

Pro-Inflammatory Priming of the Brain: The Underlying Cause of Parkinson’s Disease

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
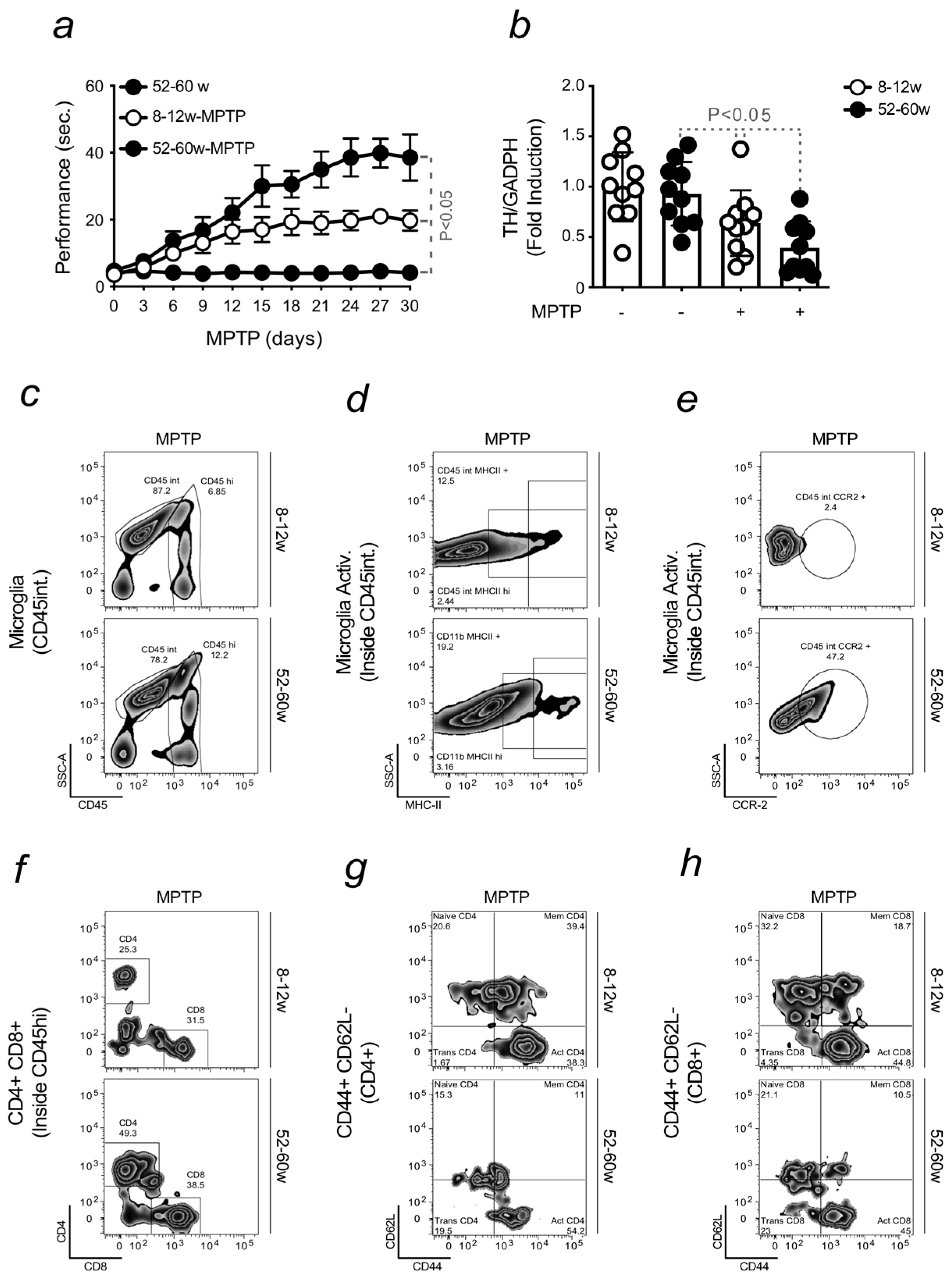
2.1. Aging Increases the Severity of Parkinson’s Disease
2.2. Aging-Associated Inflammation Promotes the Formation of Fe-Loaded Inflammatory Cells
2.3. Brain Fe Accumulation and Inflammation Sensitize to an Enhanced PD Severity
2.4. Infection-Driven Pro-Inflammatory Priming to the Brain Promotes Neuroinflammation
2.5. Pro-Inflammatory Priming to the Brain Enhances PD Severity
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schapira, A.H. Etiology of Parkinson’s Disease. Neurology 2006, 66, S10. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A. Levodopa for the Treatment of Parkinson’s Disease. N. Engl. J. Med. 2008, 359, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Beckers, M.; Bloem, B.R.; Verbeek, M.M. Mechanisms of Peripheral Levodopa Resistance in Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An Essay on the Shaking Palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G. The History of Parkinson’s Disease: Early Clinical Descriptions and Neurological Therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Parkinson Disease; World Health Organization (WHO): Geneva, Switzerland, 2022. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Wojtala, J.; Heber, I.A.; Neuser, P.; Heller, J.; Kalbe, E.; Rehberg, S.P.; Storch, A.; Linse, K.; Schneider, C.; Gräber, S.; et al. Cognitive Decline in Parkinson’s Disease: The Impact of the Motor Phenotype on Cognition. J. Neurol. Neurosurg. Psychiatry 2019, 90, 171. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Gastrointestinal Dysfunction in Parkinson’s Disease. Lancet Neurol. 2003, 2, 107–116. [Google Scholar] [CrossRef]
- Fasano, A.; Visanji, N.P.; Liu, L.W.C.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal Dysfunction in Parkinson’s Disease. Lancet Neurol. 2015, 14, 625–639. [Google Scholar] [CrossRef]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut Microbiota in Parkinson’s Disease: Temporal Stability and Relations to Disease Progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Antonietta Maselli, M.; Severi, C. The Gut-Brain Axis: Interactions between Enteric Microbiota, Central and Enteric Nervous Systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and Subsequent Risk of Parkinson’s Disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease: Molecules Implicated in the Formation and Degradation of α-Synuclein Aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease and Related Neurodegenerative Disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-Y.; Zhang, S.-P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-Analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice Deficient in TNF Receptors Are Protected against Dopaminergic Neurotoxicity: Implications for Parkinson’s Disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef]
