
Co-Catalyzed Asymmetric Hydrogenation. The Same Enantioselection Pattern for Different Mechanisms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
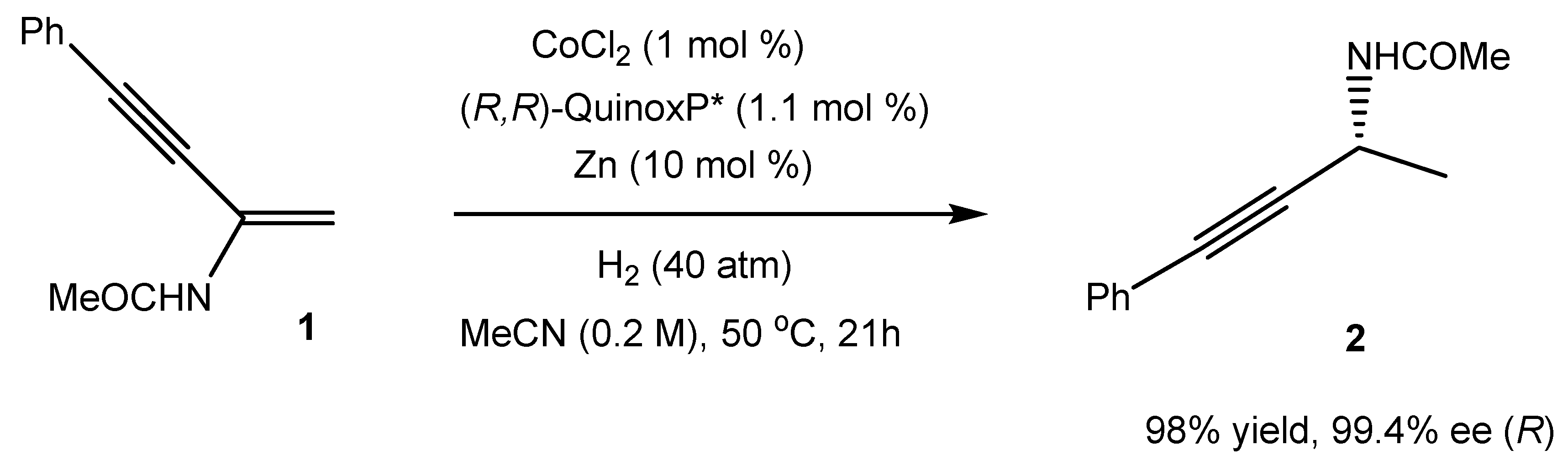{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
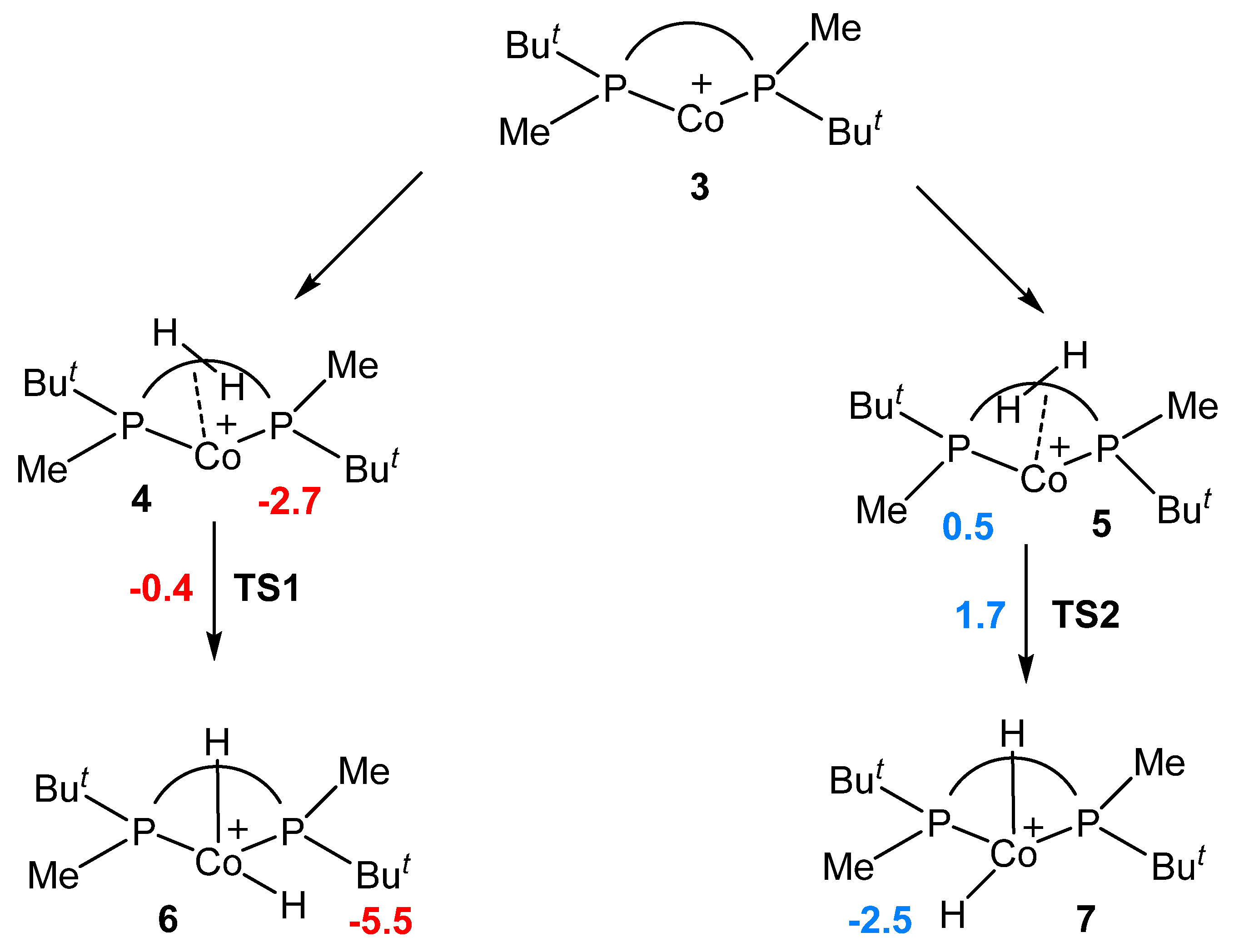
2.1. Formation of Solvate Dihydrides
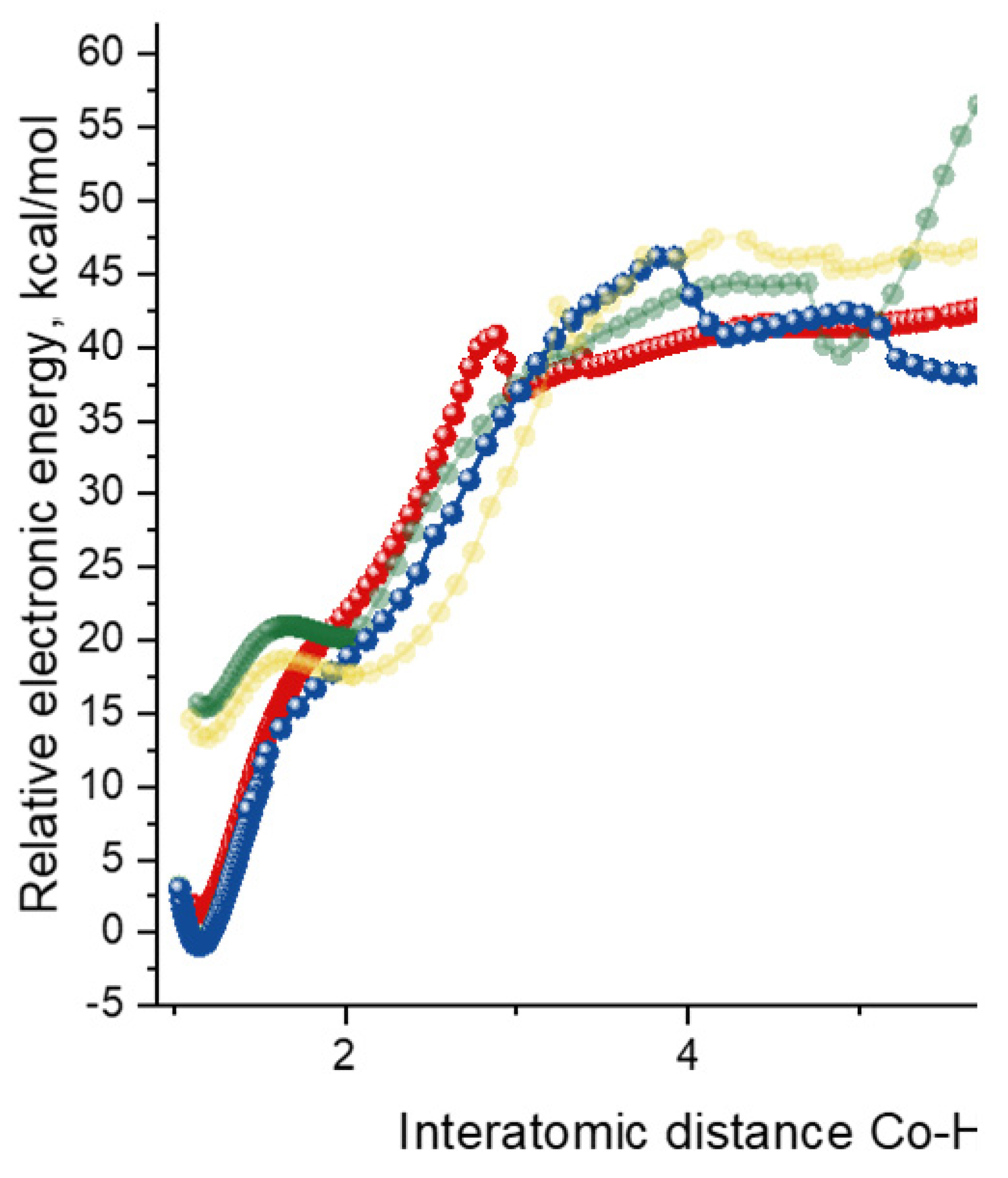
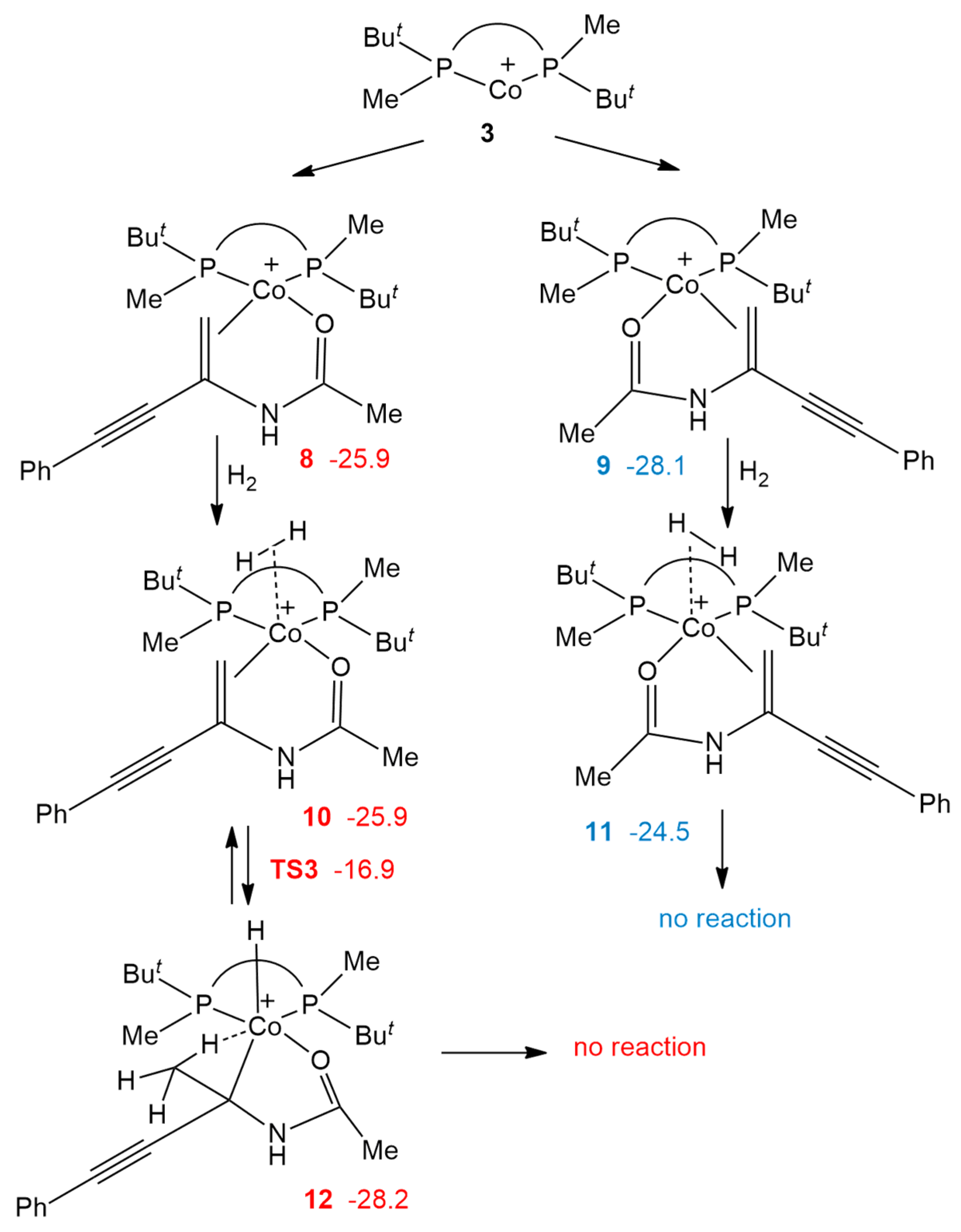
2.2. Oxidative Addition to Catalyst–Substrate Complexes
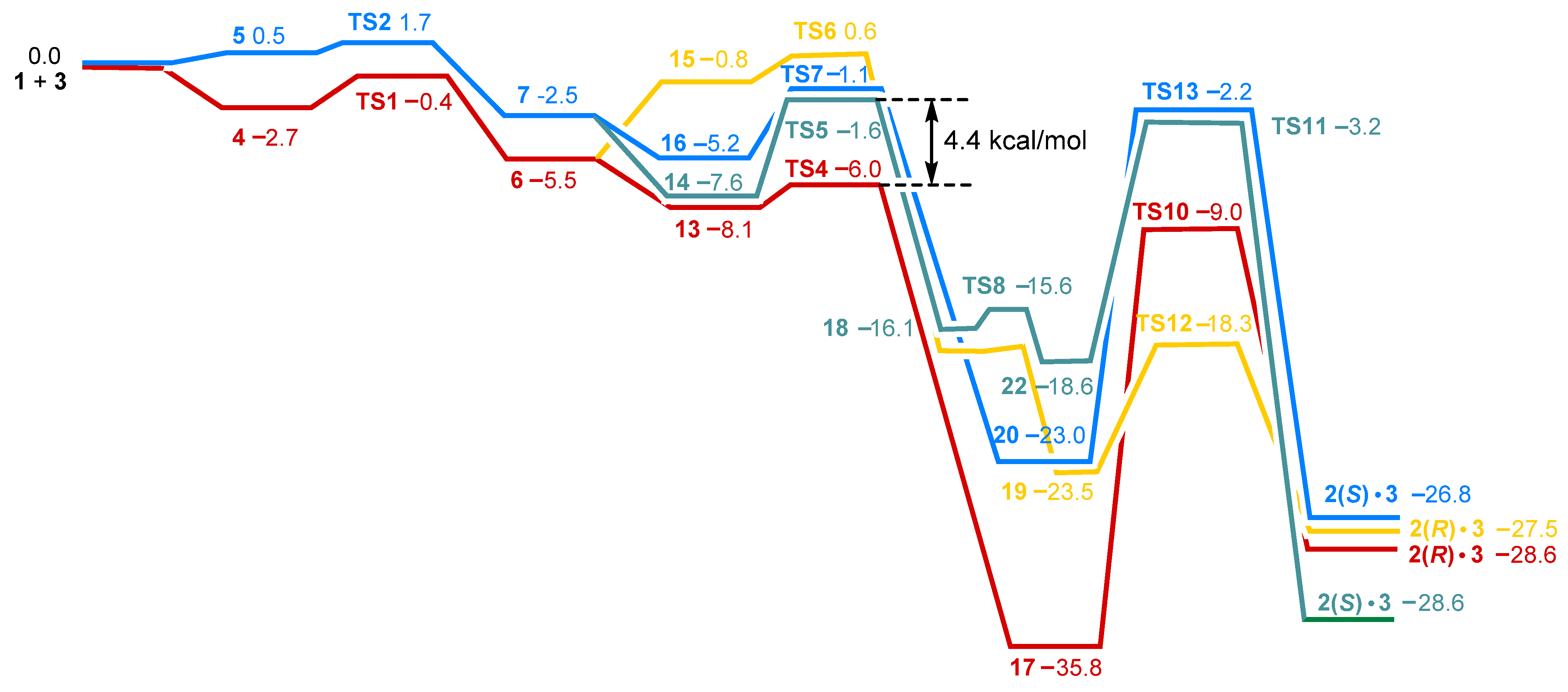
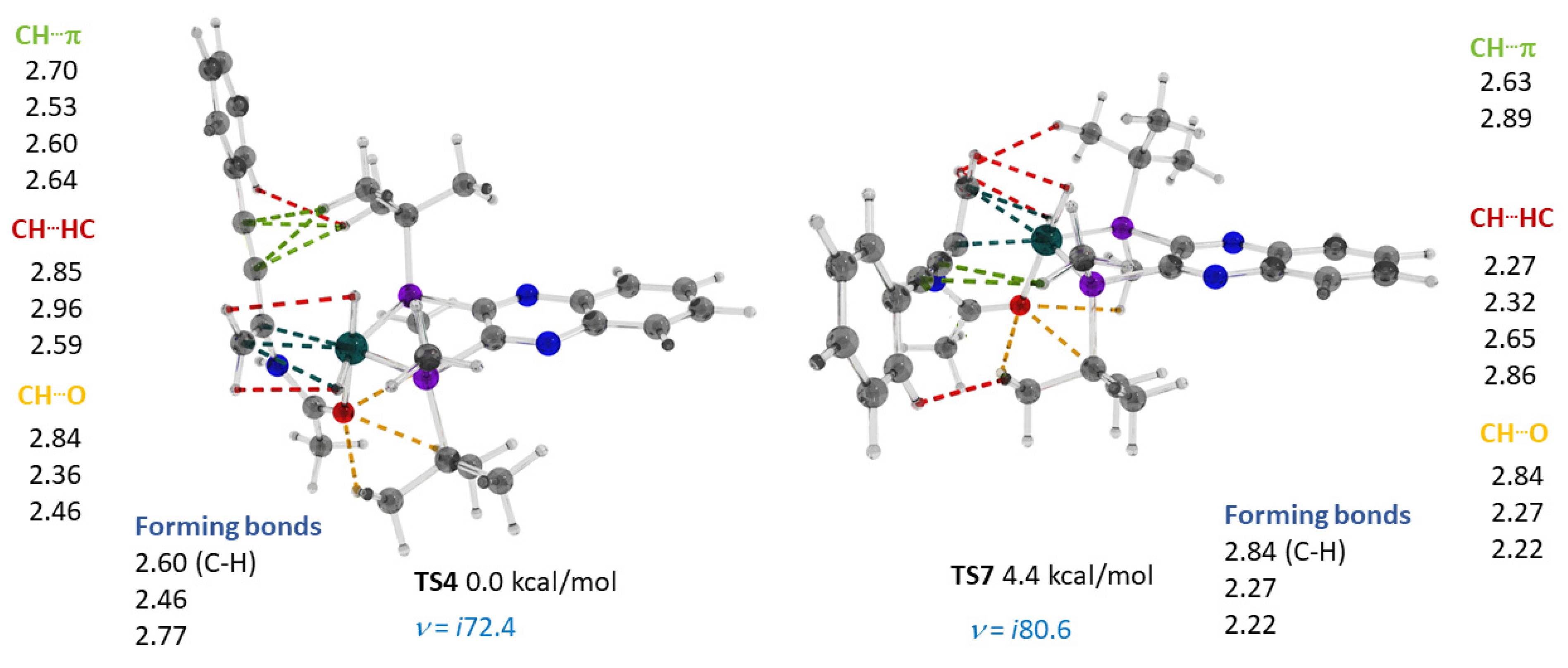
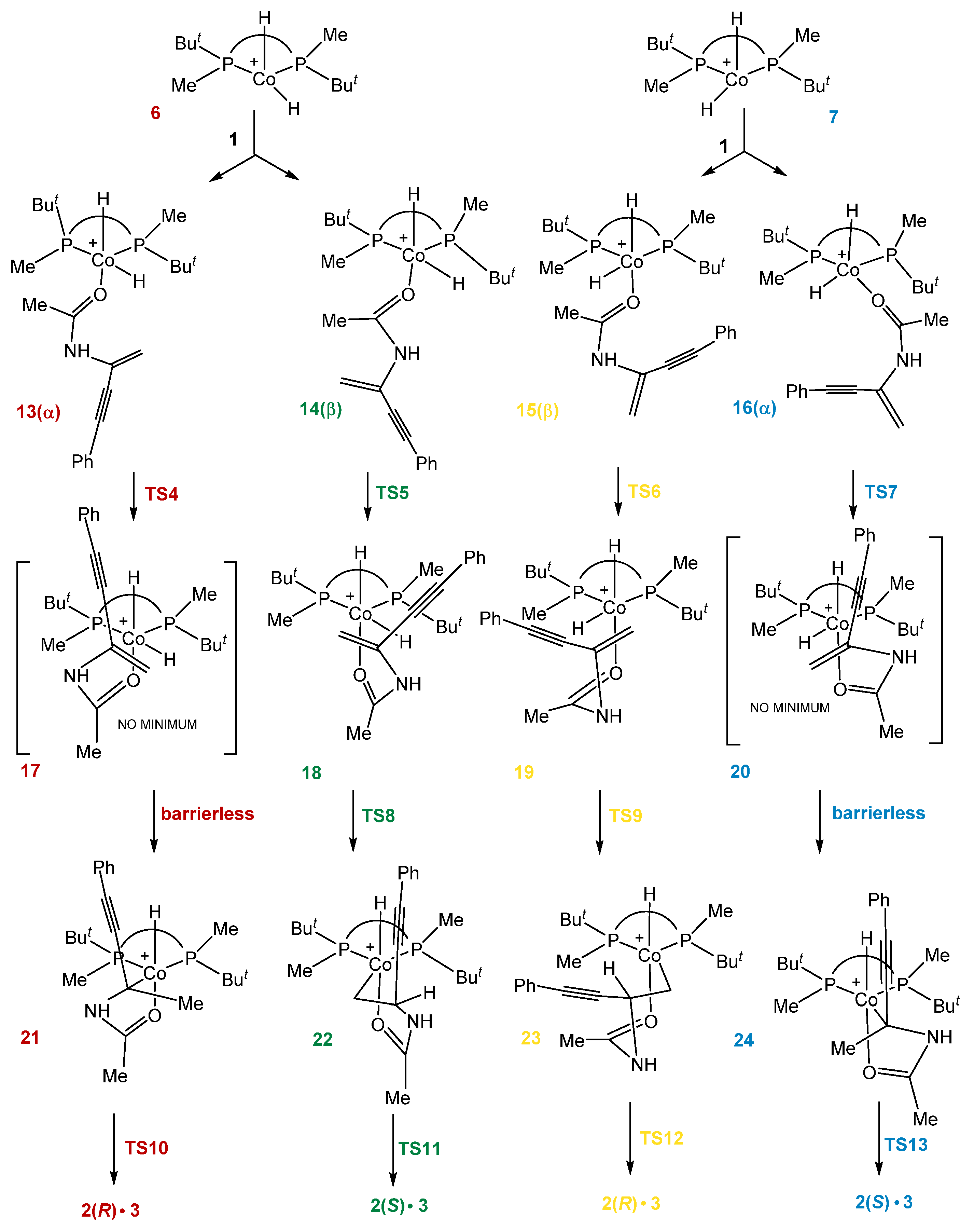
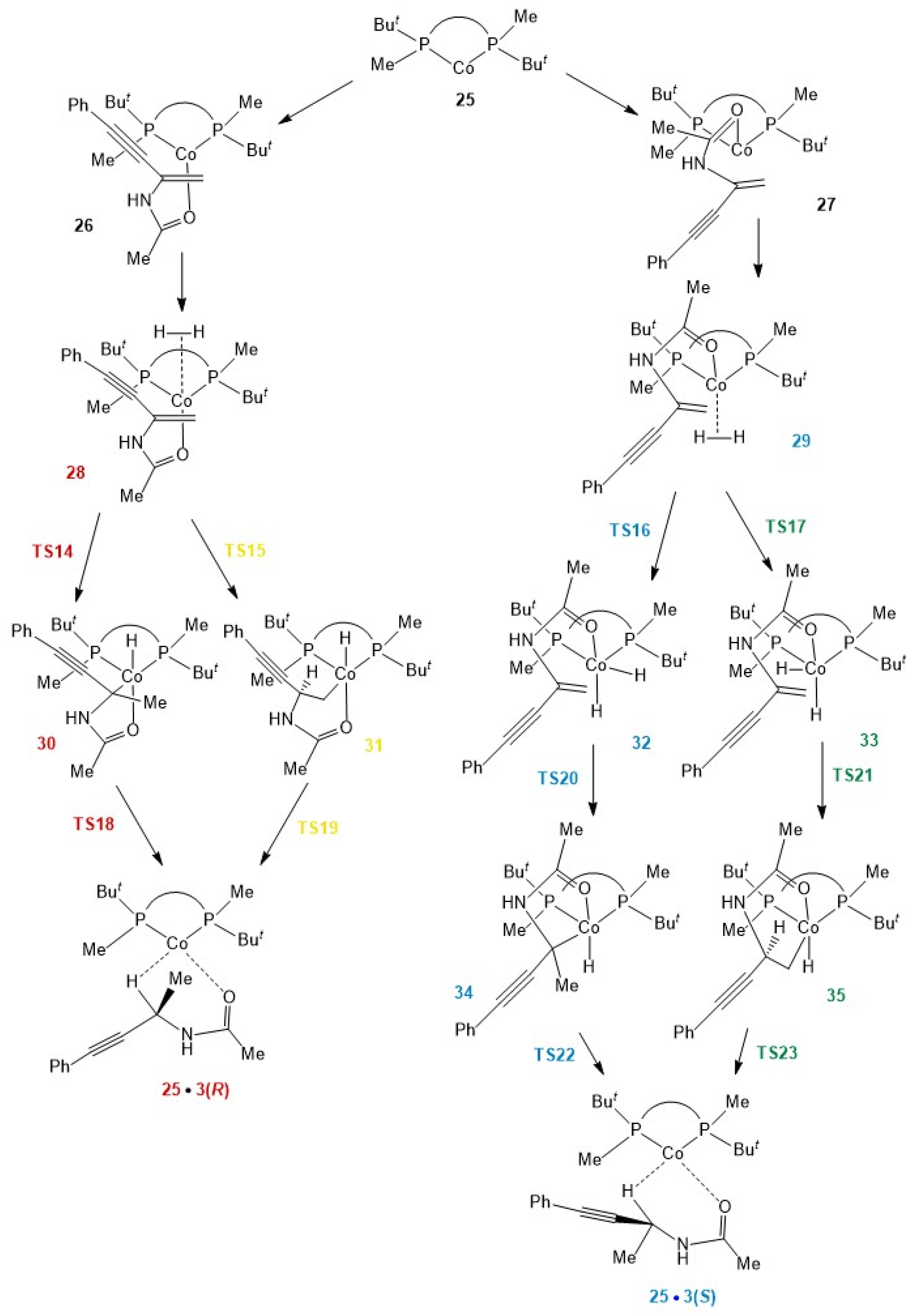
2.3. Stereoselective Formation of Chelating Dihydrides—Four Competing Catalytic Cycles
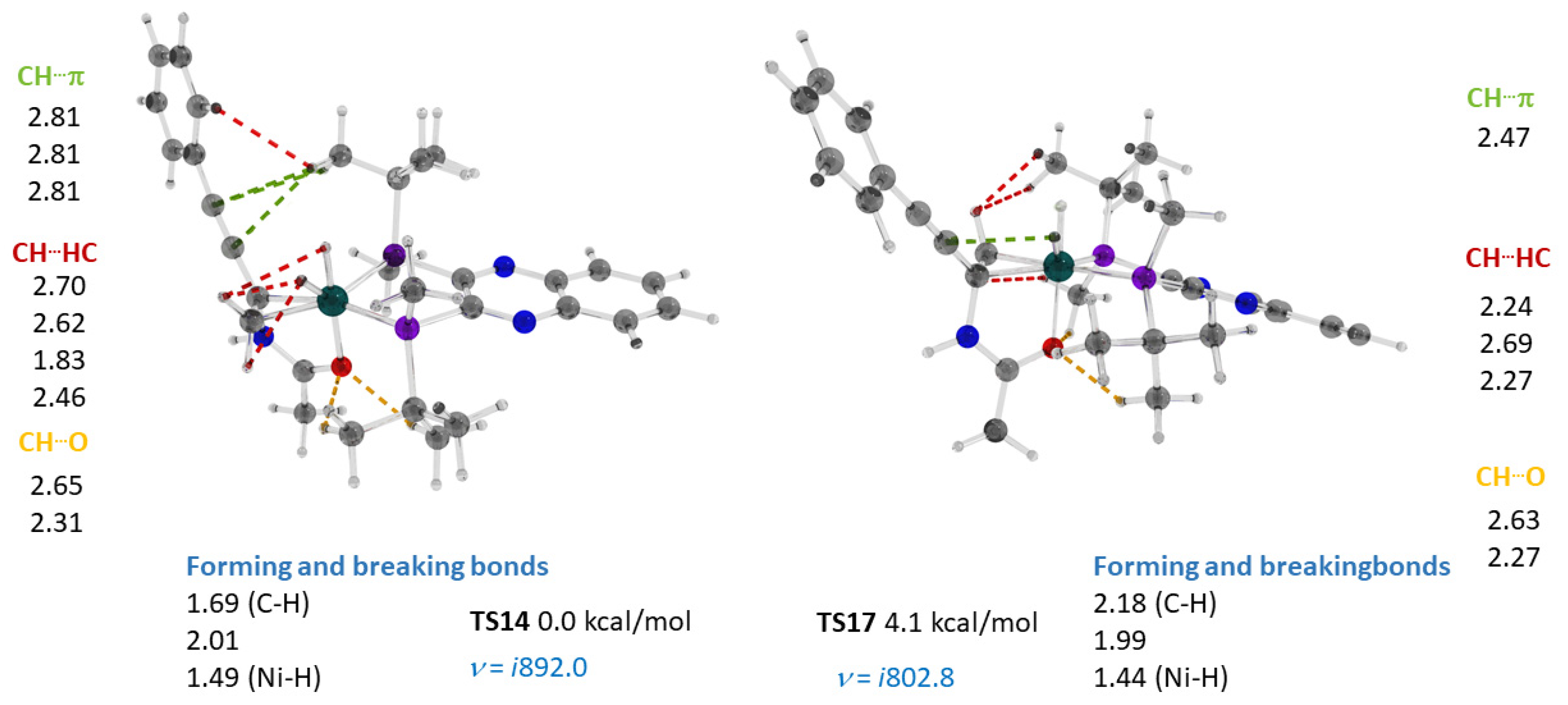
2.4. Co(0)-Co(II) Mechanism
3. Conclusions
4. Materials and Methods
Computational Details
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartwig, J. Organotransition Metal Chemistry: From Bonding to Catalysis, 1st ed.; University Science Books: Sausalito, CA, USA, 2009. [Google Scholar]
- Barton, A. Organometallic Transition Metal Catalysis: A Hostic Approach to Understanding and Predicting Their Mechanisms; Lulu: Kerala, India, 2022. [Google Scholar]
- Brown, J.M. Hydrogenation of Functionalized Carbon-Carbon Double Bonds; Jacobsen, E.N., Pfalz, A., Yamamoto, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; Volume 1, pp. 119–182. [Google Scholar]
- Halpern, J. Mechanism of Stereoselectivity of Asymmetric Hydrogenation. Science 1982, 217, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Landis, C.R.; Halpern, J. Asymmetric Hydrogenation of Methyl-(Z)-α-acetamidocinnamate Catalyzed by {1,2-Bis((phenyl-o-anisoyl)phosphino)ethane}rhodium(I): Kinetics, Mechanism, and Origin of Enantioselection. J. Am. Chem. Soc. 1987, 109, 1746–1754. [Google Scholar] [CrossRef]
