
New Pathways Identify Novel Drug Targets for the Prevention and Treatment of Alzheimer’s Disease



{kind=link}
Abstract
1. Introduction
2. Alzheimer’s Disease
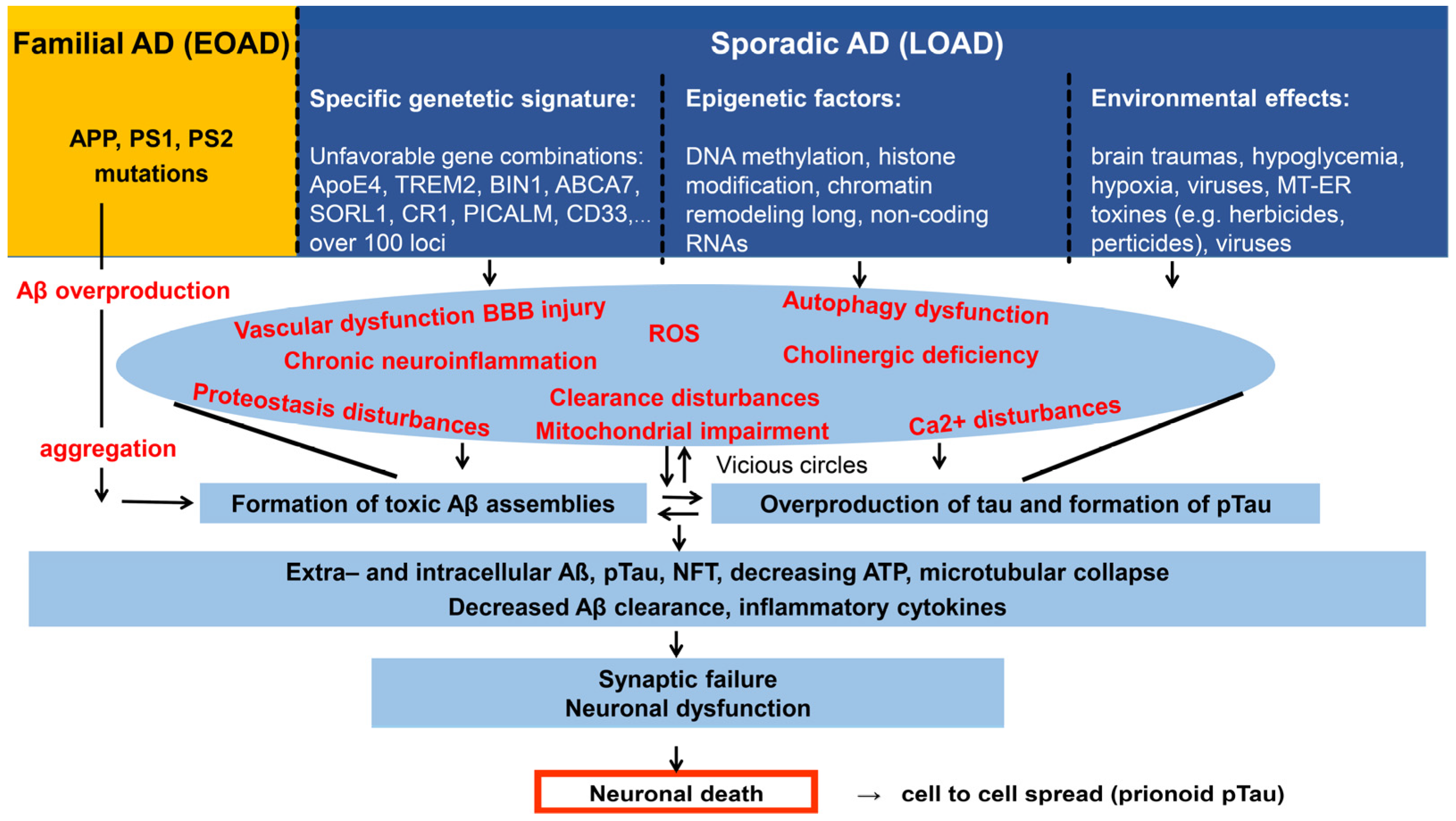
2.1. Pathology and Classification of AD. Aging and Dementia. The Main Risk Factors of AD
2.2. Genetic Background of AD, the Multiplex Model
2.3. Amyloid Structures. Physiological and Pathophysiological Role of the Aβ and Tau Proteins
- native Aβ monomer → partially folded monomer → transient oAβ → protofibrils (β-sheet → structured oAβ → fibrils (cross-β) → big aggregates, plaques.
- abnormally phosphorylated monomer (pTau) → dimer, trimer → small soluble oligomers (oTau) → granular oligomers → straight filaments → paired helical filaments → neurofibrillary tangles.
- Disaggregation and collapse of microtubules. Big tau assemblies may cause a direct physical blockade of axonal transport.
- Loss of DNA protection at the nucleus.
- Increased excitability of neurons.
- Tau may bind to synaptic vesicles and disrupt the synaptic cytoskeleton causing synaptic loss and disturbances of neural circuits.
- Tau causes neuroinflammation.
2.4. The Ever-Changing and Developing Amyloid (and Tau) Hypotheses. Alternative Hypotheses of AD
- Aβ, very probably iAβ, is the initiating factor of AD [66].
- Loss of cholinergic neurons and neurotransmission are causing factors of AD [91].
- Deficit of the glutamatergic system [88] triggers tau overproduction.
- Abnormal phosphorylation of tau proteins is in the background of AD initiation and progress [76].
- According to the dual cascade hypothesis, cellular processes in the brain cortex simultaneously drive tau and Aβ pathology [80].
