
Development of Highly Sensitive Digital Droplet PCR for Detection of cKIT Mutations in Circulating Free DNA That Mediate Resistance to TKI Treatment for Gastrointestinal Stromal Tumor (GIST)

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Partition Number, Mean Copies per Partition, Individual Partition Volume, and Total Partition Volume
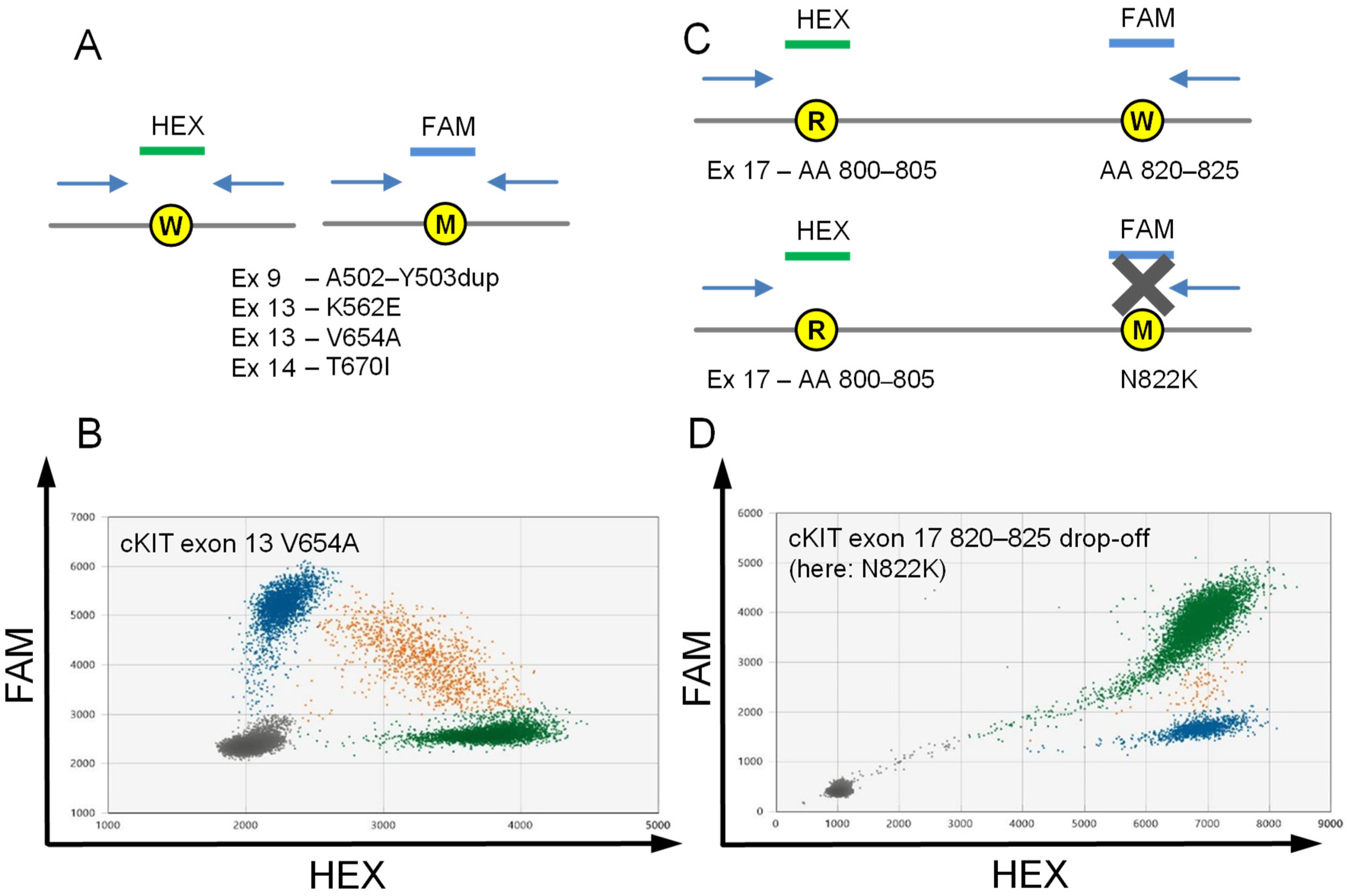
2.2. Assays to Detect Stereotypic cKIT Imatinib Resistance Mutations Exon 13 V654A and Exon 14 T670I
2.3. Drop-off Based Detection of cKIT Exon 17 Mutations
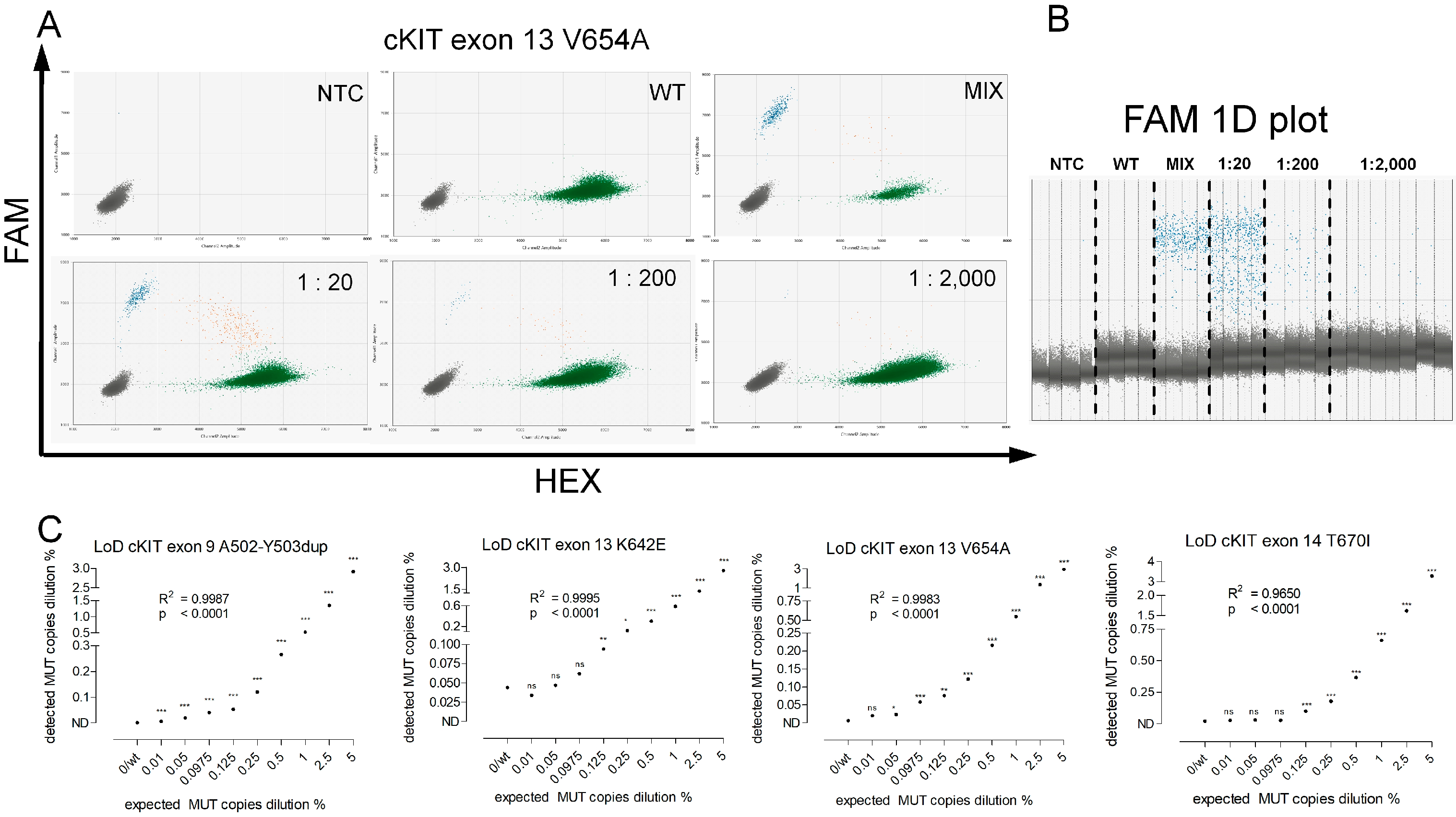
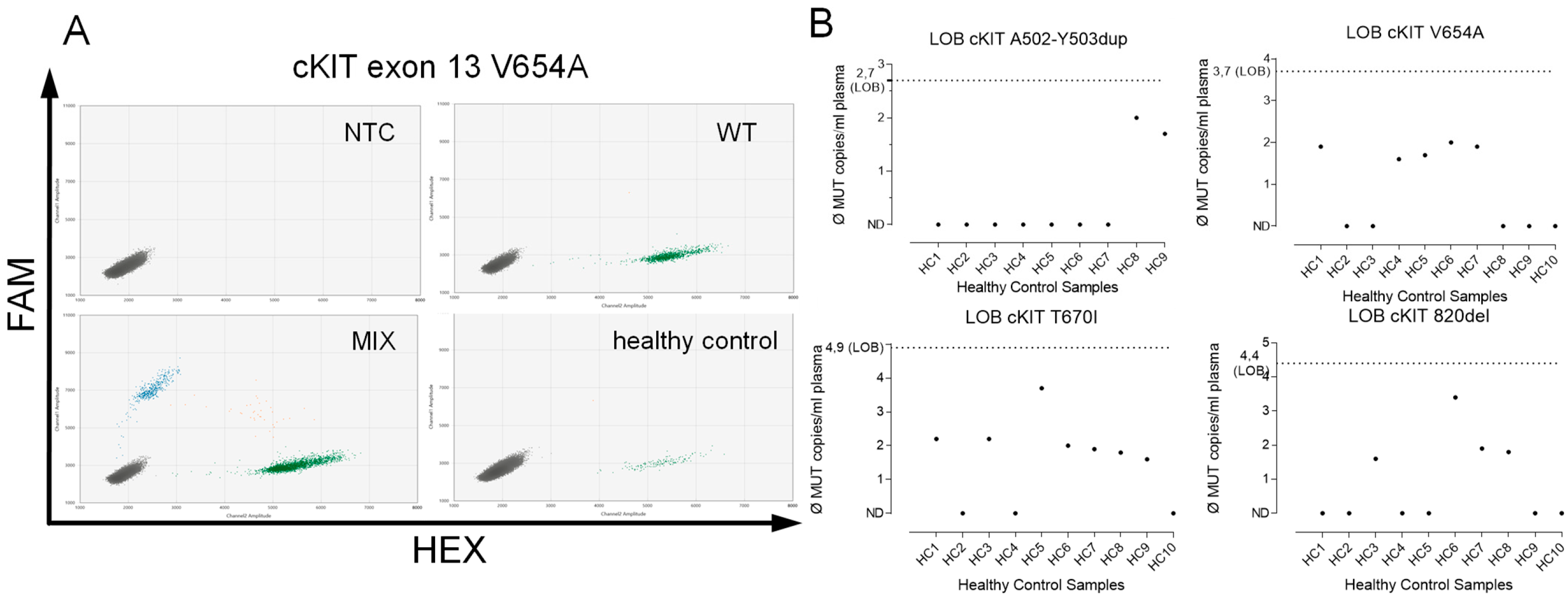
2.4. Assay Validation
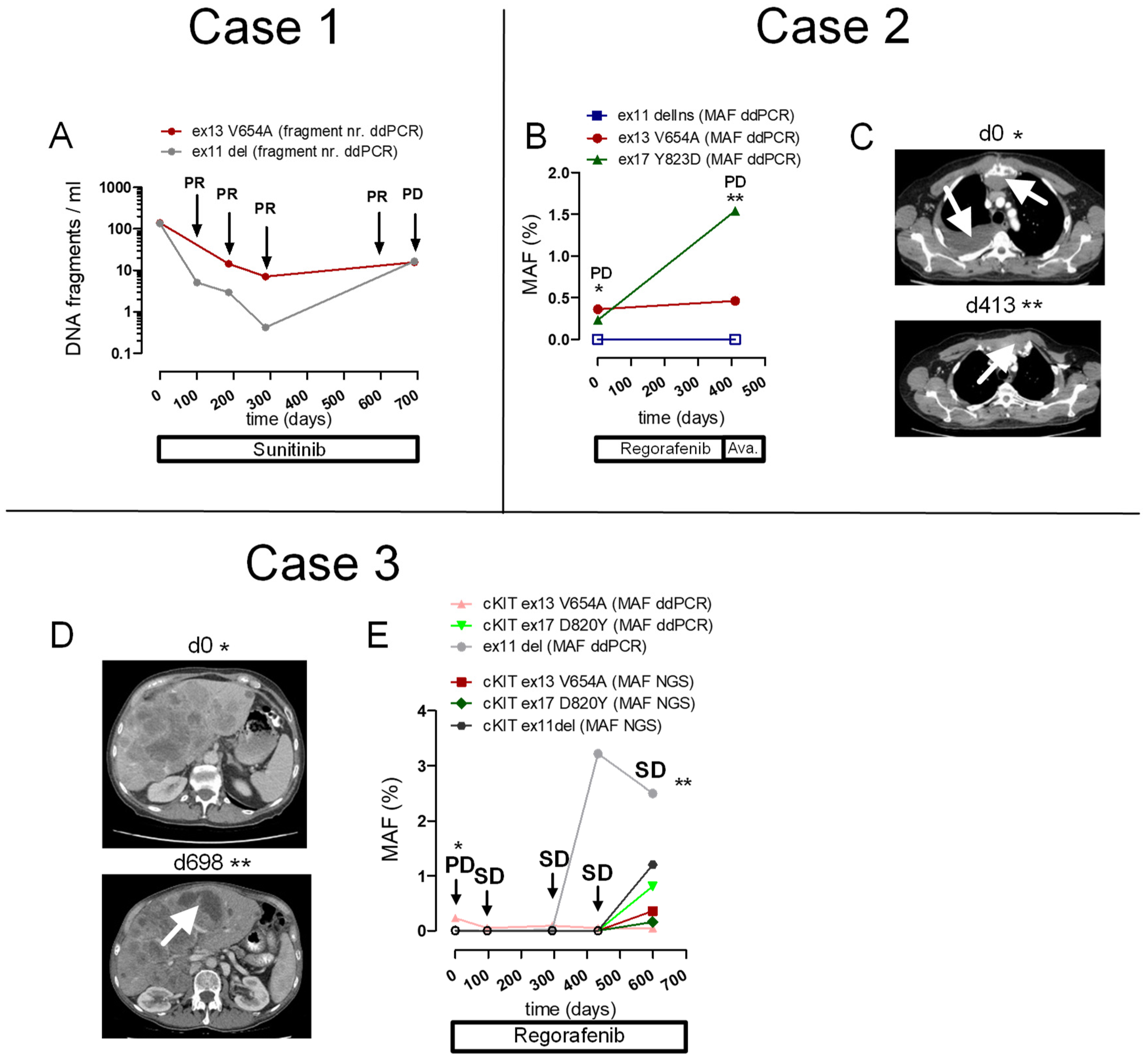
2.5. Patient Cases
2.5.1. Case 1
2.5.2. Case 2
2.5.3. Case 3
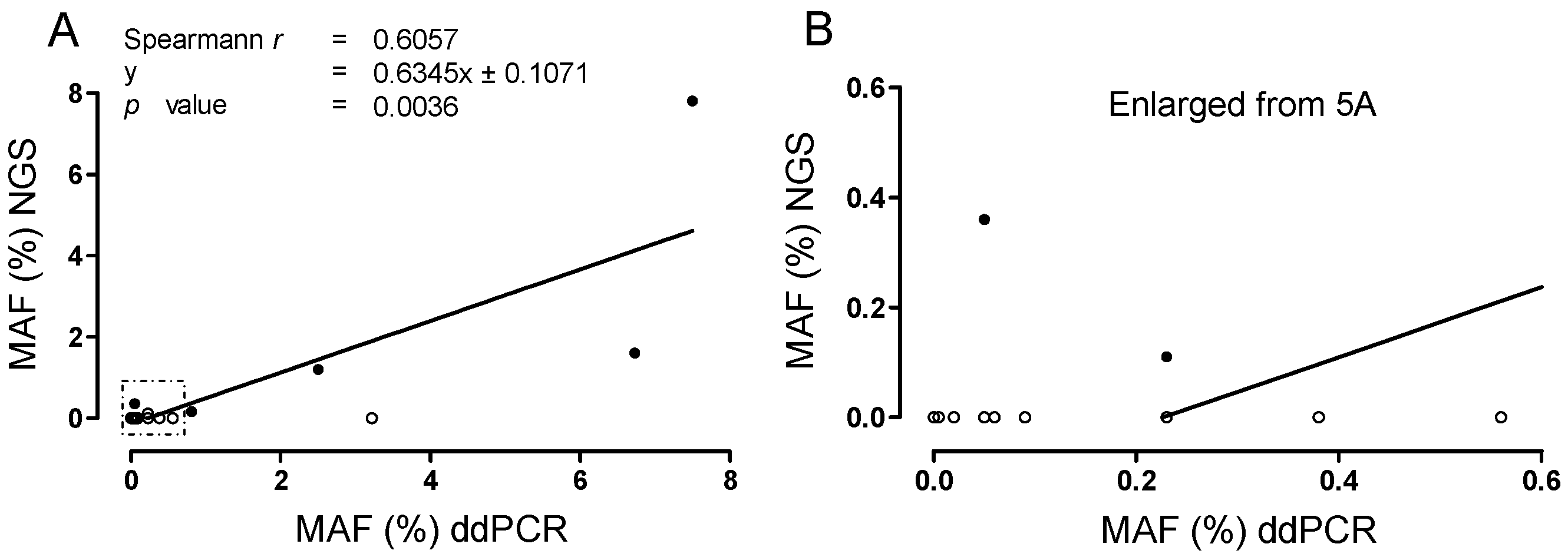
2.6. Correlation of MAF for cKIT Mutations, Determined with Targeted NGS Versus ddPCR
3. Discussion
4. Materials and Methods
4.1. Digital Droplet PCR
4.2. Assay Validation
4.3. Patients
4.4. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joensuu, H.; Rutkowski, P.; Nishida, T.; Steigen, S.E.; Brabec, P.; Plank, L.; Nilsson, B.; Braconi, C.; Bordoni, A.; Magnusson, M.K.; et al. KIT and PDGFRA Mutations and the Risk of GI Stromal Tumor Recurrence. J. Clin. Oncol. 2015, 33, 634–642. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; Desantis, D.; Singer, S.; et al. Acquired Resistance to Imatinib in Gastrointestinal Stromal Tumor Occurs through Secondary Gene Mutation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. Clinical Significance of Oncogenic KIT and PDGFRA Mutations in Gastrointestinal Stromal Tumours. Histopathology 2008, 53, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Lasota, J.; Miettinen, M. KIT Exon 11 Deletion–Inversions Represent Complex Mutations in Gastrointestinal Stromal Tumors. Cancer Genet. Cytogenet. 2007, 175, 69–72. [Google Scholar] [CrossRef]
- Lux, M.L.; Rubin, B.P.; Biase, T.L.; Chen, C.-J.; Maclure, T.; Demetri, G.; Xiao, S.; Singer, S.; Fletcher, C.D.M.; Fletcher, J.A. KIT Extracellular and Kinase Domain Mutations in Gastrointestinal Stromal Tumors. Am. J. Pathol. 2000, 156, 791–795. [Google Scholar] [CrossRef]
