
Differential Effects of Iron Chelates vs. Iron Salts on Induction of Pro-Oncogenic Amphiregulin and Pro-Inflammatory COX-2 in Human Intestinal Adenocarcinoma Cell Lines

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
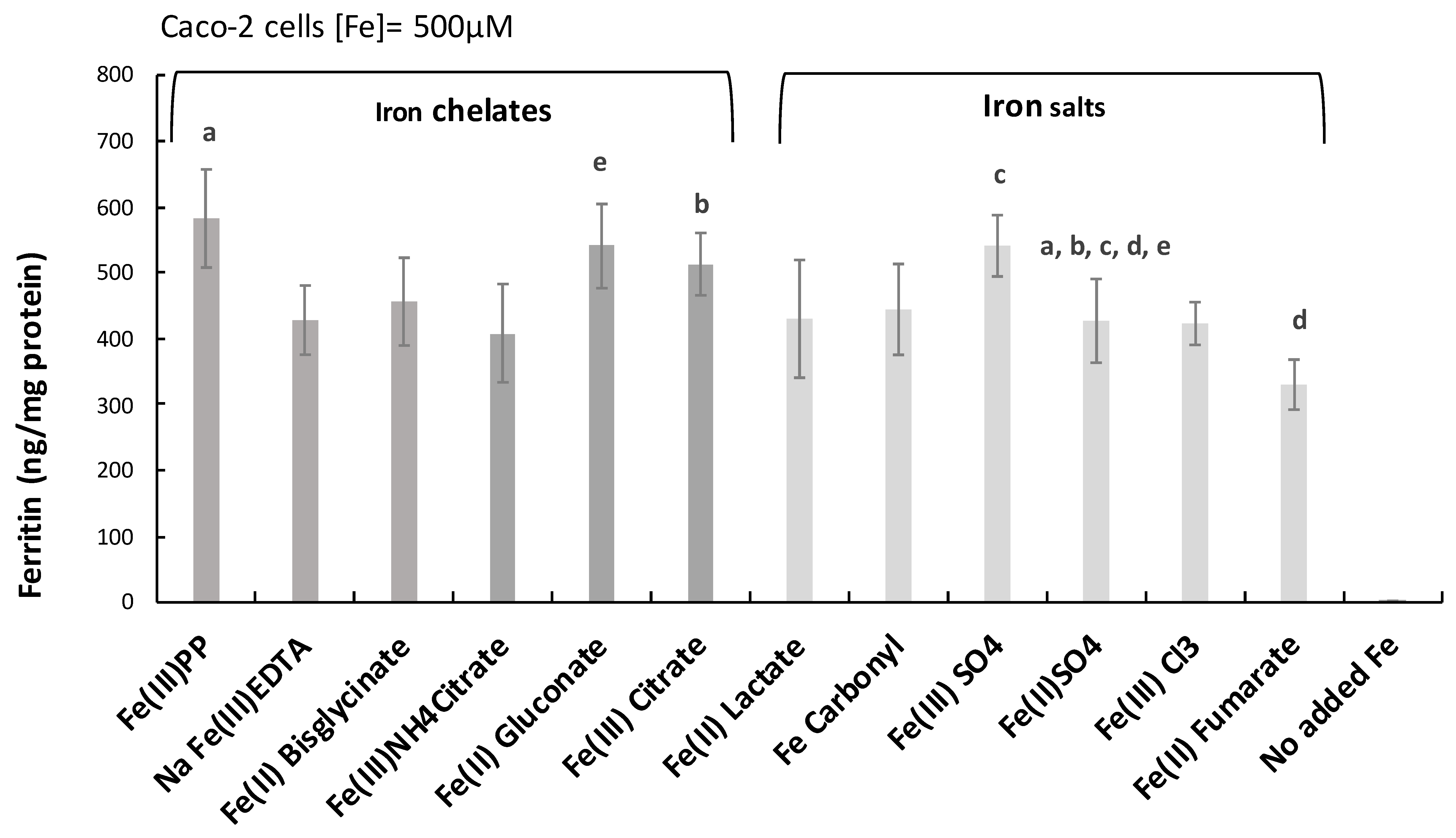
2.1. Ferric Pyrophosphate—A Major Novel Inducer of Amphiregulin
2.2. Elevation of an Amphiregulin Receptor Monomer, IGFr1
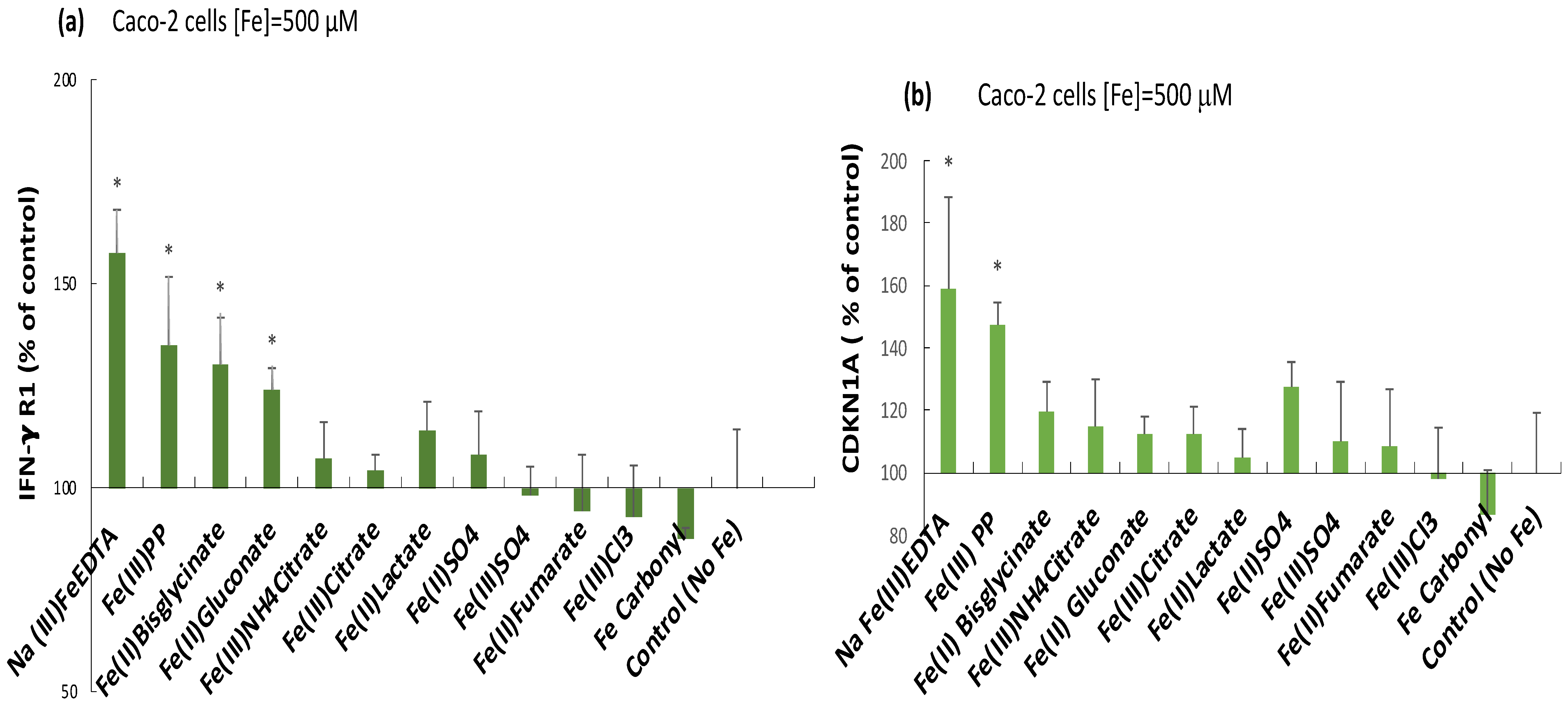
2.3. Induction of the Inflammatory Mediators IFN-γ r1, CDKNA, and IL-6 by Ferric Pyrophosphate: Engagement of the JAK/STAT Pathway
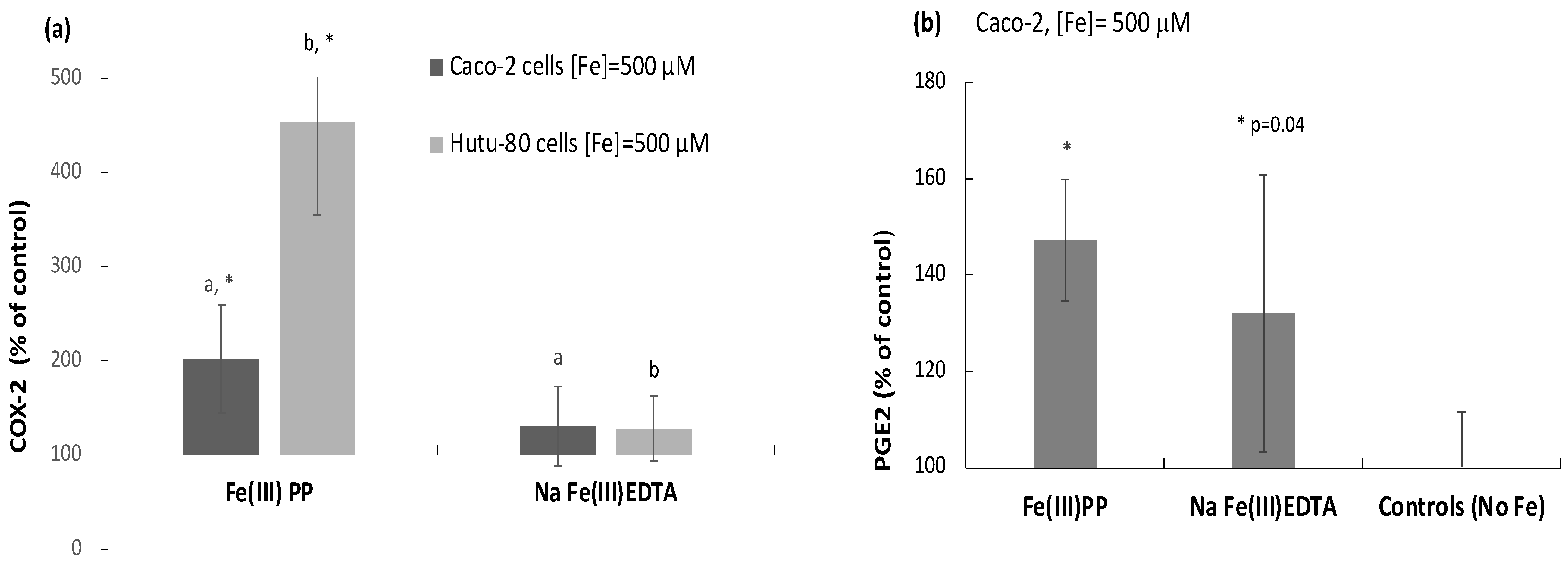
2.4. Ferric Pyrophosphate, but Not Ferric EDTA, Significantly Elevated Intracellular COX-2
2.5. COX-2 Appears to Be Downstream of IL-6 as Indicated by COX-2 Inhibition Studies
3. Discussion
3.1. Rationale for Investigating Effects of Iron Compounds in Human Adenocarcinoma Cells
3.2. The Effect of Iron Chelates and Iron Salts on Growth/Inflammatory Pathways
3.3. Iron Exposure and Load
4. Materials and Methods
4.1. Iron Compounds
4.2. Microwave Digestion and Atomic Absorption Spectroscopy (AAS)
4.3. Cell Culture
4.4. Cell Experiments
4.5. COX-2 Inhibition Studies
4.6. Biomarkers for Inflammation
4.7. Proteomic Analysis of Biomarkers for Inflammation and Cancer
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fonseca-Nunes, A.; Jakszyn, P.; Agudo, A. Iron and Cancer Risk—A Systematic Review and Meta-analysis of the Epidemiological Evidence. Cancer Epidemiol. Biomark. Prev. 2014, 23, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.L. Iron and Colorectal Cancer Risk: Human Studies. Nutr. Rev. 2001, 59, 140–148. [Google Scholar] [CrossRef]
- Pourcelot, E.; Lénon, M.; Mobilia, N.; Cahn, J.-Y.; Arnaud, J.; Fanchon, E.; Moulis, J.-M.; Mossuz, P. Iron for proliferation of cell lines and hematopoietic progenitors: Nailing down the intracellular functional iron concentration. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1596–1605. [Google Scholar] [CrossRef]