- Mitochondrial dysfunction starts a cascade of pathological events in brain cells [92].
- Chronic neuroinflammation is responsible for the initiation of damage to neurons [93].
- Impaired amyloid clearance (BBB and glymphatic clearance) is the main cause of amyloid accumulation in AD [96].
- Aβ peptides are generated in the periphery and enter the brain via the BBB [97].
- Aging is the main driver of sporadic AD pathogenesis. Each type of brain cell (microglia, astrocytes, and brain vasculature cells) participates in pathophysiological events [89].
2.5. Early Diagnosis of AD, Molecular Biomarkers
- chronic stress → subjective cognitive decline (SCD) → mild cognitive impairment (MCI) → AD dementia.
3. Conventional and Novel Targets for Slowing and/or Preventing the Progress of AD
3.1. Inhibition of the Formation of Toxic Amyloid Aggregates
3.1.1. Decreasing Aβ Production
3.1.2. Blocking Aβ Aggregation
3.1.3. Blocking Tau Biosynthesis
3.1.4. Inhibiting Tau Aggregation and Fibrillation
3.1.5. Other Tau-Directed Potential Approaches
3.2. Improvement of Amyloid Clearance. Vascular Dysfunction, BBB, and the Glymphatic System
3.2.1. Passive Immunotherapy with Monoclonal Antibodies (mAbs)
3.2.2. Clearance by the BBB and Activation of the Glymphatic System
3.3. Modulation of Chronic Neuroinflammation
3.3.1. Chronic Neuroinflammation
3.3.2. Neuroinflammation and Glial Cells as Targets for AD Drug Development
- TNFα modulation [174].
- Activation of spleen tyrosine kinase (SYK) for increasing the clearance function of MG [175].
- Activation of the CX3CR1 (C-X3-C Motif Chemokine Receptor 1) gene for improved MG phagocytotic activity [176].
- Activation of TREM2 microglial protein for slowing down pTau accumulation and cognitive loss [177].
- Activation of the PLCG (Phospholipase C γ1) enzyme for promoting the protective function of microglia [178].
- Mitigation of neuroinflammation by increasing the scavenger functions of MG by targeting INPP5 (Inositol Polyphosphate-5-Phosphatase) enzyme [179].
- Modulation of astrocyte activity for Aβ degradation and clearance, as well as BBB protection [180].
- Creating antioxidant molecules for protecting the brain from the degrading effect of ROS by activation of mucosal-associated invariant T-cells (MAIT-cells) [181].
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.I.P.; Staniforth, R.A. General Principles Underpinning Amyloid Structure. Front. Neurosci. 2022, 16, 878869. [Google Scholar] [CrossRef]
- Bogár, F.; Fülöp, L.; Penke, B. Novel Therapeutic Target for Prevention of Neurodegenerative Diseases: Modulation of Neuroinflammation with Sig-1R Ligands. Biomolecules 2022, 12, 363. [Google Scholar] [CrossRef]
- Small, G.W. Updates in the Management of Mild Cognitive Impairment and Alzheimer Disease. J. Fam. Pract. 2022, 71, S82–S87. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Tam, K. Pathological Mechanisms and Therapeutic Strategies for Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 543. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, J.; Kovacs, G.G. Prevalence of Mixed Pathologies in the Aging Brain. Alzheimers Res. Ther. 2014, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Recent Update on the Heterogeneity of the Alzheimer’s Disease Spectrum. J. Neural Transm. 2022, 129, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Capetillo-Zarate, E.; Del Tredici, K.; Braak, H. The Development of Amyloid Beta Protein Deposits in the Aged Brain. Sci. Aging Knowl. Environ. 2006, 2006, re1. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, S.H.; Feiz, F.; Ozoria, S.; Razlighi, Q.R. Alzheimer’s Disease Neuroimaging Initiative Topographical Overlapping of the Amyloid-β and Tau Pathologies in the Default Mode Network Predicts Alzheimer’s Disease with Higher Specificity. J. Alzheimers Dis. JAD 2021, 83, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-Deposition in the Human Brain and Its Relevance for the Development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.-J. Normal Aging Induces Changes in the Brain and Neurodegeneration Progress: Review of the Structural, Biochemical, Metabolic, Cellular, and Molecular Changes. Front. Aging Neurosci. 2022, 14, 931536. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A.B.; Demarez, A. Protein Aggregation as a Paradigm of Aging. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 980–996. [Google Scholar] [CrossRef] [PubMed]
- Groh, N.; Bühler, A.; Huang, C.; Li, K.W.; van Nierop, P.; Smit, A.B.; Fändrich, M.; Baumann, F.; David, D.C. Age-Dependent Protein Aggregation Initiates Amyloid-β Aggregation. Front. Aging Neurosci. 2017, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and Aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Freer, R.; Sormanni, P.; Vecchi, G.; Ciryam, P.; Dobson, C.M.; Vendruscolo, M. A Protein Homeostasis Signature in Healthy Brains Recapitulates Tissue Vulnerability to Alzheimer’s Disease. Sci. Adv. 2016, 2, e1600947. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J. Structural Alterations in the Peptide Backbone of Beta-Amyloid Core Protein May Account for Its Deposition and Stability in Alzheimer’s Disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, T.R.; Riggs, D.L.; Talbert, L.E.; Tang, J.; Coburn, E.; Kang, A.S.; Noll, J.; Augello, C.; Ford, B.D.; Julian, R.R. Spontaneous Isomerization of Long-Lived Proteins Provides a Molecular Mechanism for the Lysosomal Failure Observed in Alzheimer’s Disease. ACS Cent. Sci. 2019, 5, 1387–1395. [Google Scholar] [CrossRef]