- Tanaka, S.; Ishii, A.; Ohtaki, H.; Shioda, S.; Yoshida, T.; Numazawa, S. Activation of Microglia Induces Symptoms of Parkinson’s Disease in Wild-Type, but Not in IL-1 Knockout Mice. J. Neuroinflamm. 2013, 10, 907. [Google Scholar] [CrossRef]
- Stojakovic, A.; Paz-Filho, G.; Arcos-Burgos, M.; Licinio, J.; Wong, M.-L.; Mastronardi, C.A. Role of the IL-1 Pathway in Dopaminergic Neurodegeneration and Decreased Voluntary Movement. Mol. Neurobiol. 2017, 54, 4486–4495. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral Cytokines Profile in Parkinson’s Disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Invest. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Dobson, B.R.; Jones, G.R.; Clarke, D.T. Iron in the Basal Ganglia in Parkinson’s Disease: An in Vitro Study Using Extended X-Ray Absorption Fine Structure and Cryo-Electron Microscopy. Brain 1999, 122, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Iron, Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 7267. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Labreuche, J.; Rascol, O.; Corvol, J.-C.; Duhamel, A.; Guyon Delannoy, P.; Poewe, W.; Compta, Y.; Pavese, N.; Růžička, E.; et al. Trial of Deferiprone in Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and Cell Death Mechanisms in Parkinson’s Disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha Synuclein Aggregation Drives Ferroptosis: An Interplay of Iron, Calcium and Lipid Peroxidation. Cell. Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.-C. Ferroptosis and Its Potential Role in the Physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Lima, I.S.; Pêgo, A.C.; Martins, A.C.; Prada, A.R.; Barros, T.; Martins, G.; Gozzelino, R. Gut Dysbiosis: A Target for Protective Interventions against Parkinson’s Disease. Microorganisms 2023, 11, 880. [Google Scholar] [CrossRef]
- Martins, A.C.; Almeida, J.I.; Lima, I.S.; Kapitão, A.S.; Gozzelino, R. Iron Metabolism and the Inflammatory Response. IUBMB Life 2017, 69, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.S.; Pêgo, A.C.; Barros, J.T.; Prada, A.R.; Gozzelino, R. Cell Death-Osis of Dopaminergic Neurons and the Role of Iron in Parkinson’s Disease. Antioxid. Redox Signal. 2020, 35, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Okin, D.; Medzhitov, R. Evolution of Inflammatory Diseases. Curr. Biol. 2012, 22, R733–R740. [Google Scholar] [CrossRef] [PubMed]
