

The Endothelial Glycocalyx as a Target of Excess Soluble Fms-like Tyrosine Kinase-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
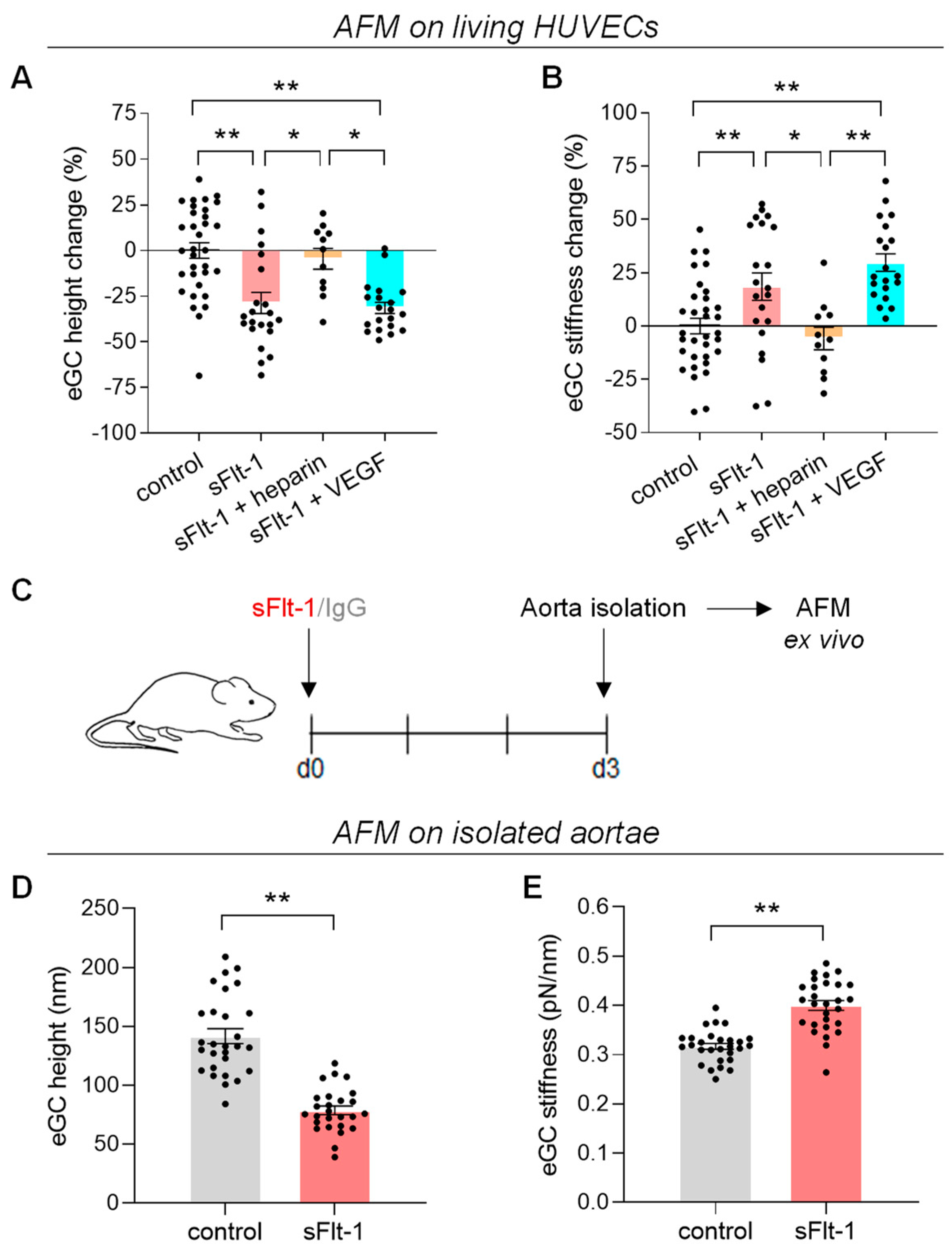
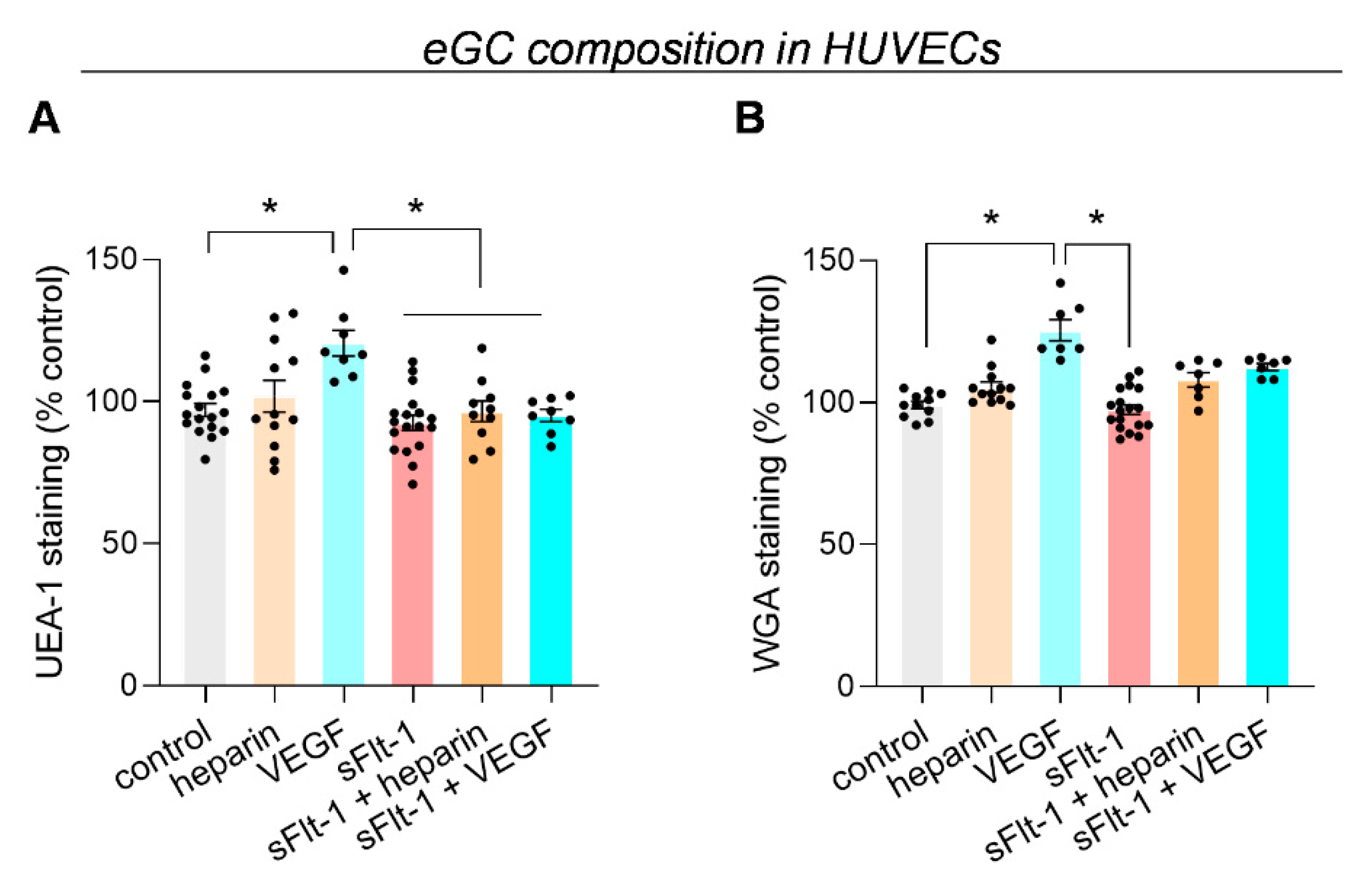
2.1. Excess sFlt-1 Leads to Conformational Changes in the eGC In Vitro and In Vivo
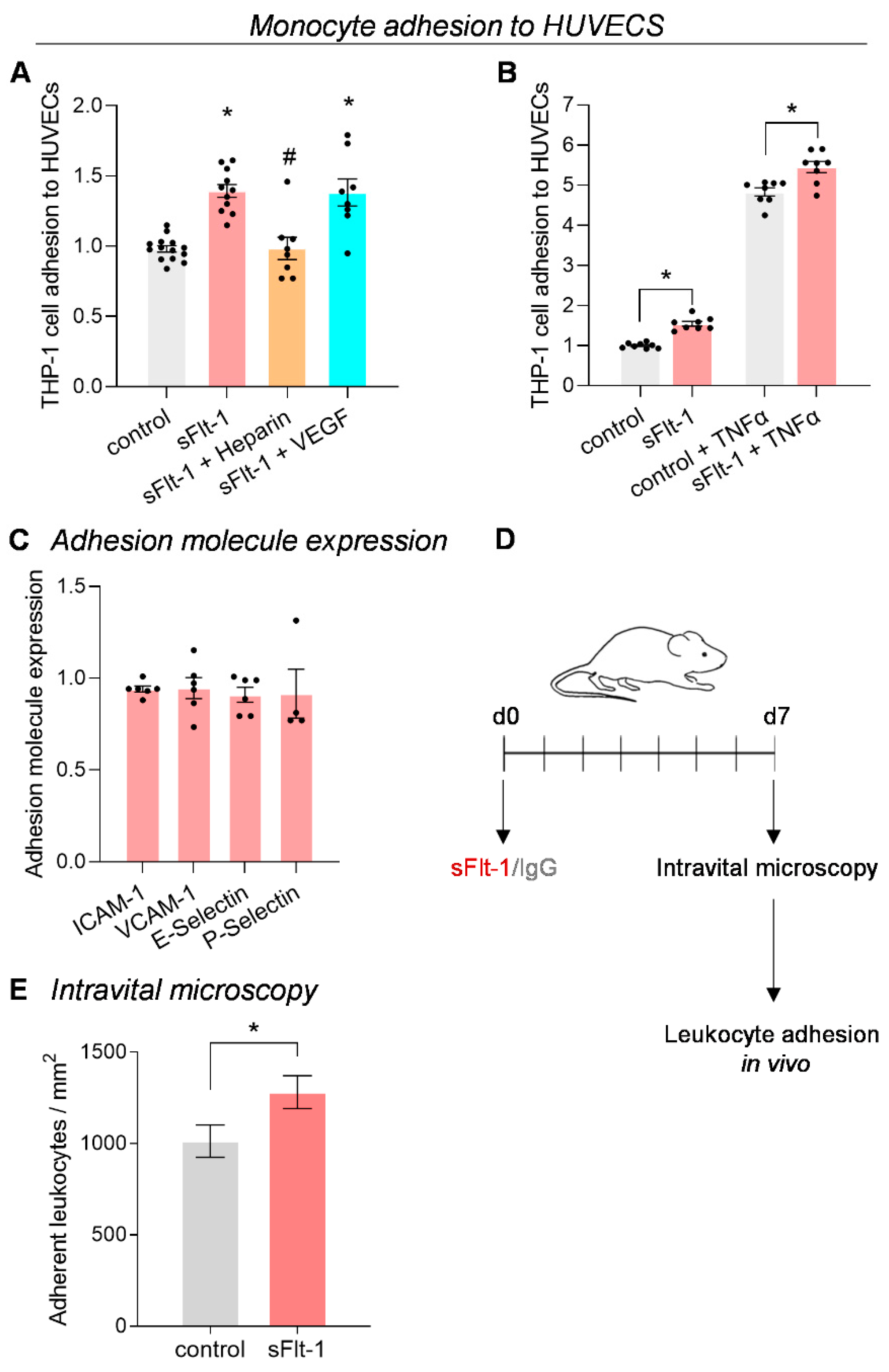
2.2. Increased Monocyte Adhesion under sFlt-1 Treatment
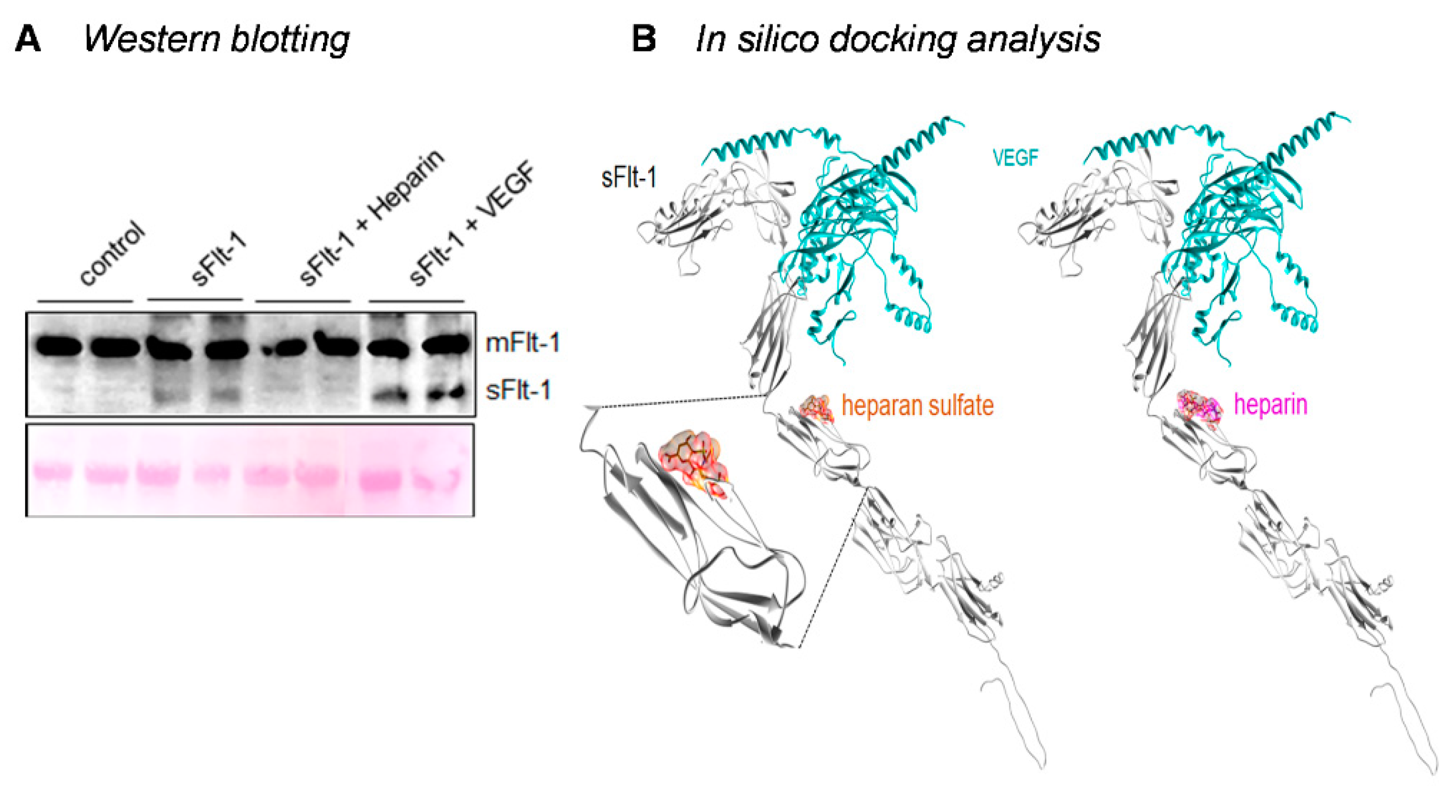
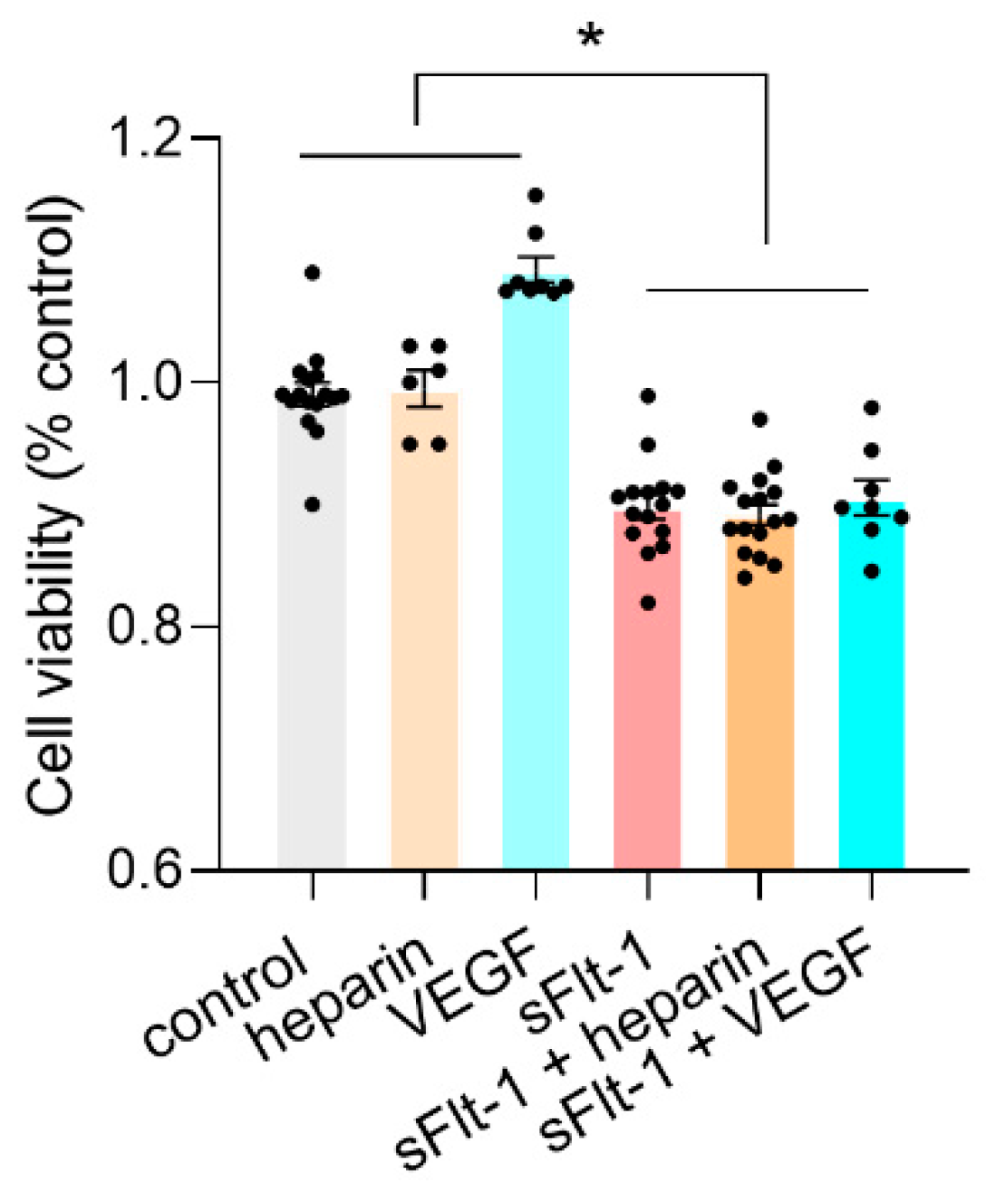
2.3. Binding of sFlt-1 to the eGC and Endothelial Cell Viability
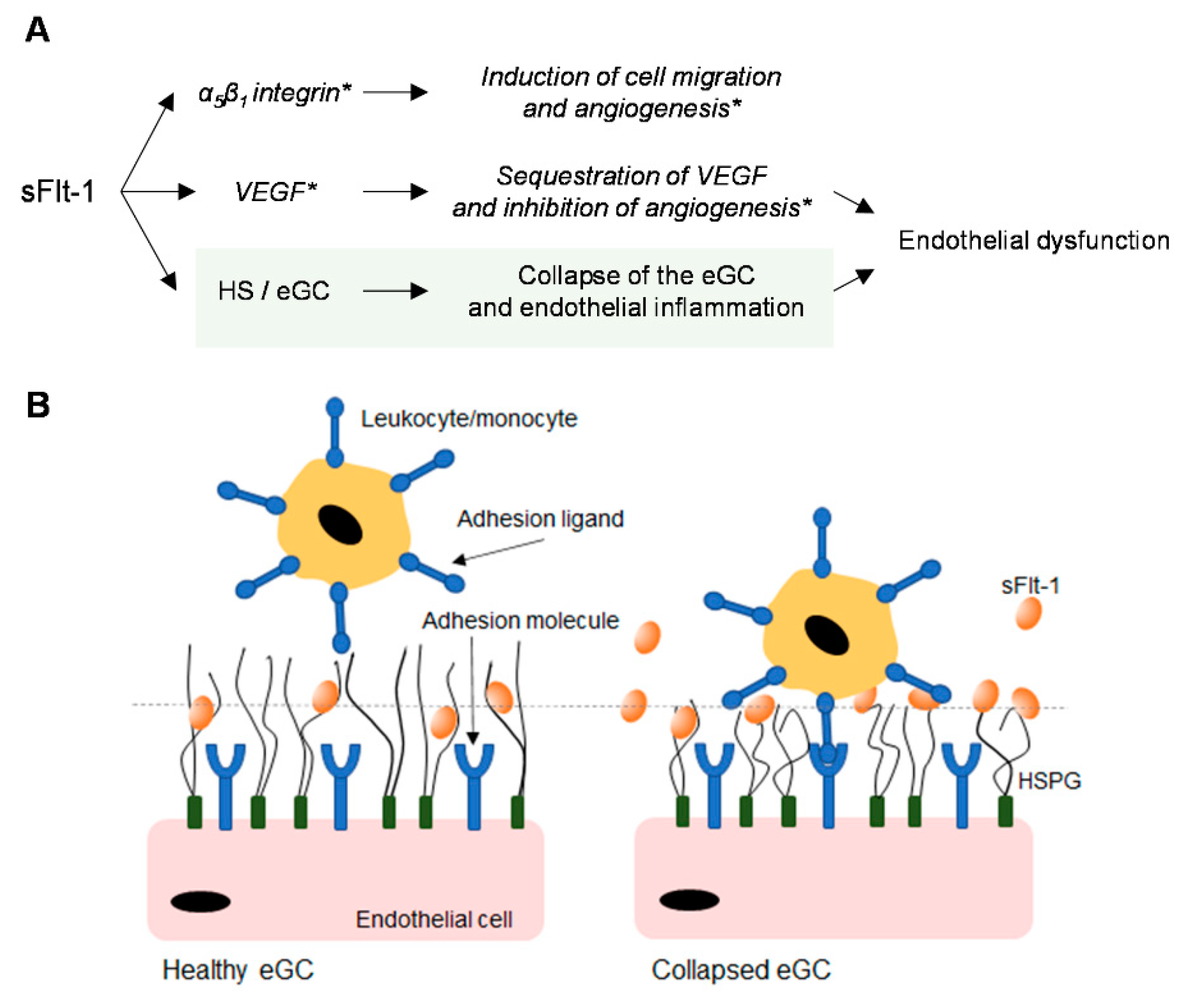
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment Protocols
4.2. eGC Analysis by Atomic Force Microscopy (AFM)
4.3. eGC Analysis by Lectin Staining
4.4. Monocyte-Endothelial Adhesion Assay
4.5. Flow Cytometric Determination of Adhesion Molecules
4.6. Western Blotting and In Silico Docking Analysis
4.7. sFlt-1 Administration In Vivo and Intravital Microscopy
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kendall, R.L.; Thomas, K.A. Inhibition of Vascular Endothelial Cell Growth Factor Activity by an Endogenously Encoded Soluble Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Wang, G.; Thomas, K.A. Identification of a Natural Soluble Form of the Vascular Endothelial Growth Factor Receptor, FLT-1, and Its Heterodimerization with KDR. Biochem. Biophys. Res. Commun. 