- Geiger, T.; Clarke, S. Deamidation, Isomerization, and Racemization at Asparaginyl and Aspartyl Residues in Peptides. Succinimide-Linked Reactions That Contribute to Protein Degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J.W.; Schey, K.L.; Friedrich, M.G. Old Proteins in Man: A Field in Its Infancy. Trends Biochem. Sci. 2016, 41, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic Architecture of Alzheimer’s Disease. Neurobiol. Dis. 2020, 143, 104976. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A Mutation in APP Protects against Alzheimer’s Disease and Age-Related Cognitive Decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; Hill, M.; Williams, J. The Multiplex Model of the Genetics of Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Blumenfeld, J.; Agrawal, A.; Traglia, M.; Grone, B.; Zilberter, M.; Yip, O.; Rao, A.; Nelson, M.R.; Hao, Y.; et al. Neuronal APOE4 Removal Protects against Tau-Mediated Gliosis, Neurodegeneration and Myelin Deficits. Nat. Aging. in press. [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Remolina Serrano, J.; Hoyle, R.; Holtzman, D.M. Microglia Drive APOE-Dependent Neurodegeneration in a Tauopathy Mouse Model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.-C. Genetics of Alzheimer’s Disease: Where We Are, and Where We Are Going. Curr. Opin. Neurobiol. 2020, 61, 40–48. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Li, Y.; Laws, S.M.; Miles, L.A.; Wiley, J.S.; Huang, X.; Masters, C.L.; Gu, B.J. Genomics of Alzheimer’s Disease Implicates the Innate and Adaptive Immune Systems. Cell Mol. Life Sci. 2021, 78, 7397–7426. [Google Scholar] [CrossRef]
- Maity, S.; Farrell, K.; Navabpour, S.; Narayanan, S.N.; Jarome, T.J. Epigenetic Mechanisms in Memory and Cognitive Decline Associated with Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12280. [Google Scholar] [CrossRef]
- Penke, B.; Bogár, F.; Fülöp, L. β-Amyloid and the Pathomechanisms of Alzheimer’s Disease: A Comprehensive View. Molecules 2017, 22, 1692. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Bogár, F.; Paragi, G.; Gera, J.; Fülöp, L. Key Peptides and Proteins in Alzheimer’s Disease. Curr. Protein Pept. Sci. 2019, 20, 577–599. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Shin, H.; Hong, S.; Kim, Y. Physiological Roles of Monomeric Amyloid-β and Implications for Alzheimer’s Disease Therapeutics. Exp. Neurobiol. 2022, 31, 65–88. [Google Scholar] [CrossRef]
- Nichols, R.A.; Gulisano, W.; Puzzo, D. Editorial: Beta Amyloid: From Physiology to Pathogenesis. Front. Mol. Neurosci. 2022, 15, 876224. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Mohamed Asik, R.; Suganthy, N.; Aarifa, M.A.; Kumar, A.; Szigeti, K.; Mathe, D.; Gulyás, B.; Archunan, G.; Padmanabhan, P. Alzheimer’s Disease: A Molecular View of β-Amyloid Induced Morbific Events. Biomedicines 2021, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Szűcs, M.; Bogár, F. Oligomerization and Conformational Change Turn Monomeric β-Amyloid and Tau Proteins Toxic: Their Role in Alzheimer’s Pathogenesis. Molecules 2020, 25, 1659. [Google Scholar] [CrossRef]
- Diociaiuti, M.; Bonanni, R.; Cariati, I.; Frank, C.; D’Arcangelo, G. Amyloid Prefibrillar Oligomers: The Surprising Commonalities in Their Structure and Activity. Int. J. Mol. Sci. 2021, 22, 6435. [Google Scholar] [CrossRef]
- Owen, M.C.; Gnutt, D.; Gao, M.; Wärmländer, S.K.T.S.; Jarvet, J.; Gräslund, A.; Winter, R.; Ebbinghaus, S.; Strodel, B. Effects of in Vivo Conditions on Amyloid Aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Lin, Y.; Lee, Y.-H.; Zheng, J.; Ramamoorthy, A. Biophysical Processes Underlying Cross-Seeding in Amyloid Aggregation and Implications in Amyloid Pathology. Biophys. Chem. 2021, 269, 106507. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pittman, J.M.; Zerweck, J.; Venkata, B.S.; Moore, P.C.; Sachleben, J.R.; Meredith, S.C. Β-Amyloid Aggregation and Heterogeneous Nucleation. Protein Sci. 2019, 28, 1567–1581. [Google Scholar] [CrossRef]
- Vicente-Zurdo, D.; Rodríguez-Blázquez, S.; Gómez-Mejía, E.; Rosales-Conrado, N.; León-González, M.E.; Madrid, Y. Neuroprotective Activity of Selenium Nanoparticles against the Effect of Amino Acid Enantiomers in Alzheimer’s Disease. Anal. Bioanal. Chem. 2022, 414, 7573–7584. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Watanabe-Nakayama, T. Aggregation and Structure of Amyloid β-Protein. Neurochem. Int. 2021, 151, 105208. [Google Scholar] [CrossRef] [PubMed]