- Le, N.T.; Richardson, D.R. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim. Biophys Acta (BBA)-Mol. Cell Res. 2002, 1603, 31–46. [Google Scholar] [CrossRef]
- Scheers, N.M.; Pereira, D.I.; Faria, N.; Powell, J.J. Ferric citrate and ferric EDTA but not ferrous sulfate drive amphiregulin-mediated activation of the MAP kinase ERK in gut epithelial cancer cells. Oncotarget 2018, 9, 17066–17077. [Google Scholar] [CrossRef]
- Seril, D.N.; Liao, J.; Ho, K.-L.K.; Warsi, A.; Yang, C.S.; Yang, G.-Y. Dietary Iron Supplementation Enhances DSS-Induced Colitis and Associated Colorectal Carcinoma Development in Mice. Dig. Dis. Sci. 2002, 47, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Radulescu, S.; Brookes, M.J.; Salgueiro, P.; Ridgway, R.A.; McGhee, E.; Anderson, K.; Ford, S.J.; Stones, D.H.; Iqbal, T.H.; Tselepis, C.; et al. Luminal Iron Levels Govern Intestinal Tumorigenesis after Apc Loss In Vivo. Cell Rep. 2016, 17, 2805–2807. [Google Scholar] [CrossRef]
- Seril, D.N.; Liao, J.; Yang, C.S.; Yang, G.-Y. Systemic Iron Supplementation Replenishes Iron Stores Without Enhancing Colon Carcinogenesis in Murine Models of Ulcerative Colitis: Comparison with Iron-Enriched Diet. Dig. Dis. Sci. 2005, 50, 696–707. [Google Scholar] [CrossRef]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.-L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2011, 1816, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Huang, M.-S.; Cheng, D.-E.; Hung, J.-Y.; Yang, C.-J.; Chou, S.-H.; Kuo, P.-L. Lung Tumor-Associated Dendritic Cell-Derived Amphiregulin Increased Cancer Progression. J. Immunol. 2011, 187, 1733–1744. [Google Scholar] [CrossRef]