1996, 226, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Structure and Dual Function of Vascular Endothelial Growth Factor Receptor-1 (Flt-1). Int. J. Biochem. Cell Biol. 2001, 33, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; de Vos, A.M. Crystal Structure at 1.7 Å Resolution of VEGF in Complex with Domain 2 of the Flt-1 Receptor. Cell 1997, 91, 695–704. [Google Scholar] [CrossRef]
- Ma, L.; Wang, X.; Zhang, Z.; Zhou, X.; Chen, A.; Yao, L. Identification of the Ligand-Binding Domain of Human Vascular-Endothelial-Growth-Factor Receptor Flt-1. Biotechnol. Appl. Biochem. 2001, 34, 199–204. [Google Scholar] [CrossRef]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-Length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Lee, S.T. The Fourth Immunoglobulin-like Loop in the Extracellular Domain of FLT-1, a VEGF Receptor, Includes a Major Heparin-Binding Site. Biochem. Biophys. Res. Commun. 1999, 264, 730–734. [Google Scholar] [CrossRef]
- Moore, K.H.; Chapman, H.; George, E.M. Unfractionated Heparin Displaces SFlt-1 from the Placental Extracellular Matrix. Biol. Sex Differ. 2020, 11, 34. [Google Scholar] [CrossRef]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt Overload Damages the Glycocalyx Sodium Barrier of Vascular Endothelium. Pflug. Arch. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Schierke, F.; Wyrwoll, M.J.; Wisdorf, M.; Niedzielski, L.; Maase, M.; Ruck, T.; Meuth, S.G.; Kusche-Vihrog, K. Nanomechanics of the Endothelial Glycocalyx Contribute to Na+—Induced Vascular Inflammation. Sci. Rep. 2017, 7, 46476. [Google Scholar] [CrossRef]