- Kotas, M.E.; Medzhitov, R. Homeostasis, Inflammation, and Disease Susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Iron in Innate Immunity: Starve the Invaders. Curr. Opin. Immunol. 2009, 21, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.C.; Lee, P.; Beutler, E. Brain Iron Metabolism and Neurodegenerative Disorders. Dev. Neurosci. 2002, 24, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Finazzi, D. Neurodegeneration with Brain Iron Accumulation: Update on Pathogenic Mechanisms. Front. Pharm. 2014, 5, 99. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef]
- Arosio, P.; Gozzelino, R.; Poli, M. Iron as Therapeutic Target in Human Diseases. Pharmaceuticals 2019, 12, 178. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Hindle, J.V. Ageing, Neurodegeneration and Parkinson’s Disease. Age Ageing 2010, 39, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or Pharmacological Iron Chelation Prevents MPTP-Induced Neurotoxicity In Vivo: A Novel Therapy for Parkinson’s Disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.P.; Dunnett, S.B. Tests to Assess Motor Phenotype in Mice: A User’s Guide. Nat. Rev. Neurosci. 2009, 10, 519–529. [Google Scholar] [CrossRef]
- Saporito, M.S.; Thomas, B.A.; Scott, R.W. MPTP Activates C-Jun NH2-Terminal Kinase (JNK) and Its Upstream Regulatory Kinase MKK4 in Nigrostriatal Neurons In Vivo. J. Neurochem. 2000, 75, 1200–1208. [Google Scholar] [CrossRef]
- Kenkhuis, B.; van Eekeren, M.; Parfitt, D.A.; Ariyurek, Y.; Banerjee, P.; Priller, J.; van der Weerd, L.; van Roon-Mom, W.M.C. Iron Accumulation Induces Oxidative Stress, While Depressing Inflammatory Polarization in Human IPSC-Derived Microglia. Stem Cell Rep. 2022, 17, 1351–1365. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s Disease: Why Is Advancing Age the Biggest Risk Factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s Disease: Different Sides of the Same Coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s Disease: Animal Models and Dopaminergic Cell Vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Singapore, 2018; ISBN 9780994438164. [Google Scholar]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Djaldetti, R.; Liberatore, G.; Vila, M.; Vukosavic, S.; Almer, G. The Parkinsonian Toxin MPTP: Action and Mechanism. Restor. Neurol. Neurosci. 2000, 16, 135–142. [Google Scholar] [PubMed]
- Sian, J.; Youdim, M.B.H.; Riederer, P.; Gerlach, M. MPTP-Induced Parkinsonian Syndrome. 1999. Available online: http://www.ncbi.nlm.nih.gov/books/NBK27974/ (accessed on 15 June 2014).
