

An In Vitro Analysis of TKI-Based Sequence Therapy in Renal Cell Carcinoma Cell Lines



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
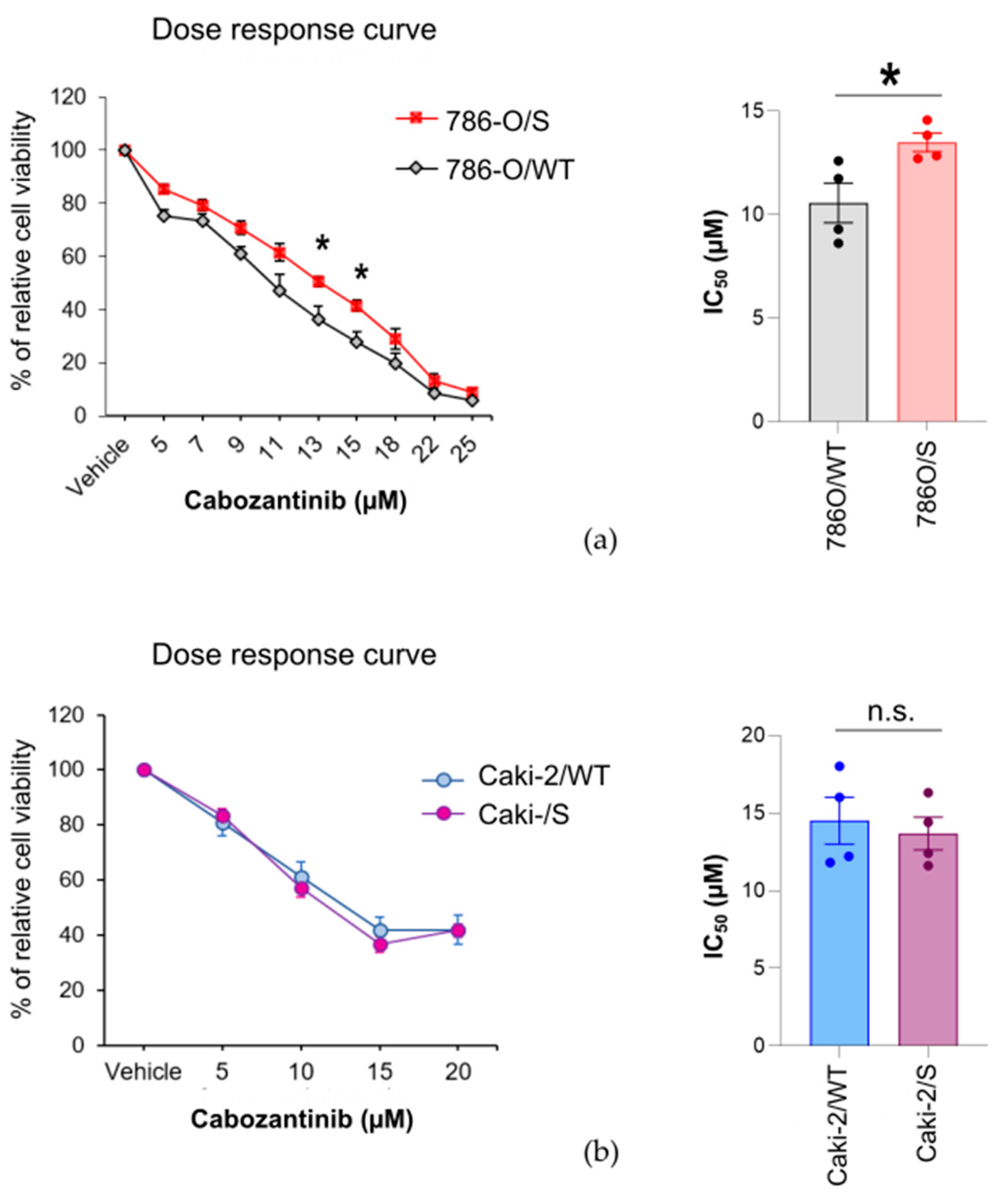
2.1. The Cytotoxic Effect of Cabozantinib on Sunitinib-Treated and Treatment-Naïve Human RCC Cell Lines Is Cell-Line-Specific
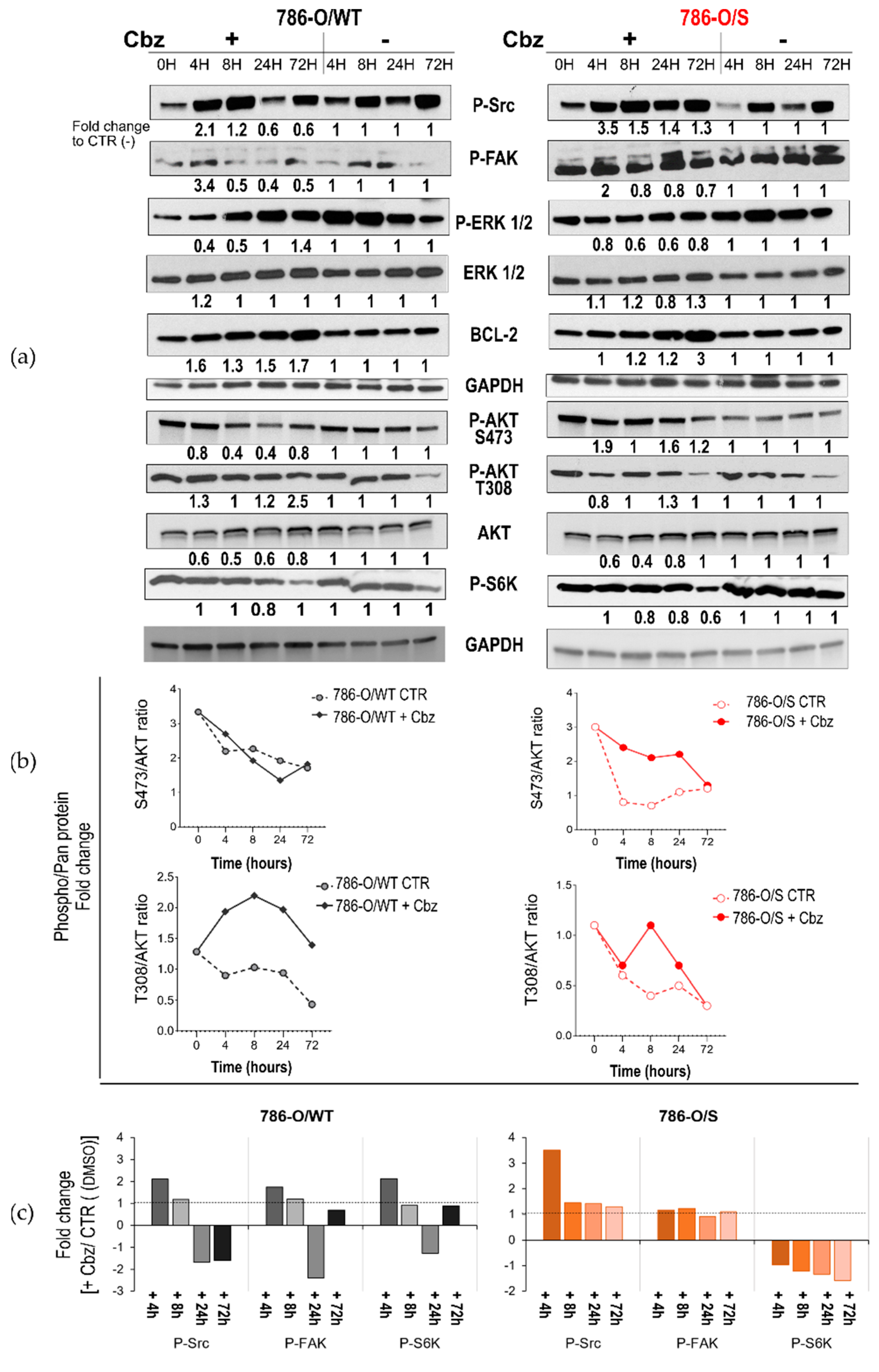
2.2. Effect of Cabozantinib on Signaling Transduction in Sunitinib-Pretreated and Treatment-Naïve 786-O Cell Lines
2.3. Effect of Cabozantinib on Signaling Transduction in Sunitinib-Pretreated and Treatment-Naïve Caki-2 Cell Lines
3. Discussion
4. Material and Methods
4.1. Compounds
4.2. Cell Culture
4.3. Establishment of Sunitinib-Resistant Cell Lines
4.4. Cell Survival (WST-1) Assay
4.5. SDS-PAGE and Immunoblotting
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Global Health Estimates: Leading Causes of Death. Available online: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death (accessed on 17 February 2021).