- Roberts, K.G.; Odell, A.F.; Byrnes, E.M.; Baleato, R.M.; Griffith, R.; Lyons, A.B.; Ashman, L.K. Resistance to C-KIT Kinase Inhibitors Conferred by V654A Mutation. Mol. Cancer Ther. 2007, 6, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal Stromal Tumours: Origin and Molecular Oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA Mutations in Gastrointestinal Stromal Tumors: Frequency, Spectrum and In Vitro Sensitivity to Imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Peregrina, D.; García-Valverde, A.; Pilco-Janeta, D.; Serrano, C. Liquid Biopsy in Gastrointestinal Stromal Tumors: Ready for Prime Time? Curr. Treat. Options Oncol. 2021, 22, 32. [Google Scholar] [CrossRef]
- Antoch, G.; Kanja, J.; Bauer, S.; Kuehl, H.; Renzing-Koehler, K.; Schuette, J.; Bockisch, A.; Debatin, J.F.; Freudenberg, L.S. Comparison of PET, CT, and Dual-Modality PET/CT Imaging for Monitoring of Imatinib (STI571) Therapy in Patients with Gastrointestinal Stromal Tumors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2004, 45, 357–365. [Google Scholar]
- Kang, G.; Bae, B.N.; Sohn, B.S.; Pyo, J.-S.; Kang, G.H.; Kim, K.-M. Detection of KIT and PDGFRA Mutations in the Plasma of Patients with Gastrointestinal Stromal Tumor. Target. Oncol. 2015, 10, 597–601. [Google Scholar] [CrossRef]
- Heydt, C.; Kumm, N.; Fassunke, J.; Künstlinger, H.; Ihle, M.A.; Scheel, A.; Schildhaus, H.-U.; Haller, F.; Büttner, R.; Odenthal, M.; et al. Massively Parallel Sequencing Fails to Detect Minor Resistant Subclones in Tissue Samples Prior to Tyrosine Kinase Inhibitor Therapy. BMC Cancer 2015, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Gutierrez Sainz, L.; Chi, P. The Management of Metastatic GIST: Current Standard and Investigational Therapeutics. J. Hematol. Oncol. 