastic anemia: Report of a case. Surgery 2001, 129, 501–503. [Google Scholar] [CrossRef]
- NTP. NTP Toxicology and Carcinogenesis Studies of Oxymetholone (CAS NO. 434-07-1) in F344/N Rats and Toxicology Studies of Oxymetholone in B6C3F1 Mice (Gavage Studies); NTP: Durham, NC, USA, 1999; pp. 1–233. [Google Scholar]
- Higashiyama, H.; Sumitomo, H.; Ozawa, A.; Igarashi, H.; Tsunekawa, N.; Kurohmaru, M.; Kanai, Y. Anatomy of the Murine Hepatobiliary System: A Whole-Organ-Level Analysis Using a Transparency Method. Anat. Rec. 2015, 299, 161–172. [Google Scholar] [CrossRef]
- Cheung, M.; Parmar, M. Reserpine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Griesemer, R.A.; Dunkel, V.C. Laboratory tests for chemical carcinogens. J. Environ. Pathol. Toxicol. 1980, 4, 565–571. [Google Scholar]
- O’Connor, J.C.; Plowchalk, D.R.; Van Pelt, C.S.; Davis, L.G.; Cook, J.C. Role of prolactin in chloro-S-triazine rat mammary tumorigenesis. Drug Chem. Toxicol. 2000, 23, 575–601. [Google Scholar] [CrossRef]
- Tikk, K.; Sookthai, D.; Fortner, R.T.; Johnson, T.; Rinaldi, S.; Romieu, I.; Tjønneland, A.; Olsen, A.; Overvad, K.; Clavel-Chapelon, F.; et al. Circulating prolactin and in situ breast cancer risk in the European EPIC cohort: A case-control study. Breast Cancer Res. 2015, 17, 49. [Google Scholar] [CrossRef]
- Wang, M.; Wu, X.; Chai, F.; Zhang, Y.; Jiang, J. Plasma prolactin and breast cancer risk: A meta-analysis. Sci. Rep. 2016, 6, 25998. [Google Scholar] [CrossRef]
- Mujagić, Z.; Srabović, N.; Mujagić, H. The role of prolactin in human breast cancer. Biochem. Med. 2009, 19, 236–249. [Google Scholar] [CrossRef]
- Skrypnyk, N.; Chen, X.; Hu, W.; Su, Y.; Mont, S.; Yang, S.; Gangadhariah, M.; Wei, S.; Falck, J.R.; Jat, J.L. PPARα Activation Can Help Prevent and Treat Non–Small Cell Lung Cancer. Cancer Res. 2014, 74, 621–631. [Google Scholar] [CrossRef]
- Torrey, C.E.; Wall, H.G.; Campbell, J.A.; Kwanyuen, P.; Hoivik, D.J.; Miller, R.T.; Allen, J.S.; Jayo, M.J.; Selinger, K.; Savina, P.M.; et al. Evaluation of the Carcinogenic Potential of Clofibrate in FVB/Tg.AC Mouse After Oral Administration—Part I. Int. J. Toxicol. 2005, 24, 313–325. [Google Scholar] [CrossRef]
- Nesfield, S.R.; Clarke, C.J.; Hoivik, D.J.; Miller, R.T.; Allen, J.S.; Selinger, K.; Santostefano, M.J. Evaluation of the Carcinogenic Potential of Clofibrate in the rasH2 Mouse. Int. J. Toxicol. 2005, 24, 301–311. [Google Scholar] [CrossRef]
- Torrey, C.E.; Wall, H.G.; Campbell, J.A.; Kwanyuen, P.; Hoivik, D.J.; Miller, R.T.; Allen, J.S.; Jayo, M.J.; Selinger, K.; Santostefano, M.J. Evaluation of the Carcinogenic Potential of Clofibrate in the FVB/Tg.AC Mouse After Dermal Application—Part II. Int. J. Toxicol. 2005, 24, 327–339. [Google Scholar] [CrossRef]
- Peters, J.M.; Taubeneck, M.W.; Keen, C.L.; Gonzalez, F.J. Di (2-Ethylhexyl) phthalate induces a functional zinc deficiency during pregnancy and teratogenesis that is independent of peroxisome proliferator-activated receptor-α. Teratology 1997, 56, 311–316. [Google Scholar] [CrossRef]
- Miki, Y.; Kidoguchi, Y.; Sato, M.; Taketomi, Y.; Taya, C.; Muramatsu, K.; Gelb, M.H.; Yamamoto, K.; Murakami, M. Dual Roles of Group IID Phospholipase A2 in Inflammation and Cancer. J. Biol. Chem. 2016, 291, 15588–15601. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, Y.; Shen, P. NF-κB and its Regulation on the Immune System. Cell. Mol. Immunol. 2004, 1, 343–350. [Google Scholar]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Balkwill, F.; Coussens, L.M. An inflammatory link. Nature 2004, 431, 405–406. [Google Scholar] [CrossRef]