- EAU Guidelines on RCC—Epidemiology Aetiology and Pathology—Uroweb. Uroweb—European Association of Urology. Available online: https://uroweb.org/guidelines/renal-cell-carcinoma/chapter/epidemiology-aetiology-and-pathology#:~:text=3.1.,Republic%20and%20Lithuania%20[14] (accessed on 17 February 2023).
- Urologic Malignancies: Advances in the Analysis and Interpretation of Clinical Findings. Available online: https://www.future-science.com/doi/epub/10.2144/fsoa-2020-0210 (accessed on 16 February 2023).
- Wang, J.; Zhang, W.; Hou, W.; Zhao, E.; Li, X. Molecular Characterization, Tumor Microenvironment Association, and Drug Susceptibility of DNA Methylation-Driven Genes in Renal Cell Carcinoma. Front. Cell Dev. Biol. 2022, 10, 510. Available online: https://www.frontiersin.org/article/10.3389/fcell.2022.837919 (accessed on 25 April 2022). [CrossRef] [PubMed]
- Hartmann, A.; Stöhr, C.G.; Junker, K. Hereditäre Nierenzellkarzinome. Pathologe 2010, 31, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, L.; Elguero, B.; Pacin, D.G.; Senin, S.; Pollak, C.; Marchiñena, P.A.G.; Jurado, A.M.; Isola, M.; Labanca, M.J.; Palazzo, M.; et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis. 2019, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Dizman, N.; Philip, E.J.; Pal, S.K. Genomic profiling in renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Zeuschner, P.; Zaccagnino, A.; Junker, K. Biomarker: Der Weg zur individualisierten Therapie bei Nierenzelltumoren. Aktuelle Urol. 2021, 52, 452–463. [Google Scholar] [CrossRef]
- Baldewijns, M.M.; van Vlodrop, I.J.; Vermeulen, P.B.; Soetekouw, P.M.; van Engeland, M.; de Bruïne, A.P. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 2010, 221, 125–138. [Google Scholar] [CrossRef]
- Shen, C.; Kaelin, W.G. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 2012, 23, 18–25. [Google Scholar] [CrossRef]
- Koochekpour, S.; Jeffers, M.; Wang, P.H.; Gong, C.; Taylor, G.A.; Roessler, L.M.; Stearman, R.; Vasselli, J.R.; Stetler-Stevenson, W.G.; Kaelin, W.G.; et al. The von Hippel-Lindau Tumor Suppressor Gene Inhibits Hepatocyte Growth Factor/Scatter Factor-Induced Invasion and Branching Morphogenesis in Renal Carcinoma Cells. Mol. Cell. Biol. 1999, 19, 5902–5912. [Google Scholar] [CrossRef]
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Alonso-Gordoa, T.; García-Bermejo, M.L.; Grande, E.; Garrido, P.; Carrato, A.; Molina-Cerrillo, J. Targeting Tyrosine kinases in Renal Cell Carcinoma: ‘New Bullets against Old Guys’. Int. J. Mol. Sci. 2019, 20, 1901. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Hoeh, B.; Flammia, R.S.; Hohenhorst, L.; Sorce, G.; Panunzio, A.; Tappero, S.; Tian, Z.; Saad, F.; Gallucci, M.; Briganti, A.; et al. IO-IO vs IO-TKI efficacy in metastatic kidney cancer patients: A structured systematic review over time. Semin. Oncol. 2022, 49, 394–399. [Google Scholar] [CrossRef]