- Fels, J.; Kusche-Vihrog, K. Endothelial Nanomechanics in the Context of Endothelial (Dys)Function and Inflammation. Antioxid. Redox Signal. 2019, 30, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, A.; Peters, W.; Chappell, D.; Kentrup, D.; Reuter, S.; Pavenstädt, H.; Oberleithner, H.; Kümpers, P. Nanomechanics of the Endothelial Glycocalyx in Experimental Sepsis. PLoS ONE 2013, 8, 80905. [Google Scholar] [CrossRef] [PubMed]
- Wewers, T.M.; Schulz, A.; Nolte, I.; Pavenstädt, H.; Brand, M.; di Marco, G.S. Circulating Soluble Fms-like Tyrosine Kinase in Renal Diseases Other than Preeclampsia. J. Am. Soc. Nephrol. 2021, 32, 1853–1863. [Google Scholar] [CrossRef]
- Greco, M.; Palumbo, C.; Sicuro, F.; Lobreglio, G. Soluble Fms-Like Tyrosine Kinase-1 Is A Marker of Endothelial Dysfunction During Sepsis. J. Clin. Med. Res. 2018, 10, 700–706. [Google Scholar] [CrossRef][Green Version]
- Giardini, V.; Carrer, A.; Casati, M.; Contro, E.; Vergani, P.; Gambacorti-Passerini, C. Increased SFLT-1/PlGF Ratio in COVID-19: A Novel Link to Angiotensin II-mediated Endothelial Dysfunction. Am. J. Hematol. 2020, 95, E188. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pacheco, J.A.; Torres-Torres, J.; Martinez-Portilla, R.J.; Solis-Paredes, J.M.; Estrada-Gutierrez, G.; Mateu-Rogell, P.; Nares-Torices, M.A.; Lopez-Marenco, M.E.; Escobedo-Segura, K.R.; Posadas-Nava, A.; et al. SFlt-1 Is an Independent Predictor of Adverse Maternal Outcomes in Women With SARS-CoV-2 Infection and Hypertensive Disorders of Pregnancy. Front. Med. 2022, 9, 894633. [Google Scholar] [CrossRef]