- Giernoth, R.; Heinrich, H.; Adams, N.J.; Deeth, R.J.; Bargon, J.; Brown, J.M. PHIP Detection of a Transient Rhodium Dihydride Intermediate in the Homogeneous Hydrogenation of Dehydroamino Acids. J. Am. Chem. Soc. 2000, 122, 12381–12382. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Imamoto, T. On the Mechanism of Stereoselection in Rh-Catalyzed Asymmetric Hydrogenation: A General Approach for Predicting the Sense of Enantioselectivity. Acc. Chem. Res. 2004, 37, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Imamoto, T. Mechanism of Enantioselection in Rh-Catalyzed Asymmetric Hydrogenation. The Origin of Utmost Catalytic Performance. Chem. Commun. 2009, 7447–7464. [Google Scholar] [CrossRef]
- Verdolino, V.; Forbes, A.; Helquist, P.; Norrby, P.-O.; Wiest, O. On the mechanism of the rhodium catalyzed acrylamide hydrogenation. J. Mol. Catal. A Chem. 2010, 324, 9–14. [Google Scholar] [CrossRef]
- Imamoto, T.; Tamura, K.; Zhang, Z.; Horiuchi, Y.; Sugiya, M.; Yoshida, K.; Yanagisawa, A.; Gridnev, I.D. Rigid P-chiral phosphine ligands with tert-butylmethylphosphino groups for rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes. J. Am. Chem. Soc. 2012, 134, 1754–1769. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Imamoto, T. Challenging the major/minor concept in Rh-catalyzed asymmetric hydrogenation. ACS Catal. 2015, 5, 2911–2915. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Dub, P.A. Enantioselection in Asymmetric Catalysis; CRC Press: Boca Raton, FL, USA, 2016; p. 234. [Google Scholar]
- Kitamura, M.; Tsukamoto, M.; Bessho, Y.; Yoshimura, M.; Kobs, U.; Widhalm, M.; Noyori, R. Mechanism of Asymmetric Hydrogenation of α-(Acylamino)acrylic Esters Catalyzed by BINAP-Ruthenium(II) Diacetate. J. Am. Chem. Soc. 2002, 124, 6649–6667. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The role of the metal-bound N–H functionality in Noyori-type molecular catalysts. Nat. Rev. Chem. 2018, 2, 396–408. [Google Scholar] [CrossRef]