- Lutter, L.; Al-Hilaly, Y.K.; Serpell, C.J.; Tuite, M.F.; Wischik, C.M.; Serpell, L.C.; Xue, W.-F. Structural Identification of Individual Helical Amyloid Filaments by Integration of Cryo-Electron Microscopy-Derived Maps in Comparative Morphometric Atomic Force Microscopy Image Analysis. J. Mol. Biol. 2022, 434, 167466. [Google Scholar] [CrossRef]
- Willbold, D.; Strodel, B.; Schröder, G.F.; Hoyer, W.; Heise, H. Amyloid-Type Protein Aggregation and Prion-like Properties of Amyloids. Chem. Rev. 2021, 121, 8285–8307. [Google Scholar] [CrossRef]
- Lövestam, S.; Scheres, S.H.W. High-Throughput Cryo-EM Structure Determination of Amyloids. Faraday Discuss. 2022, 240, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM Structures of Amyloid-β 42 Filaments from Human Brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Ito, K.; Teraoka, R.; Lambert, M.P.; Klein, W.L.; Mori, H. The E693Δ Mutation in Amyloid Precursor Protein Increases Intracellular Accumulation of Amyloid β Oligomers and Causes Endoplasmic Reticulum Stress-Induced Apoptosis in Cultured Cells. Am. J. Pathol. 2009, 174, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A Mouse Model of Amyloid Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss In Vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644.e12. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s All about Tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef]
- Sexton, C.; Snyder, H.; Beher, D.; Boxer, A.L.; Brannelly, P.; Brion, J.; Buée, L.; Cacace, A.M.; Chételat, G.; Citron, M.; et al. Current Directions in Tau Research: Highlights from Tau 2020. Alzheimers Dement. 2022, 18, 988–1007. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The Complexity of Tau in Alzheimer’s Disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Robbins, M.; Clayton, E.; Kaminski Schierle, G.S. Synaptic Tau: A Pathological or Physiological Phenomenon? Acta Neuropathol. Commun. 2021, 9, 149. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 707268. [Google Scholar] [CrossRef] [PubMed]
- Vasili, E.; Dominguez-Meijide, A.; Outeiro, T.F. Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front. Mol. Neurosci. 2019, 12, 107. [Google Scholar] [CrossRef]
- Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-Beta Peptide and Tau Protein Crosstalk in Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Mormino, E.C.; Schultz, A.P.; Betensky, R.A.; Papp, K.V.; Amariglio, R.E.; Hanseeuw, B.J.; Buckley, R.; Chhatwal, J.; Hedden, T.; et al. The Impact of Amyloid-Beta and Tau on Prospective Cognitive Decline in Older Individuals. Ann. Neurol. 2019, 85, 181–193. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Tóth, A.M.; Földi, I.; Szűcs, M.; Janáky, T. Intraneuronal β-Amyloid and Its Interactions with Proteins and Subcellular Organelles: Proteomics and 2DE. ELECTROPHORESIS 2012, 33, 3608–3616. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Hartmann, T. Intracellular Biology of Alzheimer’s Disease Amyloid Beta Peptide. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of Amyloid Plaque Formation Suggests an Intracellular Basis of Aβ Pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque Formation and the Intraneuronal Accumulation of β-Amyloid in Alzheimer’s Disease: Intraneuronal Accumulation of β-Amyloid. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C. Intracellular Mechanisms of Amyloid Accumulation and Pathogenesis in Alzheimer’s Disease. J. Mol. Neurosci. 2001, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ditaranto, K.; Tekirian, T.L.; Yang, A.J. Lysosomal Membrane Damage in Soluble Aβ-Mediated Cell Death in Alzheimer’s Disease. Neurobiol. Dis. 2001, 8, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.D.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The Revitalized Tau Hypothesis on Alzheimer’s Disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Balendra, V.; Obaid, A.A.; Esposto, J.; Tikhonova, M.A.; Gautam, N.K.; Poeggeler, B. Copper-Mediated β-Amyloid Toxicity and Its Chelation Therapy in Alzheimer’s Disease. Met. Integr. Biomet. Sci. 2022, 14, mfac018. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current Understanding of Metal Ions in the Pathogenesis of Alzheimer’s Disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef]
- Stelmashook, E.V.; Isaev, N.K.; Genrikhs, E.E.; Amelkina, G.A.; Khaspekov, L.G.; Skrebitsky, V.G.; Illarioshkin, S.N. Role of Zinc and Copper Ions in the Pathogenetic Mechanisms of Alzheimer’s and Parkinson’s Diseases. Biochem. Mosc. 2014, 79, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Fan, Y.-G.; Zhao, L.-X.; Zhang, Q.; Wang, Z.-Y. The Metal Ion Hypothesis of Alzheimer’s Disease and the Anti-Neuroinflammatory Effect of Metal Chelators. Bioorganic Chem. 2023, 131, 106301. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau Pathology Is an Initiating Factor in Sporadic Alzheimer’s Disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and Progress of Hypotheses and Clinical Trials for Alzheimer’s Disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune Attack: The Role of Inflammation in Alzheimer Disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A.F. Vascular Dysfunction in the Pathogenesis of Alzheimer’s Disease—A Review of Endothelium-Mediated Mechanisms and Ensuing Vicious Circles. Neurobiol. Dis. 2015, 82, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. [Google Scholar] [CrossRef]