- Samet, J.M.; Chiu, W.A.; Cogliano, V.; Jinot, J.; Kriebel, D.; Lunn, R.M.; Beland, F.A.; Bero, L.; Browne, P.; Fritschi, L.; et al. The IARC Monographs: Updated Procedures for Modern and Transparent Evidence Synthesis in Cancer Hazard Identification. Gynecol. Oncol. 2020, 112, 30–37. [Google Scholar] [CrossRef]
- Jones, C.; Huberman, E.; Callaham, M.; Tu, A.; Halloween, W.; Pallota, S.; Sivak, A.; Lubet, R.; Avery, M.; Kouri, R.; et al. An interlaboratory evaluation of the Syrian hamster embryo cell transformation assay using eighteen coded chemicals. Toxicol. Vitr. 1988, 2, 103–116. [Google Scholar] [CrossRef]
- Ratna, A.; Mandrekar, P. Alcohol and Cancer: Mechanisms and Therapies. Biomolecules 2017, 7, 61. [Google Scholar] [CrossRef]
- Faroon, O.; Harris, M.O. Toxicological Profile for DDT, DDE, and DDD; Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch: Atlanta, GA, USA, 2002. [Google Scholar]
- Harada, T.; Takeda, M.; Kojima, S.; Tomiyama, N. Toxicity and Carcinogenicity of Dichlorodiphenyltrichloroethane (DDT). Toxicol. Res. 2016, 32, 21–33. [Google Scholar] [CrossRef]
- Van Kesteren, P.; Pronk, M.; Heusinkveld, H.; Luijten, M.; Hakkert, B. Letter to the editor regarding the review article by Yamada et al. Critical evaluation of the human relevance of the mode of action for rodent liver tumor formation by activators of the constitutive androstane receptor (CAR). Crit. Rev. Toxicol. 2022, 52, 397–398. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Yeom, H.; Eom, S.Y.; Lee, Y.-M.; Lee, M. Genome-Wide DNA Methylation Changes in Transformed Foci Induced by Nongenotoxic Carcinogens. Environ. Mol. Mutagen. 2019, 60, 576–587. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Yeom, H.; Han, B.-I.; Ham, B.-J.; Lee, Y.-M.; Han, M.-R.; Lee, M. Predicting Carcinogenic Mechanisms of Non-Genotoxic Carcinogens via Combined Analysis of Global DNA Methylation and In Vitro Cell Transformation. Int. J. Mol. Sci. 2020, 21, 5387. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Madia, F.; Lau, P.; Paparella, M.; McGovern, T.; Luijten, M.; Jacobs, M.N. Analyses of Transcriptomics Cell Signalling for Pre-Screening Applications in the Integrated Approach for Testing and Assessment of Non-Genotoxic Carcinogens. Int. J. Mol. Sci. 2022, 23, 12718. [Google Scholar] [CrossRef] [PubMed]
- Sovadinová, I.; Upham, B.L.; Trosko, J.E.; Babica, P. Applicability of Scrape Loading-Dye Transfer Assay for Non-Genotoxic Carcinogen Testing. Int. J. Mol. Sci. 2021, 22, 8977. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, D.; Cummings-Lorbetskie, C.; Leingartner, K.; Meier, M.J.; Pickles, J.C.; Yauk, C.L. DNA methylation changes from primary cultures through senescence-bypass in Syrian hamster fetal cells initially exposed to benzo[a]pyrene. Toxicology 2023, 487, 153451. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Bult, J.M.; Cavanagh, K.; Chesne, C.; Delrue, N.; Fu, J.; Grange, E.; Langezaal, I.; Misztela, D.; Murray, J.; et al. OECD workshop consensus report: Ethical considerations with respect to human derived products, specifically human serum, in OECD test guidelines. Front. Toxicol. 2023, 5. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tests Considered | Refined Categorisation |