- Dub, P.A.; Ikariya, T. Quantum Chemical Calculations with the Inclusion of Nonspecific and Specific Solvation: Asymmetric Transfer Hydrogenation with Bifunctional Ruthenium Catalysts. J. Am. Chem. Soc. 2013, 135, 2604–2619. [Google Scholar] [CrossRef] [PubMed]
- Dub, P.A.; Gordon, J.C. The mechanism of enantioselective ketone reduction with Noyori and Noyori–Ikariya bifunctional catalysts. Dalton Trans 2016, 45, 6756–6781. [Google Scholar] [CrossRef]
- Roseblade, J.; Pfaltz, A. Iridium-catalyzed asymmetric hydrogenation of olefins. Acc. Chem. Res. 2007, 40, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Hopmann, K.H.; Bayer, A. On the Mechanism of Iridium-Catalyzed Asymmetric Hydrogenation of Imines and Alkenes: A Theoretical Study. Organometallics 2011, 30, 2483–2497. [Google Scholar] [CrossRef]
- Liu, Y.; Gridnev, I.D.; Zhang, W. Mechanism of Asymmetric Hydrogenation of Exocyclic alpha, beta-Unsaturated Carbonyl Compounds with an Iridium/BiphPhopx Catalyst: NMR and DFT Studies. Angew. Chem. Intl. Ed. 2014, 53, 1901–1905. [Google Scholar] [CrossRef]
- Sparta, M.; Riplinger, C.; Neese, F. Mechanism of Olefin Asymmetric Hydrogenation Catalyzed by Iridium Phosphino-Oxazoline: A Pair Natural Orbital Coupled Cluster Study. J. Chem. Theory Comput. 2014, 10, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Tutkowski, B.; Kerdphon, S.; Limé, E.; Helquist, P.; Andersson, P.G.; Wiest, O.; Norrby, P.-O. Revisiting the Stereodetermining Step in Enantioselective Iridium-Catalyzed Imine Hydrogenation. ACS Catal. 2018, 8, 615–623. [Google Scholar] [CrossRef]
- Cui, C.X.; Chen, H.H.; Li, S.J.; Zhang, T.; Qu, L.B.; Lan, Y. Mechanism of Ir-catalyzed hydrogenation: A theoretical view. Coord. Chem. Rev. 2020, 412, 213251. [Google Scholar] [CrossRef]