- Elbert, D.L.; Patterson, B.W.; Lucey, B.P.; Benzinger, T.L.S.; Bateman, R.J. Importance of CSF-Based Aβ Clearance with Age in Humans Increases with Declining Efficacy of Blood-Brain Barrier/Proteolytic Pathways. Commun. Biol. 2022, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.-L.; Xiang, Y.; Jin, W.-S.; Wang, J.; Shen, L.-L.; Huang, Z.-L.; Zhang, K.; Liu, Y.-H.; Zeng, F.; Liu, J.-H.; et al. Blood-Derived Amyloid-β Protein Induces Alzheimer’s Disease Pathologies. Mol. Psychiatry 2018, 23, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Fuller, J.T.; Cronin-Golomb, A.; Gatchel, J.R.; Norton, D.J.; Guzmán-Vélez, E.; Jacobs, H.I.L.; Hanseeuw, B.; Pardilla-Delgado, E.; Artola, A.; Baena, A.; et al. Biological and Cognitive Markers of Presenilin1 E280a Autosomal Dominant Alzheimer’s Disease: A Comprehensive Review of the Colombian Kindred. J. Prev. Alzheimers Dis. 2019, 6, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Villanueva, M.; Marcos Dolado, A.; Gómez-Ramírez, J.; Fernández-Blázquez, M. Brain Structural and Functional Changes in Cognitive Impairment Due to Alzheimer’s Disease. Front. Psychol. 2022, 13, 886619. [Google Scholar] [CrossRef]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimers Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef]
- van der Schaar, J.; Visser, L.N.C.; Bouwman, F.H.; Ket, J.C.F.; Scheltens, P.; Bredenoord, A.L.; van der Flier, W.M. Considerations Regarding a Diagnosis of Alzheimer’s Disease before Dementia: A Systematic Review. Alzheimers Res. Ther. 2022, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Davenport, F.; Gallacher, J.; Kourtzi, Z.; Koychev, I.; Matthews, P.M.; Oxtoby, N.P.; Parkes, L.M.; Priesemann, V.; Rowe, J.B.; Smye, S.W.; et al. Neurodegenerative Disease of the Brain: A Survey of Interdisciplinary Approaches. J. R. Soc. Interface 2023, 20, 20220406. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, Z.; Han, X.; Li, H.; Tang, X. Prediction of Alzheimer’s Disease Progression Based on Magnetic Resonance Imaging. ACS Chem. Neurosci. 2021, 12, 4209–4223. [Google Scholar] [CrossRef]
- Turner, R.S.; Stubbs, T.; Davies, D.A.; Albensi, B.C. Potential New Approaches for Diagnosis of Alzheimer’s Disease and Related Dementias. Front. Neurol. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; van der Kant, R.; Hansson, O. Tau Biomarkers in Alzheimer’s Disease: Towards Implementation in Clinical Practice and Trials. Lancet Neurol. 2022, 21, 726–734. [Google Scholar] [CrossRef]
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia 2021. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2021.pdf (accessed on 3 March 2023).
- Nemy, M.; Cedres, N.; Grothe, M.J.; Muehlboeck, J.-S.; Lindberg, O.; Nedelska, Z.; Stepankova, O.; Vyslouzilova, L.; Eriksdotter, M.; Barroso, J.; et al. Cholinergic White Matter Pathways Make a Stronger Contribution to Attention and Memory in Normal Aging than Cerebrovascular Health and Nucleus Basalis of Meynert. NeuroImage 2020, 211, 116607. [Google Scholar] [CrossRef]
- Cedres, N.; Ferreira, D.; Nemy, M.; Machado, A.; Pereira, J.B.; Shams, S.; Wahlund, L.-O.; Zettergren, A.; Stepankova, O.; Vyslouzilova, L.; et al. Association of Cerebrovascular and Alzheimer Disease Biomarkers With Cholinergic White Matter Degeneration in Cognitively Unimpaired Individuals. Neurology 2022, 99, e1619–e1629. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Pichet Binette, A.; Groot, C.; Smith, R.; Strandberg, O.; Palmqvist, S.; Stomrud, E.; Tideman, P.; Ohlsson, T.; Jögi, J.; et al. Amyloid and Tau PET-Positive Cognitively Unimpaired Individuals Are at High Risk for Future Cognitive Decline. Nat. Med. 2022, 28, 2381–2387. [Google Scholar] [CrossRef]
- Wang, C.; Xu, T.; Yu, W.; Li, T.; Han, H.; Zhang, M.; Tao, M. Early Diagnosis of Alzheimer’s Disease and Mild Cognitive Impairment Based on Electroencephalography: From the Perspective of Event Related Potentials and Deep Learning. Int. J. Psychophysiol. 2022, 182, 182–189. [Google Scholar] [CrossRef]
- Blennow, K.; Shaw, L.M.; Stomrud, E.; Mattsson, N.; Toledo, J.B.; Buck, K.; Wahl, S.; Eichenlaub, U.; Lifke, V.; Simon, M.; et al. Predicting Clinical Decline and Conversion to Alzheimer’s Disease or Dementia Using Novel Elecsys Aβ(1–42), PTau and TTau CSF Immunoassays. Sci. Rep. 2019, 9, 19024. [Google Scholar] [CrossRef]
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; DeCarli, C.; Green, R.C.; Harvey, D.; Jack, C.R.; Jagust, W.; Landau, S.M.; et al. Using the Alzheimer’s Disease Neuroimaging Initiative to Improve Early Detection, Diagnosis, and Treatment of Alzheimer’s Disease. Alzheimers Dement. 2022, 18, 824–857. [Google Scholar] [CrossRef]