|---|---|
In vitro tests
| Criteria for a non genotoxic chemical
|
| Data Source | Search Strategy | Links |
|---|---|---|
| International Agency for Research on Cancer | List of Classification of Agents from IARC Monographs 1–129 IARC Monographs | https://monographs.iarc.who.int/agents-classified-by-the-iarc/ (accessed on 26 July 2022) |
| U.S. National Toxicology Program | NTP Study Reports Collection Toxicology/Carcinogenesis 15th Report on Carcinogens (2021) | https://ntp.niehs.nih.gov/whatwestudy/testpgm/cartox/index.html (accessed on 2 November 2022) https://ntp.niehs.nih.gov/whatwestudy/assessments/cancer/roc/index.html (accessed on 2 November 2022) |
| Lhasa Ltd. | Carcinogenicity Database | https://carcdb.lhasalimited.org/study-information/44643857 (accessed on 26 July 2022) |
| National Library of Medicine | PubChem PubMed | https://pubchem.ncbi.nlm.nih.gov/compound/55784#section=Classification (accessed on 26 July 2022) https://pubmed.ncbi.nlm.nih.gov (accessed on 30 December 2022) |
| European Chemical Agency (ECHA) | ECHA C&L Inventory | https://echa.europa.eu/it/information-on-chemicals/cl-inventory-database (accessed on 30 December 2022) |
| World Health Organization (WHO) | Database of International Chemical Safety Cards (ICSCs) | https://www.ilo.org/safework/info/publications/WCMS_113134/lang--en/index.htm (accessed on 30 December 2022) |
| US Environmental Protection Agency | Integrated Risk Information System (IRIS) | https://www.epa.gov/iris (accessed on 26 July 2022) |
| Testing Chemical | ||||||||
|---|---|---|---|---|---|---|---|---|
| CAS Registry Number | Chemical Name | Experimental Conditions Test at pH | Use | IARC Classification 1 | CMR Classification | NTP (RoC) 2 | Properties of Concern 3 | Updated Information on Genotoxicity |
| 88133-11-3 | Bemitradine | 6.7 | Diuretic antihypertensive drug | NA | NA | Positive (NL) | NA | |
| 94-36-0 | Benzoyl peroxide (BPO) | 7.0 | Drug product | 3 | NA | Negative | Other concerns | |
| 85-68-7 | Butylbenzylphthalate | 6.7 | Plasticizer | 3 | R2 | IE/(NL) | CLP notification: ED | Positive Chromosomal Aberration in vivo |
| 105-60-2 | Caprolactam | 7.0 | Used in nylon manufacture | 3 | NA | Negative | Other concerns, at pH of 7.3 was positive | Clear Negative in vivo |
| 637-07-0 | Clofibrate | pH 6.7 and pH 7.0 | Antilipidemic and anticholesteremic drug | 3 | NA | NA | Other hazards | |
| 117-81-7 | di (2-Ethylhexyl) phthalate (DEHP) | pH 6.7 and pH 7.0 | Plasticizer | 2B | R1B | Reasonably anticipated to be carcinogenic (RoC) | CLP notification: ED | Positive in transgenic model |
| 111-42-2 | Diethanolamine | pH 6.7 | Emulsifier | 2B | NA | Positive in mouse both sexes, but not in rats (NL) | CLP notification: R2 | |
| 105-55-5 | N,N’-Diethylthiourea | pH 6.7 | Rubber chemical | 3 | NA | Positive in rats both sexes, but not in mouse (NL) | CLP notification: skin sensitizing | old data: Positive MLA |
| 100-41-4 | Ethyl benzene | pH 6.7 | Intermediate | 2B | NA | LE (NL) | CLP notification: C2 | |
| 72-43-5 | Methoxychlor | pH 6.7 | Pesticide | 3 | NA | Negative | No hazard | Positive MLA (old and new data), ND in vivo |