- Mas-Roselló, J.; Smejkal, T.; Cramer, N. Iridium-catalyzed acid-assisted asymmetric hydrogenation of oximes to hydroxylamines. Science 2020, 368, 1098–1102. [Google Scholar] [CrossRef]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem. Rev. 2019, 119, 2453–2523. [Google Scholar] [CrossRef]
- Zhang, Z.; Butt, N.A.; Zhou, M.; Liu, D.; Zhang, W. Asymmetric Transfer and Pressure Hydrogenation with Earth-Abundant Transition Metal Catalysts. Chin. J. Chem. 2018, 36, 443–454. [Google Scholar] [CrossRef]
- Seo, C.S.G.; Morris, R.H. Catalytic Homogeneous Asymmetric Hydrogenation: Successes and Opportunities. Organometallics 2019, 38, 47–65. [Google Scholar] [CrossRef]
- Li, B.; Chen, J.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-catalyzed asymmetric hydrogenation of N-sulfonyl imines. Angew. Chem. Int. Ed. 2019, 58, 7329–7334. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, X.-Q.; Zhang, X. Recent advances of nickel-catalyzed homogeneous asymmetric hydrogenation. Chin. J. Org. Chem. 2020, 40, 1096–1104. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Li, B.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-catalyzed asymmetric hydrogenation of 2-amidoacrylates. Angew. Chem. Int. Ed. 2020, 59, 5371–5375. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, B.; Chen, J.; Gridnev, I.D.; Yan, D.; Zhang, W. Ni-catalyzed asymmetric hydrogenation of N-aryl imino esters for the efficient synthesis of chiral α-aryl glycines. Nat. Commun. 2020, 11, 5935. [Google Scholar] [CrossRef]
- Li, B.; Chen, J.; Liu, D.; Gridnev, I.D.; Zhang, W. Nickel-catalysed asymmetric hydrogenation of oximes. Nat. Chem. 2022, 14, 920–927. [Google Scholar] [CrossRef]
- Monfette, S.; Turner, Z.R.; Semproni, S.P.; Chirik, P.J. Enantiopure C1-Symmetric Bis(imino)pyridine Cobalt Complexes for Asymmetric Alkene Hydrogenation. J. Am. Chem. Soc. 2012, 134, 4561–4564. [Google Scholar] [CrossRef]