- DeMarco, M.L.; Nguyen, Q.; Fok, A.; Hsiung, G.R.; Gugten, J.G. An Automated Clinical Mass Spectrometric Method for Identification and Quantification of Variant and Wild-type Amyloid-β 1-40 and 1-42 Peptides in CSF. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12036. [Google Scholar] [CrossRef]
- Esquivel, R.N.; Benina, N.; Hawkins, D.M.; De Simone, F.; Le Bastard, N.; Vandijck, M.; Gannon, S.; Latham, J.; Radwan, R.R.; Dickson, D. Clinical Validation of the Lumipulse G Β-amyloid Ratio (1-42/1-40) in a Subset of ADNI CSF Samples. Alzheimers Dement. 2021, 17, e055657. [Google Scholar] [CrossRef]
- Shea, D.; Colasurdo, E.; Smith, A.; Paschall, C.; Jayadev, S.; Keene, C.D.; Galasko, D.; Ko, A.; Li, G.; Peskind, E.; et al. SOBA: Development and Testing of a Soluble Oligomer Binding Assay for Detection of Amyloidogenic Toxic Oligomers. Proc. Natl. Acad. Sci. USA 2022, 119, e2213157119. [Google Scholar] [CrossRef]
- Habashi, M.; Vutla, S.; Tripathi, K.; Senapati, S.; Chauhan, P.S.; Haviv-Chesner, A.; Richman, M.; Mohand, S.-A.; Dumulon-Perreault, V.; Mulamreddy, R.; et al. Early Diagnosis and Treatment of Alzheimer’s Disease by Targeting Toxic Soluble Aβ Oligomers. Proc. Natl. Acad. Sci. USA 2022, 119, e2210766119. [Google Scholar] [CrossRef]
- Ashton, N.J.; Janelidze, S.; Mattsson-Carlgren, N.; Binette, A.P.; Strandberg, O.; Brum, W.S.; Karikari, T.K.; González-Ortiz, F.; Di Molfetta, G.; Meda, F.J.; et al. Differential Roles of Aβ42/40, p-Tau231 and p-Tau217 for Alzheimer’s Trial Selection and Disease Monitoring. Nat. Med. 2022, 28, 2555–2562. [Google Scholar] [CrossRef]
- Tatulian, S.A. Challenges and Hopes for Alzheimer’s Disease. Drug Discov. Today 2022, 27, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Wang, K.; Dong, J.; Tang, L. Alzheimer’s Disease: Updated Multi-Targets Therapeutics Are in Clinical and in Progress. Eur. J. Med. Chem. 2022, 238, 114464. [Google Scholar] [CrossRef] [PubMed]
- Gallego Villarejo, L.; Bachmann, L.; Marks, D.; Brachthäuser, M.; Geidies, A.; Müller, T. Role of Intracellular Amyloid β as Pathway Modulator, Biomarker, and Therapy Target. Int. J. Mol. Sci. 2022, 23, 4656. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, Present and Future of Therapeutic Strategies against Amyloid-β Peptides in Alzheimer’s Disease: A Systematic Review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.M.; Kabir, T.M.; Rahman, S.M.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s Disease: Inhibition of Amyloid Beta and Tau Tangle Formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef]
- Salahuddin, P.; Khan, R.H.; Furkan, M.; Uversky, V.N.; Islam, Z.; Fatima, M.T. Mechanisms of Amyloid Proteins Aggregation and Their Inhibition by Antibodies, Small Molecule Inhibitors, Nano-Particles and Nano-Bodies. Int. J. Biol. Macromol. 2021, 186, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F. Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. Int. J. Mol. Sci. 2018, 19, 2677. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, E.M.; Vigh, L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. Int. J. Mol. Sci. 2018, 19, 325. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, W.; Chen, J.; Wang, N.; Zheng, G. A Comparative Study of Resveratrol and Resveratrol-Functional Selenium Nanoparticles: Inhibiting Amyloid β Aggregation and Reactive Oxygen Species Formation Properties. J. Biomed. Mater. Res. A 2018, 106, 3034–3041. [Google Scholar] [CrossRef]
- Santos, M.A.; Chand, K.; Chaves, S. Recent Progress in Multifunctional Metal Chelators as Potential Drugs for Alzheimer’s Disease. Coord. Chem. Rev. 2016, 327, 287–303. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Hong, K.H.; Ning, Y.; Yu, P.; Ren, J.; Ji, M.; Cai, J. Design, Synthesis and Evaluation of a Novel Metal Chelator as Multifunctional Agents for the Treatment of Alzheimer’s Disease. Bioorganic Chem. 2019, 87, 720–727. [Google Scholar] [CrossRef]
- Lin, G.; Zhu, F.; Kanaan, N.M.; Asano, R.; Shirafuji, N.; Sasaki, H.; Yamaguchi, T.; Enomoto, S.; Endo, Y.; Ueno, A.; et al. Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein. Int. J. Mol. Sci. 2021, 22, 12063. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, K.; Zhu, Z.; He, Y.; Zhang, C.; Guo, Z.; Wang, X. Inhibition of Metal-Induced Amyloid β-Peptide Aggregation by a Blood–Brain Barrier Permeable Silica–Cyclen Nanochelator. RSC Adv. 2019, 9, 14126–14131. [Google Scholar] [CrossRef]
- Taş, K.; Volta, B.D.; Lindner, C.; El Bounkari, O.; Hille, K.; Tian, Y.; Puig-Bosch, X.; Ballmann, M.; Hornung, S.; Ortner, M.; et al. Designed Peptides as Nanomolar Cross-Amyloid Inhibitors Acting via Supramolecular Nanofiber Co-Assembly. Nat. Commun. 2022, 13, 5004. [Google Scholar] [CrossRef]
- Wagner, J.; Degenhardt, K.; Veit, M.; Louros, N.; Konstantoulea, K.; Skodras, A.; Wild, K.; Liu, P.; Obermüller, U.; Bansal, V.; et al. Medin Co-Aggregates with Vascular Amyloid-β in Alzheimer’s Disease. Nature 2022, 612, 123–131. [Google Scholar] [CrossRef]