2021, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary Activity of Tyrosine Kinase Inhibitors against Secondary Kit Mutations in Imatinib-Resistant Gastrointestinal Stromal Tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Jung, K.; Fleischhacker, M.; Rabien, A. Cell-Free DNA in the Blood as a Solid Tumor Biomarker—A Critical Appraisal of the Literature. Clin. Chim. Acta Int. J. Clin. Chem. 2010, 411, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non-Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS Mutations and Acquired Resistance to Anti-EGFR Therapy in Colorectal Cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Waldeck, S.; Mitschke, J.; Wiesemann, S.; Rassner, M.; Andrieux, G.; Deuter, M.; Mutter, J.; Lüchtenborg, A.; Kottmann, D.; Titze, L.; et al. Early Assessment of Circulating Tumor DNA after Curative-intent Resection Predicts Tumor Recurrence in Early-stage and Locally Advanced Non-small-cell Lung Cancer. Mol. Oncol. 2022, 16, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR -Mutant Lung Cancer Using Quantitative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.-H.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef]
- Domínguez-Vigil, I.G.; Moreno-Martínez, A.K.; Wang, J.Y.; Roehrl, M.H.A.; Barrera-Saldaña, H.A. The Dawn of the Liquid Biopsy in the Fight against Cancer. Oncotarget 2018, 9, 2912–2922. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Johansson, G.; Berndsen, M.; Lindskog, S.; Österlund, T.; Fagman, H.; Muth, A.; Ståhlberg, A. Monitoring Circulating Tumor DNA During Surgical Treatment in Patients with Gastrointestinal Stromal Tumors. Mol. Cancer Ther. 2021, 20, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Sohn, B.S.; Pyo, J.-S.; Kim, J.Y.; Lee, B.; Kim, K.-M. Detecting Primary KIT Mutations in Presurgical Plasma of Patients with Gastrointestinal Stromal Tumor. Mol. Diagn. Ther. 2016, 20, 347–351. [Google Scholar] [CrossRef]
- Malapelle, U.; Mayo de-Las-Casas, C.; Rocco, D.; Garzon, M.; Pisapia, P.; Jordana-Ariza, N.; Russo, M.; Sgariglia, R.; De Luca, C.; Pepe, F.; et al. Development of a Gene Panel for Next-Generation Sequencing of Clinically Relevant Mutations in Cell-Free DNA from Cancer Patients. Br. J. Cancer 2017, 116, 802–810. [Google Scholar] [CrossRef]