- Zhang, G.; Vasudevan, K.V.; Scott, B.L.; Hanson, S.K. Understanding the Mechanisms of Cobalt-Catalyzed Hydrogenation and Dehydrogenation Reactions. J. Am. Chem. Soc. 2013, 135, 8668–8681. [Google Scholar] [CrossRef]
- Zhong, H.; Friedfeld, M.R.; Chirik, P.J. Syntheses and Catalytic Hydrogenation Performance of Cationic Bis(phosphine) Cobalt(I) Diene and Arene Compounds. Angew. Chem. Int. Ed. 2019, 58, 9194–9198. [Google Scholar] [CrossRef] [PubMed]
- Hopmann, K.H. Cobalt–Bis(imino)pyridine-Catalyzed Asymmetric Hydrogenation: Electronic Structure, Mechanism, and Stereoselectivity. Organometallics 2013, 32, 6388–6399. [Google Scholar] [CrossRef]
- Zhong, H.; Friedfeld, M.R.; Camacho-Bunquin, J.; Sohn, H.; Yang, C.; Delferro, M.; Chirik, P.J. Exploring the Alcohol Stability of Bis(phosphine) Cobalt Dialkyl Precatalysts in Asymmetric Alkene Hydrogenation. Organometallics 2019, 38, 149–156. [Google Scholar] [CrossRef]
- Friedfeld, M.R.; Zhong, H.; Ruck, R.T.; Shevlin, M.; Chirik, P.J. Cobalt-catalyzed asymmetric hydrogenation of enamides enabled by single-electron reduction. Science 2018, 360, 888–893. [Google Scholar] [CrossRef]
- Chen, J.; Chen, C.; Ji, C.; Lu, Z. Cobalt-Catalyzed Asymmetric Hydrogenation of 1,1-Diarylethenes. Org. Lett. 2016, 18, 1594–1597. [Google Scholar] [CrossRef] [PubMed]
- Friedfeld, M.R.; Shevlin, M.; Margulieux, G.W.; Campeau, L.-C.; Chirik, P.J. Cobalt-Catalyzed Enantioselective Hydrogenation of Minimally Functionalized Alkenes: Isotopic Labeling Provides Insight into the Origin of Stereoselectivity and Alkene Insertion Preferences. J. Am. Chem. Soc. 2016, 138, 3314–3324. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lei, M. Mechanistic Insights into the Directed Hydrogenation of Hydroxylated Alkene Catalyzed by Bis(phosphine)Cobalt Dialkyl Complexes. J. Org. Chem. 2017, 82, 2703–2712. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Zhang, J.; Liu, Y.; Gridnev, I.D.; Zhang, W. Cobalt-Catalyzed Asymmetric Hydrogenation of C=N Bonds Enabled by Assisted Coordination and Nonbonding Interactions. Angew. Chem. Int. Ed. 2019, 58, 15767–15771. [Google Scholar] [CrossRef]