- Rathnasamy, G.; Ling, E.-A.; Kaur, C. Consequences of Iron Accumulation in Microglia and Its Implications in Neuropathological Conditions. CNS Neurol. Disord. Drug. Targets 2013, 12, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Nnah, I.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 129. [Google Scholar] [CrossRef]
- Angelova, D.M.; Brown, D.R. Microglia and the Aging Brain: Are Senescent Microglia the Key to Neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.-S. Microglia in Dementia with Lewy Bodies. Brain Behav. Immun. 2016, 55, 191–201. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Fenton, H.J.H. LXXIII.—Oxidation of Tartaric Acid in Presence of Iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Ke, Y.; Qian, Z.M. Iron Misregulation in the Brain: A Primary Cause of Neurodegenerative Disorders. Lancet Neurol. 2003, 2, 246–253. [Google Scholar] [CrossRef]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease Tolerance as a Defense Strategy. Science (1979) 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Andrade, B.B.; Larsen, R.; Luz, N.F.; Vanoaica, L.; Seixas, E.; Coutinho, A.; Cardoso, S.; Rebelo, S.; Poli, M.; et al. Metabolic Adaptation to Tissue Iron Overload Confers Tolerance to Malaria. Cell Host Microbe 2012, 12, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic Infections and Inflammation Affect Chronic Neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Püntener, U.; Booth, S.G.; Perry, V.H.; Teeling, J.L. Long-Term Impact of Systemic Bacterial Infection on the Cerebral Vasculature and Microglia. J. Neuroinflamm. 2012, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The Blood–Brain Barrier in Systemic Infection and Inflammation. Cell Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, Â.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme Oxygenase-1 and Carbon Monoxide Suppress the Pathogenesis of Experimental Cerebral Malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef]
- Weinert, M.; Selvakumar, T.; Tierney, T.S.; Alavian, K.N. Isolation, Culture and Long-Term Maintenance of Primary Mesencephalic Dopaminergic Neurons From Embryonic Rodent Brains. J. Vis. Exp. 2015, 19, 52475. [Google Scholar] [CrossRef]
- Seixas, E.; Gozzelino, R.; Chora, Â.; Ferreira, A.; Silva, G.; Larsen, R.; Rebelo, S.; Penido, C.; Smith, N.R.; Coutinho, A.; et al. Heme Oxygenase-1 Affords Protection against Noncerebral Forms of Severe Malaria. Proc. Natl. Acad. Sci. USA 2009, 106, 15837–15842. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, Â.; Ferreira, A.; et al. A Central Role for Free Heme in the Pathogenesis of Severe Sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, A.C.; Lima, I.S.; Pêgo, A.C.; Sá Pereira, I.; Martins, G.; Kapitão, A.; Gozzelino, R. Pro-Inflammatory Priming of the Brain: The Underlying Cause of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 7949. https://doi.org/10.3390/ijms24097949
Martins AC, Lima IS, Pêgo AC, Sá Pereira I, Martins G, Kapitão A, Gozzelino R. Pro-Inflammatory Priming of the Brain: The Underlying Cause of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(9):7949. https://doi.org/10.3390/ijms24097949
Chicago/Turabian StyleMartins, Ana Catarina, Illyane Sofia Lima, Ana Catarina Pêgo, Inês Sá Pereira, Gracelino Martins, Antonino Kapitão, and Raffaella Gozzelino. 2023. "Pro-Inflammatory Priming of the Brain: The Underlying Cause of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 9: 7949. https://doi.org/10.3390/ijms24097949
APA StyleMartins, A. C., Lima, I. S., Pêgo, A. C., Sá Pereira, I., Martins, G., Kapitão, A., & Gozzelino, R. (2023). Pro-Inflammatory Priming of the Brain: The Underlying Cause of Parkinson’s Disease. International Journal of Molecular Sciences, 24(9), 7949. https://doi.org/10.3390/ijms24097949