- Ziganshina, M.M.; Yarotskaya, E.L.; Bovin, N.V.; Sukhikh, G.T. Endothelial Dysfunction as a Consequence of Endothelial Glycocalyx Damage: A Role in the Pathogenesis of Preeclampsia. In Endothelial Dysfunction—Old Concepts and New Challenges; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Padberg, J.S.; Wiesinger, A.; di Marco, G.S.; Reuter, S.; Grabner, A.; Kentrup, D.; Lukasz, A.; Oberleithner, H.; Pavenstädt, H.; Brand, M.; et al. Damage of the Endothelial Glycocalyx in Chronic Kidney Disease. Atherosclerosis 2014, 234, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Weissgerber, T.L.; Garcia-Valencia, O.; Milic, N.M.; Codsi, E.; Cubro, H.; Nath, M.C.; White, W.M.; Nath, K.A.; Garovic, V.D. Early Onset Preeclampsia Is Associated with Glycocalyx Degradation and Reduced Microvascular Perfusion. J. Am. Heart Assoc. 2019, 8, e010647. [Google Scholar] [CrossRef]
- Sela, S.; Natanson-Yaron, S.; Zcharia, E.; Vlodavsky, I.; Yagel, S.; Keshet, E. Local Retention versus Systemic Release of Soluble VEGF Receptor-1 Are Mediated by Heparin-Binding and Regulated by Heparanase. Circ. Res. 2011, 108, 1063–1070. [Google Scholar] [CrossRef]
- Dragovich, M.A.; Genemaras, K.; Dailey, H.L.; Jedlicka, S.; Frank Zhang, X. Dual Regulation of L-Selectin-Mediated Leukocyte Adhesion by Endothelial Surface Glycocalyx. Cell. Mol. Bioeng. 2017, 10, 102–113. [Google Scholar] [CrossRef]
- Pollmann, S.; Scharnetzki, D.; Manikowski, D.; Lenders, M.; Brand, E. Endothelial Dysfunction in Fabry Disease Is Related to Glycocalyx Degradation. Front. Immunol. 2021, 12, 789142. [Google Scholar] [CrossRef] [PubMed]