- Zhong, H.; Shevlin, M.; Chirik, P.J. Cobalt-Catalyzed Asymmetric Hydrogenation of α,β-Unsaturated Carboxylic Acids by Homolytic H2 Cleavage. J. Am. Chem. Soc. 2020, 142, 5272–5281. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xiao, Y.; Yang, Y.; Duan, Y.-N.; Li, F.; Hu, Q.; Chung, L.W.; Chen, G.-Q.; Zhang, X. Enantioselective Hydrogenation of Tetrasubstituted α,β-Unsaturated Carboxylic Acids Enabled by Cobalt(II) Catalysis: Scope and Mechanistic Insights. Angew. Chem. Int. Ed. 2021, 60, 11384–11390. [Google Scholar] [CrossRef]
- Du, X.; Xiao, Y.; Huang, J.-M.; Zhang, Y.; Duan, Y.-N.; Wang, H.; Shi, C.; Chen, G.-Q.; Zhang, X. Cobalt-catalyzed highly enantioselective hydrogenation of α,β-unsaturated carboxylic acids. Nat. Commun. 2020, 11, 3239. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, Z.; Liu, Y.; Zhang, W. Cobalt-Catalyzed Chemo- and Enantioselective Hydrogenation of Conjugated Enynes. Angew. Chem. Int. Ed. 2021, 60, 16989–16993. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, L.N.; Pavlovic, L.; Zhong, H.; Friedfeld, M.R.; Shevlin, M.; Hopmann, K.H.; Chirik, P.J. Mechanistic Investigations of the Asymmetric Hydrogenation of Enamides with Neutral Bis(phosphine) Cobalt Precatalysts. J. Am. Chem. Soc. 2022, 144, 15764–15778. [Google Scholar] [CrossRef]
- Pavlovich, L.; Mendelsohn, L.N.; Zhong, H.Y.; Chirik, P.J.; Hopmann, K.H. Cobalt-Catalyzed Asymmetric Hydrogenation of Enamides: Insights into Mechanisms and Solvent Effects. Organometallics 2022, 41, 1872–1882. [Google Scholar] [CrossRef]