| 93-15-2 | Methyl eugenol | pH 6.7 | Flavoring agent | 2B | NA | Reasonably anticipated to be carcinogenic (RoC) | CLP notification: M2, C2 | Positive evidence in transgenic model Positive comet in vivo |
| 21340-68-1 | Methylclofenapate | pH 7.0 | Antilipidemic drug | NA | NA | NA | NA | |
| 434-07-1 | Oxymetholone | pH 6.7 | Anabolic steroid | NA | NA | Reasonably anticipated to be carcinogenic (RoC) | No notification | |
| 50-55-5 | Reserpine | pH 6.7 | Antihypertensive drug | 3 | NA | Reasonably anticipated to be carcinogenic (RoC) | Other hazards | Positive in vivo Micronucleus |
| 16561-29-8 | 12-O-Tetradecanoyl phobol 13-acetate (TPA) | pH 6.7 and pH 7.0 | Phorbol ester | NA | NA | Positive (NL) | Other hazards | |
| 50892-23-4 | Wyeth-14,643 | pH 6.7 | Pharmaceutical | NA | NA | Negative | CLP notification: C1B | Positive CA in vitro, Positive transgenic rodent |
| Group A Chemicals | Target Organs in Rodents | Initiation/Promotion In Vivo and/or In Vitro Tests | Epidemiological Evidence | Mechanisms |
|---|---|---|---|---|
| di (2-Ethylhexyl) phthalate (DEHP) | Liver, hepatocarcinoma (RM, RF, MM, MF) Liver (PPAR-null transgenic mice) Benign testicular tumors (RM) Benign pancreatic tumors (RM) [129] | Acting as a promoter in two different strains of mice | An association with breast cancer has recently been reported [130] | Activation of PPARα in rodents. Alternative mechanisms in PPAR-null transgenic mice may include CAR activation and peroxisome proliferation. None of these mechanisms were confirmed in humanized PPAR-null transgenic mice [131]. Possible implication of AhR-mediated signaling and activation of CYP 1B1 [132]. Antiandrogenic activity (identified on list 1 as an Endocrine Disruptor at the EU level) and possible implication of epigenetic DNA methylation disruption properties of DEHP, with links with cancer requiring to be elucidated [74] |
| Diethanolamine (DEA) | Liver neoplasms (hepatocarcinoma and hepatoblastoma) (MM, MF) [133] | No data | Perturbation of choline homeostasis [134] | |
| N,N′-Diethylthiourea (DETU) | Carcinoma of Thyroid Gland Follicular Cells (RM, RF) [135] | No data | Possible inhibition of thyroid hormone biosynthetic enzyme by the thiourea/thiocarbonyl moiety via formation of disulfide bridge [136]. DETU also affects the metabolic profile of cholesterol (increase), arachidonic acid (decrease), long chain carnitine contents (decrease) [137] | |
| Ethylbenzene | Kidney neoplasms (RM) (inhalation) Testis neoplasms (RM) (inhalation) [138,139] | No data | Associated with rat nephropathy, following an accumulation of α2u-globulin [140]. This mechanism is considered not human relevant [141] | |
| Methyl eugenol | Liver adenomas and carcinomas (RM, RF MM, MF) Stomach neuroendocrine tumors (RM, RF, MM) [129] | No data | Oxidative stress [142,143] | |
| Oxymetholone | Liver adenomas and carcinomas (RF) [129] | Limited evidence of leukemia, liver cancer, or esophageal cancer following oxymetholone treatment. A case of ampullary carcinoma (bile-duct) has been also reported [129] A case of hepatocarcinoma in a AAS abuser has been described [144] | Modulation of androgen receptor [145] | |