- Namløs, H.M.; Boye, K.; Mishkin, S.J.; Barøy, T.; Lorenz, S.; Bjerkehagen, B.; Stratford, E.W.; Munthe, E.; Kudlow, B.A.; Myklebost, O.; et al. Noninvasive Detection of CtDNA Reveals Intratumor Heterogeneity and Is Associated with Tumor Burden in Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2018, 17, 2473–2480. [Google Scholar] [CrossRef]
- Wada, N.; Kurokawa, Y.; Takahashi, T.; Hamakawa, T.; Hirota, S.; Naka, T.; Miyazaki, Y.; Makino, T.; Yamasaki, M.; Nakajima, K.; et al. Detecting Secondary C-KIT Mutations in the Peripheral Blood of Patients with Imatinib-Resistant Gastrointestinal Stromal Tumor. Oncology 2016, 90, 112–117. [Google Scholar] [CrossRef]
- Xu, H.; Chen, L.; Shao, Y.; Zhu, D.; Zhi, X.; Zhang, Q.; Li, F.; Xu, J.; Liu, X.; Xu, Z. Clinical Application of Circulating Tumor DNA in the Genetic Analysis of Patients with Advanced GIST. Mol. Cancer Ther. 2018, 17, 290–296. [Google Scholar] [CrossRef]
- Jilg, S.; Rassner, M.; Maier, J.; Waldeck, S.; Kehl, V.; Follo, M.; Philipp, U.; Sauter, A.; Specht, K.; Mitschke, J.; et al. Circulating CKIT and PDGFRA DNA Indicates Disease Activity in Gastrointestinal Stromal Tumor (GIST). Int. J. Cancer 2019, 145, 2292–2303. [Google Scholar] [CrossRef]
- Hudecova, I. Digital PCR Analysis of Circulating Nucleic Acids. Clin. Biochem. 2015, 48, 948–956. [Google Scholar] [CrossRef]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-Free DNA (CfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Ryu, M.-H.; Na, Y.S.; Ryoo, B.-Y.; Park, S.R.; Kang, Y.-K. Analysis of Serum Protein Biomarkers, Circulating Tumor DNA, and Dovitinib Activity in Patients with Tyrosine Kinase Inhibitor-Refractory Gastrointestinal Stromal Tumors. Ann. Oncol. 2014, 25, 2272–2277. [Google Scholar] [CrossRef]
- Day, E.; Dear, P.H.; McCaughan, F. Digital PCR Strategies in the Development and Analysis of Molecular Biomarkers for Personalized Medicine. Methods 2013, 59, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, Y.; Li, J.; Sun, N.; Yuan, J.; Shen, L. Secondary Mutations of C-KIT Contribute to Acquired Resistance to Imatinib and Decrease Efficacy of Sunitinib in Chinese Patients with Gastrointestinal Stromal Tumors. Med. Oncol. 2013, 30, 522. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.M.; Sandau, K.; et al. Molecular Correlates of Imatinib Resistance in Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Parab, T.M.; DeRogatis, M.J.; Boaz, A.M.; Grasso, S.A.; Issack, P.S.; Duarte, D.A.; Urayeneza, O.; Vahdat, S.; Qiao, J.-H.; Hinika, G.S. Gastrointestinal Stromal Tumors: A Comprehensive Review. J. Gastrointest. Oncol. 2019, 10, 144–154. [Google Scholar] [CrossRef]
- Casali, P.G.; Le Cesne, A.; Poveda Velasco, A.; Kotasek, D.; Rutkowski, P.; Hohenberger, P.; Fumagalli, E.; Judson, I.R.; Italiano, A.; Martin Broto, J.; et al. Imatinib Failure-Free Survival (IFS) in Patients with Localized Gastrointestinal Stromal Tumors (GIST) Treated with Adjuvant Imatinib (IM): The EORTC/AGITG/FSG/GEIS/ISG Randomized Controlled Phase III Trial. J. Clin. Oncol. 2013, 31, 10500. [Google Scholar] [CrossRef]
- Dematteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Maki, R.G.; Pisters, P.W.T.; Demetri, G.D.; Blackstein, M.E.; Blanke, C.D.; von Mehren, M.; Brennan, M.F.; et al. Adjuvant Imatinib Mesylate after Resection of Localised, Primary Gastrointestinal Stromal Tumour: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Lond. Engl. 2009, 373, 1097–1104. [Google Scholar] [CrossRef]
- Joensuu, H.; Eriksson, M.; Sundby Hall, K.; Hartmann, J.T.; Pink, D.; Schütte, J.; Ramadori, G.; Hohenberger, P.; Duyster, J.; Al-Batran, S.-E.; et al. One vs Three Years of Adjuvant Imatinib for Operable Gastrointestinal Stromal Tumor: A Randomized Trial. JAMA 2012, 307, 1265–1272. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.M.; Roberts, P.J.; Heinz, D.; et al. Long-Term Results From a Randomized Phase II Trial of Standard- Versus Higher-Dose Imatinib Mesylate for Patients With Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Rankin, C.; Demetri, G.D.; Ryan, C.W.; von Mehren, M.; Benjamin, R.S.; Raymond, A.K.; Bramwell, V.H.C.; Baker, L.H.; Maki, R.G.; et al. Phase III Randomized, Intergroup Trial Assessing Imatinib Mesylate at Two Dose Levels in Patients with Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing the Kit Receptor Tyrosine Kinase: S0033. J. Clin. Oncol. 2008, 26, 626–632. [Google Scholar] [CrossRef]