- McBryan, J.; Howlin, J.; Napoletano, S.; Martin, F. Amphiregulin: Role in Mammary Gland Development and Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2008, 13, 159–169. [Google Scholar] [CrossRef]
- D’Antonio, A.; Losito, S.; Pignata, S.; Grassi, M.; Perrone, F.; De Luca, A.; Tambaro, R.; Bianco, C.; Gullick, W.; Johnson, G.; et al. Transforming growth factor α, amphiregulin and cripto-1 are frequently expressed in advanced human ovarian carcinomas. Int. J. Oncol. 2002, 21, 941–948. [Google Scholar] [CrossRef]
- Chayangsu, C.; Khunsri, S.; Sriuranpong, V.; Tanasanvimon, S. The correlations between serum amphiregulin and other clinicopathological factors in colorectal cancer. J. Gastrointest. Oncol. 2017, 8, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Park, H.; Kim, K.J.; Kim, J.W.; Sung, J.H.; Nam, M.; Lee, J.H.; Jung, E.H.; Suh, K.J.; Lee, J.Y.; et al. Amphiregulin can predict treatment resistance to palliative first-line cetuximab plus FOLFIRI chemotherapy in patients with RAS wild-type metastatic colorectal cancer. Sci. Rep. 2021, 11, 23803. [Google Scholar] [CrossRef]
- Chen, J.-C.; Huang, C.; Lee, I.-N.; Wu, Y.-P.; Tang, C.-H. Amphiregulin enhances cell migration and resistance to doxorubicin in chondrosarcoma cells through the MAPK pathway. Mol. Carcinog. 2018, 57, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Kim, T.R.; Kim, S.-H.; Kim, I.-H.; Lim, J.-O.; Park, J.H.; Yun, S.; Lee, I.-C.; Park, H.-O.; Kim, J.-C. Four-Week Repeated Intravenous Dose Toxicity of Self-Assembled-Micelle Inhibitory RNA-Targeting Amphiregulin in Mice. Int. J. Toxicol. 2021, 40, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Putoczki, T. JAK-STAT Signalling Pathway in Cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Lee, S.; Guo, H.; Evers, B.M.; Sheng, H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003, 63, 5218–5223. [Google Scholar] [PubMed]