| Reserpine | Mammary gland neoplasms (MF) Seminal vesicles undifferentiated carcinoma (MM) Adrenal gland pheocromocytoma (RM) [129] | Acting as a promoter in rats | Increased risk of breast cancer among individuals who had used reserpine for over 10 years [146] Small but significant increase in the risk of breast cancer with reserpine. This finding was not confirmed by prospective studies [147] | Chromaffin cell proliferation is the postulated mechanism for pheocromocytoma [148]. Increase of serum levels of prolactin could be responsible for mammary gland tumors [149]. Both mechanisms are related to the ability of reserpine to affect the neural response |
| Group B | Target Organs in Rodents | Initiation/Promotion In Vivo and/or In Vitro Tests | Epidemiological Evidence | Mechanisms |
| Wyeth-14,643 | Liver adenomas and carcinomas (RM, MM) [150,151,152,153,154] No neoplastic formation in p53+/− mice [155] | No data | PPARα dependent peroxisome proliferation | |
| Group C | Target Organs in Rodents | Initiation/Promotion In Vivo and/or In Vitro Tests | Epidemiological Evidence | Mechanisms |
| Bemitradine (BEM) | Liver adenoma and carcinoma (RM) Mammary neoplasms (RF) [156] | Positive in rat altered hepatic foci model [156] | CAR-activator and related increase of Cyp3b1 [157] | |
| Butylbenzyl phthalate (BBP) | Pancreas: Adenoma- acinar cell [158] | Increased incidence of prostate intraepithelial neoplasm in rats treated with 3,2′-dimethyl-4-aminobiphenol (DMAB) [159] | No significant association with breast cancer risk in occupationally exposed women [160,161] | A PPARα-dependent mode of action (has been proposed for the induction of pancreatic acinar cell tumors [162] BBP can also act as a weak AhR agonist and modulate AhR-mediated signaling pathway [132,163] |
| Clofibrate | Liver adenomas and carcinomas [164] Pancreas carcinoma acinar cells [165] No neoplastic formation in p53+/− mice. Non-neoplastic findings in the adrenals, pancreas, and prostate [155]. | Patients given clofibrate developed several adverse health conditions but not cancer. | PPARα dependent peroxisome proliferation | |
| Methylclofenapate | Liver hepatocarcinoma (RM, RF, MM, MF) Pancreas adenoma (RM) Testes adenoma Leyding cells (RM) [166,167] | |||
| 12-O-Tetradecanoyl phobol 13-acetate (TPA) | Potent promoter of the skin carcinogenesis in mouse Complete carcinogen | Used as a promoter in in vitro and in vivo initiation-promotion tests TPA alone induces a significant incidence (p value 0.05) of papillomas and some carcinomas in mouse skin [95] | No data | TPA mimics the action of diacylglycerol (DAG), thus activating several receptors downstream of the signaling pathway |
| Group D | Target Organs in Rodents | Initiation/Promotion In Vivo and/or In Vitro Tests | Epidemiological Evidence | Mechanisms |
| Benzoyl peroxide (BPO) | Negative results | Acts as a promoter in mouse skin initiated with dimethylbenz(a)anthracene (DMBA) [168] | No data | As a free radical generating chemical, it was found to induce direct oxidative activation of protein kinase C [169,170] |
| Methoxychlor | Liver: hemagiosarcomas (RM) | An association between leukemia and farmers using methoxychlor has been reported (OR 2.2) [171] | Methoxychlor metabolites interact with estrogen and androgen receptors [172] |
| Test Chemical | Carcinogenicity Evidence | ||||||
|---|---|---|---|---|---|---|---|