- Ganne, A.; Balasubramaniam, M.; Mainali, N.; Atluri, P.; Shmookler Reis, R.J.; Ayyadevara, S. Physiological Consequences of Targeting 14-3-3 and Its Interacting Partners in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15457. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh, J.; Ruchti, E.; Jiao, W.; Limoni, G.; MacLachlan, C.; Small, S.A.; Knott, G.; Santa-Maria, I.; McCabe, B.D. Retromer Deficiency in Tauopathy Models Enhances the Truncation and Toxicity of Tau. Nat. Commun. 2022, 13, 5049. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.E.; Smith, T.; Yu, D.; Praticò, D. Association of Retromer Deficiency and Tau Pathology in Down Syndrome. Ann. Neurol. 2022, 91, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Patel, H.; You, Y.; Jury, N.; Perkins, A.; Lee-Gosselin, A.; Taylor, X.; You, Y.; Viana Di Prisco, G.; Huang, X.; et al. Bassoon Contributes to Tau-Seed Propagation and Neurotoxicity. Nat. Neurosci. 2022, 25, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble Pool of Abeta Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Holscher, C.; Gengler, S.; Gault, V.A.; Harriott, P.; Mallot, H.A. Soluble Beta-Amyloid[25–35] Reversibly Impairs Hippocampal Synaptic Plasticity and Spatial Learning. Eur. J. Pharmacol. 2007, 561, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.-L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally Administered Antibodies against Amyloid β-Peptide Enter the Central Nervous System and Reduce Pathology in a Mouse Model of Alzheimer Disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef]
- Song, C.; Shi, J.; Zhang, P.; Zhang, Y.; Xu, J.; Zhao, L.; Zhang, R.; Wang, H.; Chen, H. Immunotherapy for Alzheimer’s Disease: Targeting β-Amyloid and Beyond. Transl. Neurodegener. 2022, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; De Strooper, B. The Amyloid Hypothesis in Alzheimer Disease: New Insights from New Therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell Neurosci. 2021, 15, 695479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Wang, J.; Zhang, Z.-N.; Su, Q.; Guo, J.-H. The Relationship between Amyloid-Beta and Brain Capillary Endothelial Cells in Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 2355. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Behl, T.; Kumar, A.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. Targeting Endothelin in Alzheimer’s Disease: A Promising Therapeutic Approach. BioMed Res. Int. 2021, 2021, 7396580. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Verheggen, I.C.M.; Van Boxtel, M.P.J.; Verhey, F.R.J.; Jansen, J.F.A.; Backes, W.H. Interaction between Blood-Brain Barrier and Glymphatic System in Solute Clearance. Neurosci. Biobehav. Rev. 2018, 90, 26–33. [Google Scholar] [CrossRef]
- Mogensen, F.L.-H.; Delle, C.; Nedergaard, M. The Glymphatic System (En)during Inflammation. Int. J. Mol. Sci. 2021, 22, 7491. [Google Scholar] [CrossRef] [PubMed]
- Riba, M.; del Valle, J.; Molina-Porcel, L.; Pelegrí, C.; Vilaplana, J. Wasteosomes (Corpora Amylacea) as a Hallmark of Chronic Glymphatic Insufficiency. Proc. Natl. Acad. Sci. USA 2022, 119, e2211326119. [Google Scholar] [CrossRef]
- Cunningham, C.; Hennessy, E. Co-Morbidity and Systemic Inflammation as Drivers of Cognitive Decline: New Experimental Models Adopting a Broader Paradigm in Dementia Research. Alzheimers Res. Ther. 2015, 7, 33. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2017, 16, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.A.; Kareem, O.; Khushtar, M.; Akbar, M.; Haque, M.R.; Iqubal, A.; Haider, M.F.; Pottoo, F.H.; Abdulla, F.S.; Al-Haidar, M.B.; et al. Neuroinflammation: A Potential Risk for Dementia. Int. J. Mol. Sci. 2022, 23, 616. [Google Scholar] [CrossRef]
- de Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef]
- Szabo, A.; O’Connell, K.S.; Ueland, T.; Sheikh, M.A.; Agartz, I.; Andreou, D.; Aukrust, P.; Boye, B.; Bøen, E.; Drange, O.K.; et al. Increased Circulating IL-18 Levels in Severe Mental Disorders Indicate Systemic Inflammasome Activation. Brain. Behav. Immun. 2022, 99, 299–306. [Google Scholar] [CrossRef]
- Cheng, X.; Wei, Y.; Qian, Z.; Han, L. Autophagy Balances Neuroinflammation in Alzheimer’s Disease. Cell Mol. Neurobiol. in press. [CrossRef] [PubMed]
- Panda, C.; Mahapatra, R.K. Bi-Directional Relationship Between Autophagy and Inflammasomes in Neurodegenerative Disorders. Cell Mol. Neurobiol. 2023, 43, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Katzmarski, N.; Kipnis, J.; Meyer-Luehmann, M. Microglia as a Critical Player in Both Developmental and Late-Life CNS Pathologies. Acta Neuropathol. 2014, 128, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia Control the Spread of Neurotropic Virus Infection via P2Y12 Signalling and Recruit Monocytes through P2Y12-Independent Mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef]