- Patel, S. Long-Term Efficacy of Imatinib for Treatment of Metastatic GIST. Cancer Chemother. Pharmacol. 2013, 72, 277–286. [Google Scholar] [CrossRef] [PubMed]
- von Mehren, M.; Heinrich, M.C.; Joensuu, H.; Blanke, C.D.; Wehrle, E.; Demetri, G.D. Follow-up Results after 9 Years (Yrs) of the Ongoing, Phase II B2222 Trial of Imatinib Mesylate (IM) in Patients (Pts) with Metastatic or Unresectable KIT+ Gastrointestinal Stromal Tumors (GIST). J. Clin. Oncol. 2011, 29, 10016. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Rankin, C.; Blanke, C.D.; Demetri, G.D.; Borden, E.C.; Ryan, C.W.; von Mehren, M.; Blackstein, M.E.; Priebat, D.A.; Tap, W.D.; et al. Correlation of Long-Term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017, 3, 944–952. [Google Scholar] [CrossRef]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.M.; Corless, C.L.; Fletcher, J.A. Heterogeneity of Kinase Inhibitor Resistance Mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.; Lange, T.; Kerle, I.; Specht, K.; Bruegel, M.; Wickenhauser, C.; Jost, P.; Niederwieser, D.; Peschel, C.; Duyster, J.; et al. Detection of Mutant Free Circulating Tumor DNA in the Plasma of Patients with Gastrointestinal Stromal Tumor Harboring Activating Mutations of CKIT or PDGFRA. Clin. Cancer Res. 2013, 19, 4854–4867. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, S.; Wang, R.; Wang, S.-Y.; Han, Q.; Xu, H.-T.; Yang, P.; Liu, Y. Identifying Secondary Mutations in Chinese Patients with Imatinib-Resistant Gastrointestinal Stromal Tumors (GISTs) by Next Generation Sequencing (NGS). Pathol. Oncol. Res. POR 2020, 26, 91–100. [Google Scholar] [CrossRef]
- Serrano, C.; Leal, A.; Kuang, Y.; Morgan, J.A.; Barysauskas, C.M.; Phallen, J.; Triplett, O.; Mariño-Enríquez, A.; Wagner, A.J.; Demetri, G.D.; et al. Phase I Study of Rapid Alternation of Sunitinib and Regorafenib for the Treatment of Tyrosine Kinase Inhibitor Refractory Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2019, 25, 7287–7293. [Google Scholar] [CrossRef]