| CAS | Chemical Name | Clone/Experimental Protocol | Use | IARC Classification 1 | CMR Classification | NTP (RoC) 2 | Properties of Concern 3 |
| Registry Number | |||||||
| 87-29-6 | Cinnamyl anthranilate | A31-1 | Flavoring substance | 3 | NC | Positive (NL) | No hazards |
| 56-53-1 | Diethylstilbestrol (DES) | A31 | Synthetic nonsteroidal estrogen. Former use: miscarriage prevention, hormone replacement therapy. Current use drug in clinical trials for the treatment of prostate and breast cancer | Known to be a human carcinogen | |||
| First listed in the First Annual Report on Carcinogens [129] | |||||||
| 50-29-3 | Dichlorodiphenyl trichloroethane (DDT) | A31 | Pesticide | 2A | NA | Reasonably anticipated to be a human carcinogen [129] | CLP notification: C2 |
| ED | |||||||
| 50-28-2 | Estradiol | A31-1-13 | Hormone replacement therapy; combined oral contraceptives | NA | R 1A | Known to be human carcinogen [129] | CLP notification: C2 |
| 64-17-5 | Ethyl Alcohol Ethanol | A31-1-13 | Industrial use in the manufacture of drugs, plastics, lacquers, polishes, plasticizers, and cosmetics; medical uses as a topical anti-infective, and as an antidote for ethylene glycol or methanol overdose. Commercial use in beverages | 1 | NC | NA | CLP notification: C 1B |
| 598-55-0 | Methylcarbamate | A31-1-1 | Primarily used in the textile and polymer industries as a reactive intermediate | 3 | NA | Clear evidence in rats both sexes, negative in mouse (NL) | CLP notification: C2 |
| 34807-41-5 | Mezerein | A31-1-13 | Daphnetoxin, folk medicine plant used in cancer treatment | NA | NA | NA | Other hazards |
| 139-13-9 | Nitrilotriacetic acid, trisodium salt | A31-1-13 | Boiler feedwater additive, in water and textile treatment, in metal plating and cleaning and in pulp and paper processing | 2B | NA | Reasonably anticipated to be a human carcinogen [129] | CLP notification: C2 |
| 50-55-5 | Reserpine | A31-1-1 | Antihypertensive drug | 3 | NA | Reasonably anticipated to be carcinogenic [129] | Other hazards |
| 128-44-9 | Sodium saccharin | A31-1-13 | Artificial sweetener | NA | NA | Delisted from RoC [129] | NA |
| SHE CTA Transforming Chemicals | BALB/c 3T3 Transforming Chemicals |
|---|---|
| Group A: Chemicals for which a clear correlation exists between transforming properties and in vivo carcinogenesis | |
| Di (2-Ethylhexyl) phthalate (DEHP) | Di (2-Ethylhexyl) phthalate (DEHP) |
| Reserpine | Reserpine |
| Diethanolamine | Diethylstilbestrol (DES) |
| N,N′-Diethylthiourea (DETU) | Dichlorodiphenyl trichloroethane (DDT) |
| Ethylbenzene | Estradiol |
| Methyl eugenol | Nitrilotriacetic acid, trisodium salt |
| Oxymetholone | Ethanol |
| Group B: Chemicals for which available data indicate a possible correlation between transforming properties and in vivo carcinogenesis | |
| Wyeth-14,643 | Methylcarbamate |
| Group C: Chemicals for which data are suggestive for a possible correlation between transforming properties and in vivo carcinogenesis | |
| Bemitradine | Cinnamyl anthranilate |
| Butylbenzoylphthalate | |
| Clofibrate | |
| Methylclofenapate | |
| 12-O-Tetradecanoyl phobol 13-acetate (TPA) | |
| Group D: Chemicals for which not enough data are available to show a correlation between transforming properties and in vivo carcinogenesis | |
| Benzoyl peroxide | Mezerein |
| Methoxychlor | |