- Colotti, G.; Failla, C.M.; Lacal, P.M.; Ungarelli, M.; Ruffini, F.; di Micco, P.; Orecchia, A.; Morea, V. Neuropilin-1 Is Required for Endothelial Cell Adhesion to Soluble Vascular Endothelial Growth Factor Receptor 1. FEBS J. 2022, 289, 183–198. [Google Scholar] [CrossRef]
- Jin, J.; Sison, K.; Li, C.; Tian, R.; Wnuk, M.; Sung, H.K.; Jeansson, M.; Zhang, C.; Tucholska, M.; Jones, N.; et al. Soluble FLT1 Binds Lipid Microdomains in Podocytes to Control Cell Morphology and Glomerular Barrier Function. Cell 2012, 151, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Lacal, P.M.; Schietroma, C.; Morea, V.; Zambruno, G.; Failla, C.M. Vascular Endothelial Growth Factor Receptor-1 Is Deposited in the Extracellular Matrix by Endothelial Cells and Is a Ligand for the A5β1 Integrin. J. Cell Sci. 2003, 116, 3479–3489. [Google Scholar] [CrossRef]
- Soro, S.; Orecchia, A.; Morbidelli, L.; Lacal, P.M.; Morea, V.; Ballmer-Hofer, K.; Ruffini, F.; Ziche, M.; D’Atri, S.; Zambruno, G.; et al. A Proangiogenic Peptide Derived from Vascular Endothelial Growth Factor Receptor-1 Acts through Alpha5beta1 Integrin. Blood 2008, 111, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Cindrova-Davies, T.; Sanders, D.A.; Burton, G.J.; Charnock-Jones, D.S. Soluble FLT1 Sensitizes Endothelial Cells to Inflammatory Cytokines by Antagonizing VEGF Receptor-Mediated Signalling. Cardiovasc. Res. 2011, 89, 671–679. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Hynes, R.O. Enrichment of Extracellular Matrix Proteins from Tissues and Digestion into Peptides for Mass Spectrometry Analysis. J. Vis. Exp. 2015, 101, e53057. [Google Scholar] [CrossRef]