- Serrano, C.; Vivancos, A.; López-Pousa, A.; Matito, J.; Mancuso, F.M.; Valverde, C.; Quiroga, S.; Landolfi, S.; Castro, S.; Dopazo, C.; et al. Clinical Value of next Generation Sequencing of Plasma Cell-Free DNA in Gastrointestinal Stromal Tumors. BMC Cancer 2020, 20, 99. [Google Scholar] [CrossRef]
- Garg, S.; Grenier, S.; Misyura, M.; Sukhai, M.A.; Thomas, M.; Kamel-Reid, S.; Stockley, T. Assessing the Diagnostic Yield of Targeted Next-Generation Sequencing for Melanoma and Gastrointestinal Tumors. J. Mol. Diagn. JMD 2020, 22, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, P.A.; ter Elst, A.; Tibbesma, M.; Bosman, L.J.; Mathijssen, R.; Atrafi, F.; van Coevorden, F.; Steeghs, N.; Farag, S.; Gelderblom, H.; et al. A Single Digital Droplet PCR Assay to Detect Multiple KIT Exon 11 Mutations in Tumor and Plasma from Patients with Gastrointestinal Stromal Tumors. Oncotarget 2018, 9, 13870–13883. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The Digital MIQE Guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- Armbruster, D.A.; Pry, T. Limit of Blank, Limit of Detection and Limit of Quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. S1), S49–S52. [Google Scholar]
- Parikh, R.; Mathai, A.; Parikh, S.; Chandra Sekhar, G.; Thomas, R. Understanding and Using Sensitivity, Specificity and Predictive Values. Indian J. Ophthalmol. 2008, 56, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Gupta, V. Methods for the Determination of Limit of Detection and Limit of Quantitation of the Analytical Methods. Chron. Young Sci. 2011, 2, 21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cKIT | ||||||||||
| Mut | Exon | Temperature | Annealing Time | Pos. Signals Neg. Controls | Mean Copies Per Partition (+Variation) | Number of Partitions (Mean) | Total Volume of Partitions Measured in µL (Mean) | Limit of Detection (LoD) | Limit of Blank (LoB) Fragments/mL | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A502-Y503 dup | 9 | 55 °C | 60 s 50 cycles | WT: 0 MUT: 0 | 1.33 ± 0.5 | 15,652.1 | 15.3 | 1:17,322 (0.006%) | 2.7 |
| 2 | K642E | 13 | 55 °C | 60 s 40 cycles | WT: 0.3 MUT: 0.3 | 0.80 ± 0.4 | 18,029.6 | 17.1 | 1:1068 (0.09%) | 6.70 |