| Group A Chemicals | Target Organs in Rodents | Initiation/Promotion In Vivo and/or In Vitro Tests | Epidemiological Evidence | Mechanisms |
|---|---|---|---|---|
| Diethylstilbestrol (DES) | Several different tissue sites (primarily estrogen-sensitive organs and tissues). Such as: mammary gland, carcinoma, adenocarcinoma (MM, MF) cervix and uterus, adenocarcinoma, vagina (squamous-cell-carcinoma (MF)) [129] | No data | Sufficient evidence of carcinogenicity from studies in humans. | As a synthetic estrogen, DES can react with the estrogen receptors, but the molecular mechanisms triggering and sustaining the pathway to cancer are still poorly understood [173] |
| Di (2-Ethylhexyl) phthalate (DEHP) | See Table 5 | See Table 5 | See Table 5 | See Table 5 |
| Dichlorodiphenyl trichloroethane (DDT) | Liver, hepatocellular (MM, MF, R) | Positive in CTA BALB/c mouse embryo [175,176] | Epidemiological studies gave mixed results, showing positive associations with breast cancer in women subgroups exposed to high levels of DDT, multiple myeloma in farmers, and liver cancer in high-level exposed population. Negative associations were also reported | DDT behaves as an estrogen receptor agonist and/or androgen receptor antagonist. Due to their long half-life and its lipophilic nature, DDT and its metabolite DDE (dichlorodiphenyl dichloroethylene) are still detected in the serum of western pregnant and lactating mothers [177,178] |
| Multiple sites, sarcoma-reticulum cell (MF) [174] | ||||
| Estrogen | Endometrial, cervical, and mammary-gland tumors in mice, mammary and pituitary-gland tumors in rats, and kidney tumors in hamsters [129] | Positive in CTA BALB/c A31-1-13 | Increased incidence of endometrial and ovarian cancer as a consequence of the therapy [129] | Estrogen receptor interaction causing cell proliferation, affecting cell differentiation and gene expression [129]. AhR interaction leading to Cyp1A1 and Cyp1B1 mediated modulation of estrogen metabolism [10,67,179] |
| Nitrilotriacetic acid (NTA), trisodium salt | Kidney: adenocarinoma–tubular (RM, RF, MM, MF) [129]; | Positive in CTA BALB/c mouse embryo cells [180] | Data available from epidemiological studies are inadequate [129] | It has been suggested that the nephron-carcinogenic properties of NTA are related to dose-dependent changes in intracellular zinc ion homeostasis, due to the chelating properties of NTA |
| Urinary bladder: Carcinoma-squamous cell; Carcinoma-transitional cell (RF) | ||||
| Ureter: Adenoma-papillary; Papilloma (RM) [129] | ||||
| Reserpine | See Table 6 | See Table 6 | See Table 6 | See Table 6 |
| CAS Registry Number | Chemical Name | Carcinogenicity | Genotoxicity Studies with Negative or Equivocal Results |
|---|---|---|---|
| 10108-64-2 | Cadmium chloride | IARC class 1 | Ames, in vitro CA |
| 120-80-9 | Catechol | IARC class 2B | Ames, in vivo MN |
| 474-25-9 | Chenodeoxicholic acid | Colon cancer promoter | Ames |
| 3165-93-3 | 4-Chloro-o-toluidine hydrochloride | IARC class 2A | Ames |
| 81-25-4 | Cholic acid | Colon cancer promoter | Ames |
| 83-44-3 | Deoxycholic acid | Colon cancer promoter | Ames |
| 50-29-3 | DDT | IARC class 2A | Ames in vitro CA, in vivo MN |
| 111-42-2 | Diethanolamine | IARC class 2B | Ames, in vitro CA in vivo MN |
| 117-81-7 | DEHP | IARC class 2B | Ames, in vivo MN, in vitro MN, MLA |