- Vlahu, C.A.; Lemkes, B.A.; Struijk, D.G.; Koopman, M.G.; Krediet, R.T.; Vink, H. Damage of the Endothelial Glycocalyx in Dialysis Patients. J. Am. Soc. Nephrol. 2012, 23, 1900–1908. [Google Scholar] [CrossRef]
- Hesse, B.; Rovas, A.; Buscher, K.; Kusche-Vihrog, K.; Brand, M.; di Marco, G.S.; Kielstein, J.T.; Pavenstädt, H.; Linke, W.A.; Nofer, J.R.; et al. Symmetric Dimethylarginine in Dysfunctional High-Density Lipoprotein Mediates Endothelial Glycocalyx Breakdown in Chronic Kidney Disease. Kidney Int. 2020, 97, 502–515. [Google Scholar] [CrossRef]
- Cosgun, Z.C.; Fels, B.; Kusche-Vihrog, K. Nanomechanics of the Endothelial Glycocalyx: From Structure to Function. Am. J. Pathol. 2020, 190, 732–741. [Google Scholar] [CrossRef]
- Searle, J.; Mockel, M.; Gwosc, S.; Datwyler, S.A.; Qadri, F.; Albert, G.I.; Holert, F.; Isbruch, A.; Klug, L.; Muller, D.N.; et al. Heparin Strongly Induces Soluble Fms-like Tyrosine Kinase 1 Release in Vivo and in Vitro-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2972–2974. [Google Scholar] [CrossRef]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.C.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal Avascularity Is Due to Soluble VEGF Receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef]
- Gandhi, J.G.; Koch, D.L.; Paszek, M.J. Equilibrium Modeling of the Mechanics and Structure of the Cancer Glycocalyx. Biophys. J. 2019, 116, 694–708. [Google Scholar] [CrossRef]