| 3 | V654A | 13 | 52 °C | 90 s 50 cycles + 1 µL MgCl2 | WT: 0 MUT: 0 | 0.82 ± 0.6 | 15,280.3 | 15.6 | 1:4337 (0.02%) | 3.70 |
| 4 | T670I | 14 | 55 °C | 90 s 50 cycles | WT: 0 MUT: 1.5 | 1.35 ± 0.5 | 16,380.5 | 16.7 | 1:1009 (0.10%) | 4.90 |
| 5 | 820f -> drop off | 17 | 55 °C | 90 s 50 cycles | WT: 0 MUT: 0 | 1.12 ± 0.4 | 15,493.7 | 16.7 | 1:610 (0.16%) | 4.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rassner, M.; Waldeck, S.; Follo, M.; Jilg, S.; Philipp, U.; Jolic, M.; Wehrle, J.; Jost, P.J.; Peschel, C.; Illert, A.L.; et al. Development of Highly Sensitive Digital Droplet PCR for Detection of cKIT Mutations in Circulating Free DNA That Mediate Resistance to TKI Treatment for Gastrointestinal Stromal Tumor (GIST). Int. J. Mol. Sci. 2023, 24, 5411. https://doi.org/10.3390/ijms24065411
Rassner M, Waldeck S, Follo M, Jilg S, Philipp U, Jolic M, Wehrle J, Jost PJ, Peschel C, Illert AL, et al. Development of Highly Sensitive Digital Droplet PCR for Detection of cKIT Mutations in Circulating Free DNA That Mediate Resistance to TKI Treatment for Gastrointestinal Stromal Tumor (GIST). International Journal of Molecular Sciences. 2023; 24(6):5411. https://doi.org/10.3390/ijms24065411
Chicago/Turabian StyleRassner, Michael, Silvia Waldeck, Marie Follo, Stefanie Jilg, Ulrike Philipp, Martina Jolic, Julius Wehrle, Philipp J. Jost, Christian Peschel, Anna Lena Illert, and et al. 2023. "Development of Highly Sensitive Digital Droplet PCR for Detection of cKIT Mutations in Circulating Free DNA That Mediate Resistance to TKI Treatment for Gastrointestinal Stromal Tumor (GIST)" International Journal of Molecular Sciences 24, no. 6: 5411. https://doi.org/10.3390/ijms24065411
APA StyleRassner, M., Waldeck, S., Follo, M., Jilg, S., Philipp, U., Jolic, M., Wehrle, J., Jost, P. J., Peschel, C., Illert, A. L., Duyster, J., Scherer, F., & von Bubnoff, N. (2023). Development of Highly Sensitive Digital Droplet PCR for Detection of cKIT Mutations in Circulating Free DNA That Mediate Resistance to TKI Treatment for Gastrointestinal Stromal Tumor (GIST). International Journal of Molecular Sciences, 24(6), 5411. https://doi.org/10.3390/ijms24065411