- Zhou, S.; Fleischer, S.; Junge, K.; Das, S.; Addis, D.; Beller, M. Enantioselective Synthesis of Amines: General, Efficient Iron-Catalyzed Asymmetric Transfer Hydrogenation of Imines. Angew. Chem. Int. Ed. 2010, 49, 8121–8125. [Google Scholar] [CrossRef] [PubMed]
- Lagaditis, P.O.; Sues, P.E.; Sonnenberg, J.F.; Wan, K.Y.; Lough, A.J.; Morris, R.H. Iron(II) Complexes Containing Unsymmetrical P–N–P′ Pincer Ligands for the Catalytic Asymmetric Hydrogenation of Ketones and Imines. J. Am. Chem. Soc. 2014, 136, 1367–1380. [Google Scholar] [CrossRef]
- Smith, S.A.M.; Lagaditis, P.O.; Lüpke, A.; Lough, A.J.; Morris, R.H. Unsymmetrical Iron P-NH-P′ Catalysts for the Asymmetric Pressure Hydrogenation of Aryl Ketones. Chem.-Eur. J. 2017, 23, 7212–7216. [Google Scholar] [CrossRef]
- Sonnenberg, J.F.; Wan, K.Y.; Sues, P.E.; Morris, R.H. Ketone Asymmetric Hydrogenation Catalyzed by P-NH-P′ Pincer Iron Catalysts: An Experimental and Computational Study. ACS Catal. 2017, 7, 316–326. [Google Scholar] [CrossRef]
- Seo, C.S.G.; Tannoux, T.; Smith, S.A.M.; Lough, A.J.; Morris, R.H. Enantioselective hydrogenation of activated aryl imines catalyzed by an iron(II) P-NH-P′ complex. J. Org. Chem. 2019, 84, 12040–12049. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, Y.; Han, Z.; Ding, K. Lutidine-based chiral pincer manganese catalysts for enantioselective hydrogenation of ketones. Angew. Chem. Int. Ed. 2019, 58, 4973–4977. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Han, Z.; Ding, K. Manganese-catalyzed anti-selective asymmetric hydrogenation of α-substituted β-ketoamides. Angew. Chem. Int. Ed. 2020, 59, 15565–15569. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric Hydrogenation. Acc. Chem. Res. 1983, 16, 106–112. [Google Scholar] [CrossRef]
- Gridnev, I.D. Attraction versus Repulsion in Rhodium-Catalyzed Asymmetric Hydrogenation. ChemCatChem 2016, 8, 3463–3468. [Google Scholar] [CrossRef]