- Shao, J.; Evers, B.M.; Sheng, H. Prostaglandin E2 Synergistically Enhances Receptor Tyrosine Kinase-dependent Signaling System in Colon Cancer Cells. J. Biol. Chem. 2004, 279, 14287–14293. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, I.; Brinckerhoff, E.C.; Sinclair, J.F.; Sinclair, P.R.; Bonkowsky, H.L.; Harris, E.D. Iron increases collagenase production by rabbit synovial fibroblasts. J. Lab. Clin. Med. 1981, 97, 396–402. [Google Scholar]
- Glover, J.; Jacobs, A. Observations on iron in the jejunal lumen after a standard meal. Gut 1971, 12, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Flørenes, V.A.; Lu, C.; Bhattacharya, N.; Rak, J.; Sheehan, C.; Slingerland, J.M.; Kerbel, R.S. Interleukin-6 dependent induction of the cyclin dependent kinase inhibitor p21WAF1/CIP1 is lost during progression of human malignant melanoma. Oncogene 1999, 18, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Mergler, B.I.; Roth, E.; Bruggraber, S.F.; Powell, J.J.; Pereira, D.I. Development of the Caco-2 Model for Assessment of Iron Absorption and Utilisation at Supplemental Levels. J. Pharm. Nutr. Sci. 2012, 2, 27–34. [Google Scholar] [CrossRef]
- Lund, E.K.; Wharf, S.G.; Fairweather-Tait, S.J.; Johnson, I.T. Oral ferrous sulfate supplements increase the free radical–generating capacity of feces from healthy volunteers. Am. J. Clin. Nutr. 1999, 69, 250–255. [Google Scholar] [CrossRef]
- Block, G.A.; Fishbane, S.; Rodriguez, M.; Smits, G.; Shemesh, S.; Pergola, P.E.; Wolf, M.; Chertow, G.M. A 12-Week, Double-Blind, Placebo-Controlled Trial of Ferric Citrate for the Treatment of Iron Deficiency Anemia and Reduction of Serum Phosphate in Patients with CKD Stages 3-5. Am. J. Kidney Dis. 2015, 65, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Glahn, R.P.; Lee, O.A.; Yeung, A.; Goldman, M.I.; Miller, D.D. Caco-2 Cell Ferritin Formation Predicts Nonradiolabeled Food Iron Availability in an In Vitro Digestion/Caco-2 Cell Culture Model. J. Nutr. 1998, 128, 1555–1561. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Dickens, E.R.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef]