- Tenold, M.; Ravi, P.; Kumar, M.; Bowman, A.; Hammers, H.; Choueiri, T.K.; Lara, P.N. Current Approaches to the Treatment of Advanced or Metastatic Renal Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 187–196. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Gagliano, T.; Giamas, G. Direct Effects of Anti-Angiogenic Therapies on Tumor Cells: VEGF Signaling. Trends Mol. Med. 2017, 23, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Rovithi, M.; De Haas, R.R.; Honeywell, R.J.; Dekker, H.; Poel, D.; Azijli, K.; Peters, G.J.; Broxterman, H.J.; Verheul, H.M.W. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cell. Oncol. 2015, 38, 119–129. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Budka, J.; Damayanti, N.P.; Arrington, J.; Ferris, M.; Hsu, C.-C.; Chintala, S.; Orillion, A.; Miles, K.M.; Shen, L.; et al. EZH2 Modifies Sunitinib Resistance in Renal Cell Carcinoma by Kinome Reprogramming. Cancer Res. 2017, 77, 6651–6666. [Google Scholar] [CrossRef]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.-J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.-F.; et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Liu, W.; Zhan, Y. Long Non-coding RNA CCAT1 Acts as an Oncogene and Promotes Sunitinib Resistance in Renal Cell Carcinoma. Front. Oncol. 2020, 10, 516552. [Google Scholar] [CrossRef]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Marona, P.; Górka, J.; Kotlinowski, J.; Majka, M.; Jura, J.; Miekus, K. C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas. Cells 2019, 8, 272. [Google Scholar] [CrossRef]
- Nakaigawa, N.; Yao, M.; Baba, M.; Kato, S.; Kishida, T.; Hattori, K.; Nagashima, Y.; Kubota, Y. Inactivation of von Hippel-Lindau Gene Induces Constitutive Phosphorylation of MET Protein in Clear Cell Renal Carcinoma. Cancer Res. 2006, 66, 3699–3705. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Powles, T. Recent eUpdate to the ESMO Clinical Practice Guidelines on renal cell carcinoma on cabozantinib and nivolumab for first-line clear cell renal cancer: Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up1. Ann. Oncol. 2021, 32, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.-L.; Peltola, K.; et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- George, D.J.; Hessel, C.; Halabi, S.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Picus, J.; Small, E.J.; Dakhil, S.; Feldman, D.R.; et al. Cabozantinib Versus Sunitinib for Untreated Patients with Advanced Renal Cell Carcinoma of Intermediate or Poor Risk: Subgroup Analysis of the Alliance A031203 CABOSUN trial. Oncologist 2019, 24, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Hessel, C.; Halabi, S.; Sanford, B.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur. J. Cancer 2018, 94, 115–125. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.M.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Jia, P.; Li, X.; Pei, G.; Wang, C.; Fang, X.; Zhao, Z.; Cai, Z.; Yi, X.; et al. Clonal architectures predict clinical outcome in clear cell renal cell carcinoma. Nat. Commun. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Kim, B.; Wang, S.; Lee, J.M.; Jeong, Y.; Ahn, T.; Son, D.-S.; Park, H.W.; Yoo, H.-S.; Song, Y.-J.; Lee, E.; et al. Synthetic lethal screening reveals FGFR as one of the combinatorial targets to overcome resistance to Met-targeted therapy. Oncogene 2014, 34, 1083–1093. [Google Scholar] [CrossRef]