- Siren, E.M.J.; Chapanian, R.; Constantinescu, I.; Brooks, D.E.; Kizhakkedathu, J.N. Oncotically Driven Control over Glycocalyx Dimension for Cell Surface Engineering and Protein Binding in the Longitudinal Direction. Sci. Rep. 2018, 8, 7581. [Google Scholar] [CrossRef]
- Delgadillo, L.F.; Lomakina, E.B.; Kuebel, J.; Waugh, R.E. Changes in Endothelial Glycocalyx Layer Protective Ability after Inflammatory Stimulus. Am. J. Physiol. Cell Physiol. 2021, 320, C216–C224. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.; Gossen, M.; Lendlein, A.; Jung, F. Venous and Arterial Endothelial Cells from Human Umbilical Cords: Potential Cell Sources for Cardiovascular Research. Int. J. Mol. Sci. 2021, 22, 978. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves Problems That Endothelial Cells Do Not Know Exist. Trends Pharmacol. Sci. 2017, 38, 322. [Google Scholar] [CrossRef]
- di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; König, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstädt, H.; Brand, M. The Soluble VEGF Receptor SFlt1 Contributes to Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The Vascular Marker Soluble Fms-Like Tyrosine Kinase 1 Is Associated with Disease Severity and Adverse Outcomes in Chronic Heart Failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Kentrup, D.; Reuter, S.; Mayer, A.B.; Golle, L.; Tiemann, K.; Fobker, M.; Engelbertz, C.; Breithardt, G.; Brand, E.; et al. Soluble Flt-1 Links Microvascular Disease with Heart Failure in CKD. Basic Res. Cardiol. 