- Burk, M.J.; Casy, G.; Johnson, N.B. A Three-Step Procedure for Asymmetric Catalytic Reductive Amidation of Ketones. J. Org. Chem. 1998, 63, 6084–6085. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Higashi, N.; Imamoto, T. On the Origin of opposite Stereoselection in the Asymmetric Hydrogenation of Phenyl- and tert-Butyl-Substituted Enamides. J. Am. Chem. Soc. 2000, 122, 10486–10487. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Yasutake, M.; Higashi, N.; Imamoto, T. Asymmetric Hydrogenation of Enamides with Rh-BisP* and Rh-MiniPHOS Catalysts. Scope, Limitations, and Mechanism. J. Am. Chem. Soc. 2001, 123, 5268–5276. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, rev. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Moleculesl. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar]
- Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; Schleyer, P.V.R.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W. Highly correlated systems. Excitation energies of first row transition metals Sc–Cu. J. Chem. Phys. 1989, 91, 1062–1065. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gridnev, I.D. Co-Catalyzed Asymmetric Hydrogenation. The Same Enantioselection Pattern for Different Mechanisms. Int. J. Mol. Sci. 2023, 24, 5568. https://doi.org/10.3390/ijms24065568
Gridnev ID. Co-Catalyzed Asymmetric Hydrogenation. The Same Enantioselection Pattern for Different Mechanisms. International Journal of Molecular Sciences. 2023; 24(6):5568. https://doi.org/10.3390/ijms24065568
Chicago/Turabian StyleGridnev, Ilya D. 2023. "Co-Catalyzed Asymmetric Hydrogenation. The Same Enantioselection Pattern for Different Mechanisms" International Journal of Molecular Sciences 24, no. 6: 5568. https://doi.org/10.3390/ijms24065568
APA StyleGridnev, I. D. (2023). Co-Catalyzed Asymmetric Hydrogenation. The Same Enantioselection Pattern for Different Mechanisms. International Journal of Molecular Sciences, 24(6), 5568. https://doi.org/10.3390/ijms24065568