- Shinh, Y.-S.; Lai, C.-Y.; Kao, Y.-R.; Shiah, S.-G.; Chu, Y.-W.; Lee, H.-S.; Wu, C.-W. Expression of Axl in Lung Adenocarcinoma and Correlation with Tumor Progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, B.J.; Kim, H.S. Clinicopathological impacts of high c-Met expression in renal cell carcinoma: A meta-analysis and review. Oncotarget 2017, 8, 75478–75487. [Google Scholar] [CrossRef]
- Ko, B.; He, T.; Gadgeel, S.; Halmos, B. MET/HGF pathway activation as a paradigm of resistance to targeted therapies. Ann. Transl. Med. 2017, 5, 1245. [Google Scholar] [CrossRef]
- Kammerer-Jacquet, S.-F.; Medane, S.; Bensalah, K.; Bernhard, J.-C.; Yacoub, M.; Dupuis, F.; Ravaud, A.; Verhoest, G.; Mathieu, R.; Peyronnet, B.; et al. Correlation of c-MET Expression with PD-L1 Expression in Metastatic Clear Cell Renal Cell Carcinoma Treated by Sunitinib First-Line Therapy. Target. Oncol. 2017, 12, 487–494. [Google Scholar] [CrossRef]
- Ravaud, A.; Gross-Goupil, M. Overcoming resistance to tyrosine kinase inhibitors in renal cell carcinoma. Cancer Treat. Rev. 2012, 38, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Lee, Y.H.; Boeke, M.; Jilaveanu, L.B.; Liu, Z.; Bottaro, D.P.; Kluger, H.M.; Shuch, B. MET Inhibition in Clear Cell Renal Cell Carcinoma. J. Cancer 2016, 7, 1205–1214. [Google Scholar] [CrossRef]
- Pan, T.; Martinez, M.; Hubka, K.M.; Song, J.H.; Lin, S.C.; Yu, G.; Lee, Y.-C.; Gallick, G.E.; Tu, S.-M.; Harrington, D.A.; et al. Cabozantinib Reverses Renal Cell Carcinoma-Mediated Osteoblast Inhibition in Three-Dimensional Co-culture In Vitro and Reduces Bone Osteolysis In Vivo. Mol. Cancer Ther. 2020, 19, 1266. [Google Scholar] [CrossRef]
- Ravaud, A.; Digue, L.; Trufflandier, N.; Smith, D. VEGFR TKI ‘resistance’ or transient clinical insensitivity to VEGFR TKI in metastatic renal cell carcinoma. Ann. Oncol. 2009, 21, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Small, E.J. Treatment of advanced renal cell carcinoma patients with cabozantinib, an oral multityrosine kinase inhibitor of MET, AXL and VEGF receptors. Futur. Oncol. 2019, 15, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Aurilio, G.; Massari, F.; Grande, E.; Matrana, M.R.; Rizzo, M.; De Giorgi, U.; Incorvaia, L.; Martignetti, A.; Molina-Cerrillo, J.; et al. Nivolumab VERSUS Cabozantinib as Second-Line Therapy in Patients With Advanced Renal Cell Carcinoma: A Real-World Comparison. Clin. Genitourin. Cancer 2022, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Aimudula, A.; Nasier, H.; Yang, Y.; Zhang, R.; Lu, P.; Hao, J.; Bao, Y. PPARα mediates sunitinib resistance via NF-κB activation in clear cell renal cell carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 2389–2400. [Google Scholar] [PubMed]
- Butz, H.; Ding, Q.; Nofech-Mozes, R.; Lichner, Z.; Ni, H.; Yousef, G.M. Elucidating mechanisms of sunitinib resistance in renal cancer: An integrated pathological-molecular analysis. Oncotarget 2017, 9, 4661–4674. [Google Scholar] [CrossRef]
- DepMap: The Cancer Dependency Map Project at Broad Institute. Available online: https://depmap.org/portal/depmap/ (accessed on 2 November 2022).