| 116355-83-0 | Fumonisin B1 | IARC class 2B | Ames |
| 5989-27-5 | D-Limonene | IARC class 3, Male rat kidney tumors | Ames, in vivo comet, in vitro CA |
| 434-13-9 | Lithocholic acid | Colon cancer promoter | Ames |
| 135-23-9 | Methapyrilene HCl | Hepatocarcinogen in rats | Ames, in vitro CA, SCE, in vivo CA, in vivo MN |
| 124-58-3 | Methylarsonic acid | IARC class 2B | Ames |
| 34807-41-5 | Mezerein | Tumor promoter on mouse skin | Ames |
| 78111-17-8 | Okadaic acid | Tumor promoter on mouse skin | Ames, in vitro CHO/HGPRT |
| 57-83-0 | Progesterone | IARC class 2B | Ames in vitro CA, SCE, in vivo CA |
| 16561-29-8 | 12-O-Tetradecanoyl phobol 13-acetate (TPA) | Tumor promoter on mouse skin, Carcinogen on heirless mouse skin, Carcinogen in NTP | Ames |
| 7631-89-2 | Sodium arsenate | IARC class 1 | Ames, MLA |
| 7784-46-5 | Sodium arsenite | IARC class 1 | Ames |
| 82385-42-0 | Sodium saccharin | IARC class 3, Rat and mouse bladder tumors | Ames, in vivo CA, in vivo comet, in vitro MLA |
| 1746-01-6 | 2,3,7,8-Tetrachlorodibenzo-P-dioxin (TCDD) | IARC class 1 | Ames, in vitro MLA, in vitro CA, SCE, in vivo CA |
| Degree of Cell Confluence at Different Time after Cell Seeding | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Seeding Density | 24 h | 48 h | 72 h | 96 h | ||||||||
| C | MIX | XF | C | MIX | XF | C | MIX | XF | C | MIX | XF | |
| 5 × 105 | 50 | 50 | 20 | 70–80 | 60–70 | 20 | >90 | 80–90 | 10 | / | / | 10 |
| 2.5 × 105 | 20–30 | 30 | 10–20 | 50–60 | 50 | 10 | 80 | 50–60 | 10 | / | / | 10 |
| 1 × 105 | 10 | 10 | <10 | 40 | 10–20 | <10 | 50–60 | 10–15 | <10 | >90 | 10–15 | <10 |
| 0.5 × 105 | <10 | <10 | ND | 20 | 10 | <10 | 30–40 | 10 | <10 | 70–80 | 10 | <10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colacci, A.; Corvi, R.; Ohmori, K.; Paparella, M.; Serra, S.; Da Rocha Carrico, I.; Vasseur, P.; Jacobs, M.N. The Cell Transformation Assay: A Historical Assessment of Current Knowledge of Applications in an Integrated Approach to Testing and Assessment for Non-Genotoxic Carcinogens. Int. J. Mol. Sci. 2023, 24, 5659. https://doi.org/10.3390/ijms24065659
Colacci A, Corvi R, Ohmori K, Paparella M, Serra S, Da Rocha Carrico I, Vasseur P, Jacobs MN. The Cell Transformation Assay: A Historical Assessment of Current Knowledge of Applications in an Integrated Approach to Testing and Assessment for Non-Genotoxic Carcinogens. International Journal of Molecular Sciences. 2023; 24(6):5659. https://doi.org/10.3390/ijms24065659
Chicago/Turabian StyleColacci, Annamaria, Raffaella Corvi, Kyomi Ohmori, Martin Paparella, Stefania Serra, Iris Da Rocha Carrico, Paule Vasseur, and Miriam Naomi Jacobs. 2023. "The Cell Transformation Assay: A Historical Assessment of Current Knowledge of Applications in an Integrated Approach to Testing and Assessment for Non-Genotoxic Carcinogens" International Journal of Molecular Sciences 24, no. 6: 5659. https://doi.org/10.3390/ijms24065659
APA StyleColacci, A., Corvi, R., Ohmori, K., Paparella, M., Serra, S., Da Rocha Carrico, I., Vasseur, P., & Jacobs, M. N. (2023). The Cell Transformation Assay: A Historical Assessment of Current Knowledge of Applications in an Integrated Approach to Testing and Assessment for Non-Genotoxic Carcinogens. International Journal of Molecular Sciences, 24(6), 5659. https://doi.org/10.3390/ijms24065659