- Herdy, J.R.; Traxler, L.; Agarwal, R.K.; Karbacher, L.; Schlachetzki, J.C.M.; Boehnke, L.; Zangwill, D.; Galasko, D.; Glass, C.K.; Mertens, J.; et al. Increased Post-Mitotic Senescence in Aged Human Neurons Is a Pathological Feature of Alzheimer’s Disease. Cell Stem Cell 2022, 29, 1637–1652.e6. [Google Scholar] [CrossRef] [PubMed]
- McNamara, N.B.; Munro, D.A.D.; Bestard-Cuche, N.; Uyeda, A.; Bogie, J.F.J.; Hoffmann, A.; Holloway, R.K.; Molina-Gonzalez, I.; Askew, K.E.; Mitchell, S.; et al. Microglia Regulate Central Nervous System Myelin Growth and Integrity. Nature 2023, 613, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia Monitor and Protect Neuronal Function through Specialized Somatic Purinergic Junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained Microglial Depletion with CSF1R Inhibitor Impairs Parenchymal Plaque Development in an Alzheimer’s Disease Model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buée, L.; De Strooper, B.; et al. Novel Alzheimer Risk Genes Determine the Microglia Response to Amyloid-β but Not to TAU Pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, K.I.; Bachstetter, A.D.; Colonna, M.; Ginhoux, F.; Holmes, C.; Lamb, B.; Landreth, G.; Lee, D.C.; Low, D.; Lynch, M.A.; et al. Targeting Innate Immunity for Neurodegenerative Disorders of the Central Nervous System. J. Neurochem. 2016, 138, 653–693. [Google Scholar] [CrossRef]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s Disease at Single-Cell Level. Are There Common Patterns in Humans and Mice? J. Exp. Med. 2021, 218, e20202717. [Google Scholar] [CrossRef]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent Advances in Molecular Pathways and Therapeutic Implications Targeting Neuroinflammation for Alzheimer’s Disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α Signalling and Inflammation: Interactions between Old Acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Ennerfelt, H.; Frost, E.L.; Shapiro, D.A.; Holliday, C.; Zengeler, K.E.; Voithofer, G.; Bolte, A.C.; Lammert, C.R.; Kulas, J.A.; Ulland, T.K.; et al. SYK Coordinates Neuroprotective Microglial Responses in Neurodegenerative Disease. Cell 2022, 185, 4135–4152.e22. [Google Scholar] [CrossRef]
- Puntambekar, S.S.; Moutinho, M.; Lin, P.B.-C.; Jadhav, V.; Tumbleson-Brink, D.; Balaji, A.; Benito, M.A.; Xu, G.; Oblak, A.; Lasagna-Reeves, C.A.; et al. CX3CR1 Deficiency Aggravates Amyloid Driven Neuronal Pathology and Cognitive Decline in Alzheimer’s Disease. Mol. Neurodegener. 2022, 17, 47. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Strandberg, O.; Whelan, C.D.; Zetterberg, H.; Blennow, K.; Palmqvist, S.; Stomrud, E.; Mattsson-Carlgren, N.; Hansson, O. Microglial Activation Protects against Accumulation of Tau Aggregates in Nondemented Individuals with Underlying Alzheimer’s Disease Pathology. Nat. Aging 2022, 2, 1138–1144. [Google Scholar] [CrossRef]
- Claes, C.; England, W.E.; Danhash, E.P.; Kiani Shabestari, S.; Jairaman, A.; Chadarevian, J.P.; Hasselmann, J.; Tsai, A.P.; Coburn, M.A.; Sanchez, J.; et al. The P522R Protective Variant of PLCG2 Promotes the Expression of Antigen Presentation Genes by Human Microglia in an Alzheimer’s Disease Mouse Model. Alzheimers Dement. 2022, 18, 1765–1778. [Google Scholar] [CrossRef]
- Castranio, E.L.; Hasel, P.; Haure-Mirande, J.; Ramirez Jimenez, A.V.; Hamilton, B.W.; Kim, R.D.; Glabe, C.G.; Wang, M.; Zhang, B.; Gandy, S.; et al. Microglial INPP5D Limits Plaque Formation and Glial Reactivity in the PSAPP Mouse Model of Alzheimer’s Disease. Alzheimers Dement. in press. [CrossRef]
- Birch, A.M. The Contribution of Astrocytes to Alzheimer’s Disease. Biochem. Soc. Trans. 2014, 42, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bailey, J.T.; Xu, E.; Singh, K.; Lavaert, M.; Link, V.M.; D’Souza, S.; Hafiz, A.; Cao, J.; Cao, G.; et al. Mucosal-Associated Invariant T Cells Restrict Reactive Oxidative Damage and Preserve Meningeal Barrier Integrity and Cognitive Function. Nat. Immunol. 2022, 23, 1714–1725. [Google Scholar] [CrossRef]
- Sasaguri, H.; Hashimoto, S.; Watamura, N.; Sato, K.; Takamura, R.; Nagata, K.; Tsubuki, S.; Ohshima, T.; Yoshiki, A.; Sato, K.; et al. Recent Advances in the Modeling of Alzheimer’s Disease. Front. Neurosci. 2022, 16, 807473. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penke, B.; Szűcs, M.; Bogár, F. New Pathways Identify Novel Drug Targets for the Prevention and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 5383. https://doi.org/10.3390/ijms24065383
Penke B, Szűcs M, Bogár F. New Pathways Identify Novel Drug Targets for the Prevention and Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(6):5383. https://doi.org/10.3390/ijms24065383
Chicago/Turabian StylePenke, Botond, Mária Szűcs, and Ferenc Bogár. 2023. "New Pathways Identify Novel Drug Targets for the Prevention and Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 6: 5383. https://doi.org/10.3390/ijms24065383
APA StylePenke, B., Szűcs, M., & Bogár, F. (2023). New Pathways Identify Novel Drug Targets for the Prevention and Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(6), 5383. https://doi.org/10.3390/ijms24065383