- Brodaczewska, K.K.; Szczylik, C.; Fiedorowicz, M.; Porta, C.; Czarnecka, A.M. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016, 15, 83. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, A.; Miyazaki, T.; Tanaka, H.; Morimoto, M.; Nakai, K.; Soeda, J. Cabozantinib inhibits AXL- and MET-dependent cancer cell migration induced by growth-arrest-specific 6 and hepatocyte growth factor. Biochem. Biophys. Rep. 2020, 21, 100726. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, E.; Shen, H.; Wang, X.; Tang, T.; Zhang, X.; Xu, J.; Tang, Z.; Guo, C.; Bai, X.; et al. Targeting the HGF/MET Axis in Cancer Therapy: Challenges in Resistance and Opportunities for Improvement. Front. Cell Dev. Biol. 2020, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Cepero, V.; Sierra, J.R.; Corso, S.; Ghiso, E.; Casorzo, L.; Perera, T.; Comoglio, P.M.; Giordano, S. MET and KRAS Gene Amplification Mediates Acquired Resistance to MET Tyrosine Kinase Inhibitors. Cancer Res. 2010, 70, 7580–7590. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Dang, H.; You, H.; Rountree, C.B.; Ding, W. The EGFR/ErbB3 Pathway Acts as a Compensatory Survival Mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0128159. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Vargas, J.C.S.; Biondani, P.; Maggi, C.; Gariboldi, M.; Gloghini, A.; Inno, A.; Volpi, C.C.; Gualeni, A.V.; Di Bartolomeo, M.; De Braud, F.; et al. Role of cMET in the Development and Progression of Colorectal Cancer. Int. J. Mol. Sci. 2013, 14, 18056–18077. [Google Scholar] [CrossRef]
- Antoniades, I.; Kyriakou, M.; Charalambous, A.; Kalalidou, K.; Christodoulou, A.; Christoforou, M.; Skourides, P.A. FAK displacement from focal adhesions: A promising strategy to target processes implicated in cancer progression and metastasis. Cell Commun. Signal. 2021, 19, 3. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Chuang, H.-H.; Zhen, Y.-Y.; Tsai, Y.-C.; Chuang, C.-H.; Hsiao, M.; Huang, M.-S.; Yang, C.-J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 1726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Stockwell, S.R.; Elbanna, M.; Ketteler, R.; Freeman, J.; Al-Lazikani, B.; Eccles, S.; Brandon, A.D.H.; Raynaud, F.; Hayes, A.; et al. Signalling involving MET and FAK supports cell division independent of the activity of the cell cycle-regulating CDK4/6 kinases. Oncogene 2019, 38, 5905–5920. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Chen, H.-C. Direct Interaction of Focal Adhesion Kinase (FAK) with Met Is Required for FAK To Promote Hepatocyte Growth Factor-Induced Cell Invasion. Mol. Cell. Biol. 2006, 26, 5155–5167. [Google Scholar] [CrossRef]
- Xu, A.M.; Huang, P.H. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef] [PubMed]
- CT-707, A Novel FAK Inhibitor, Synergizes with Cabozantinib to Suppress Hepatocellular Carcinoma by Blocking Cabozantinib-Induced FAK Activation|Molecular Cancer Therapeutics|American Association for Cancer Research. Available online: https://aacrjournals.org/mct/article/15/12/2916/147598/CT-707-a-Novel-FAK-Inhibitor-Synergizes-with (accessed on 19 April 2022).