- Antunes, N.; Kundu, B.; Kundu, S.C.; Reis, R.L.; Correlo, V. In Vitro Cancer Models: A Closer Look at Limitations on Translation. Bioengineering 2022, 9, 166. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarczykowska, A.; Engström, N.; Dobermann, D.; Powell, J.; Scheers, N. Differential Effects of Iron Chelates vs. Iron Salts on Induction of Pro-Oncogenic Amphiregulin and Pro-Inflammatory COX-2 in Human Intestinal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2023, 24, 5507. https://doi.org/10.3390/ijms24065507
Tarczykowska A, Engström N, Dobermann D, Powell J, Scheers N. Differential Effects of Iron Chelates vs. Iron Salts on Induction of Pro-Oncogenic Amphiregulin and Pro-Inflammatory COX-2 in Human Intestinal Adenocarcinoma Cell Lines. International Journal of Molecular Sciences. 2023; 24(6):5507. https://doi.org/10.3390/ijms24065507
Chicago/Turabian StyleTarczykowska, Agata, Niklas Engström, Darja Dobermann, Jonathan Powell, and Nathalie Scheers. 2023. "Differential Effects of Iron Chelates vs. Iron Salts on Induction of Pro-Oncogenic Amphiregulin and Pro-Inflammatory COX-2 in Human Intestinal Adenocarcinoma Cell Lines" International Journal of Molecular Sciences 24, no. 6: 5507. https://doi.org/10.3390/ijms24065507
APA StyleTarczykowska, A., Engström, N., Dobermann, D., Powell, J., & Scheers, N. (2023). Differential Effects of Iron Chelates vs. Iron Salts on Induction of Pro-Oncogenic Amphiregulin and Pro-Inflammatory COX-2 in Human Intestinal Adenocarcinoma Cell Lines. International Journal of Molecular Sciences, 24(6), 5507. https://doi.org/10.3390/ijms24065507