- Hiscox, S.; Jordan, N.J.; Morgan, L.; Green, T.P.; Nicholson, R.I. Src kinase promotes adhesion-independent activation of FAK and enhances cellular migration in tamoxifen-resistant breast cancer cells. Clin. Exp. Metastasis 2007, 24, 157–167. [Google Scholar] [CrossRef]
- Del Mistro, G.; Riemann, S.; Schindler, S.; Beissert, S.; Kontermann, R.E.; Ginolhac, A.; Halder, R.; Presta, L.; Sinkkonen, L.; Sauter, T.; et al. Focal adhesion kinase plays a dual role in TRAIL resistance and metastatic outgrowth of malignant melanoma. Cell Death Dis. 2022, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.C.; Serrels, A.; Stupack, D.G.; Schlaepfer, D.D.; Frame, M.C. Targeting FAK in anticancer combination therapies. Nat. Rev. Cancer 2021, 21, 313–324. [Google Scholar] [CrossRef]
- Halder, J.; Landen, C.N.; Lutgendorf, S.K.; Li, Y.; Jennings, N.B.; Fan, D.; Nelkin, G.M.; Schmandt, R.; Schaller, M.D.; Sood, A.K. Focal Adhesion Kinase Silencing Augments Docetaxel-Mediated Apoptosis in Ovarian Cancer Cells. Clin. Cancer Res. 2005, 11, 8829–8836. [Google Scholar] [CrossRef]
- Roy-Luzarraga, M.; Reynolds, L.E.; de Luxán-Delgado, B.; Maiques, O.; Wisniewski, L.; Newport, E.; Rajeeve, V.; Drake, R.J.; Gómez-Escudero, J.; Richards, F.M.; et al. Suppression of Endothelial Cell FAK Expression Reduces Pancreatic Ductal Adenocarcinoma Metastasis after Gemcitabine Treatment. Cancer Res. 2022, 82, 1909–1925. [Google Scholar] [CrossRef]
- Higuchi, M.; Ishiyama, K.; Maruoka, M.; Kanamori, R.; Takaori-Kondo, A.; Watanabe, N. Paradoxical activation of c-Src as a drug-resistant mechanism. Cell Rep. 2021, 34, 108876. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Molina, A.M. PI3K/AKT inhibitors in patients with refractory renal cell carcinoma: What have we learnt so far? Ann. Oncol. 2017, 28, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Martinez Calejman, C.; Trefely, S.; Entwisle, S.W.; Luciano, A.; Jung, S.M.; Hsiao, W.; Torres, A.; Hung, C.M.; Li, H.; Snyder, N.W.; et al. mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat. Commun. 2020, 11, 575. [Google Scholar] [CrossRef] [PubMed]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Kumar, K.A.; Reddanna, P. The Paradox of Akt-mTOR Interactions. Front. Oncol. 2013, 3, 165. [Google Scholar] [CrossRef]
- Goncharova, E.A.; Goncharov, D.A.; Li, H.; Pimtong, W.; Lu, S.; Khavin, I.; Krymskaya, V.P. mTORC2 Is Required for Proliferation and Survival of TSC2-Null Cells. Mol. Cell. Biol. 2011, 31, 2484–2498. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaccagnino, A.; Vynnytska-Myronovska, B.; Stöckle, M.; Junker, K. An In Vitro Analysis of TKI-Based Sequence Therapy in Renal Cell Carcinoma Cell Lines. Int. J. Mol. Sci. 2023, 24, 5648. https://doi.org/10.3390/ijms24065648
Zaccagnino A, Vynnytska-Myronovska B, Stöckle M, Junker K. An In Vitro Analysis of TKI-Based Sequence Therapy in Renal Cell Carcinoma Cell Lines. International Journal of Molecular Sciences. 2023; 24(6):5648. https://doi.org/10.3390/ijms24065648
Chicago/Turabian StyleZaccagnino, Angela, Bozhena Vynnytska-Myronovska, Michael Stöckle, and Kerstin Junker. 2023. "An In Vitro Analysis of TKI-Based Sequence Therapy in Renal Cell Carcinoma Cell Lines" International Journal of Molecular Sciences 24, no. 6: 5648. https://doi.org/10.3390/ijms24065648
APA StyleZaccagnino, A., Vynnytska-Myronovska, B., Stöckle, M., & Junker, K. (2023). An In Vitro Analysis of TKI-Based Sequence Therapy in Renal Cell Carcinoma Cell Lines. International Journal of Molecular Sciences, 24(6), 5648. https://doi.org/10.3390/ijms24065648