2015, 110, 30. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess Placental Soluble Fms-like Tyrosine Kinase 1 (SFlt1) May Contribute to Endothelial Dysfunction Hypertension, and Proteinuria in Preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Wikström, A.K.; Larsson, A.; Eriksson, U.J.; Nash, P.; Nordén-Lindeberg, S.; Olovsson, M. Placental Growth Factor and Soluble FMS-like Tyrosine Kinase-1 in Early-Onset and Late-Onset Preeclampsia. Obstet. Gynecol. 2007, 109, 1368–1374. [Google Scholar] [CrossRef]
- Wewers, T.M.; Mayer, A.B.; Pfleiderer, A.; Beul, K.; Schmidt, R.; Heitplatz, B.; van Marck, V.; Nolte, I.; Pavenstädt, H.; Reuter, S.; et al. Increased Soluble Fms-like Tyrosine Kinase 1 after Ischemia Reperfusion Contributes to Adverse Clinical Outcomes Following Kidney Transplantation. Kidney Int. 2019, 95, 1091–1102. [Google Scholar] [CrossRef]
- Hagmann, H.; Bossung, V.; Ali Belaidi, A.; Fridman, A.; Karumanchi, S.A.; Thadhani, R.; Schermer, B.; Mallmann, P.; Schwarz, G.; Benzing, T.; et al. Low-Molecular Weight Heparin Increases Circulating SFlt-1 Levels and Enhances Urinary Elimination. PLoS ONE 2014, 9, e85258. [Google Scholar] [CrossRef]
- Kornacki, J.; Wirstlein, P.; Wender-Ozegowska, E. Serum Levels of Soluble FMS-like Tyrosine Kinase 1 and Endothelial Glycocalyx Components in Early- and Late-Onset Preeclampsia. J. Matern. Fetal Neonatal Med. 2021, 32, 7466–7470. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, I.; Swirski, F.K.; Robbins, C.S. Monocyte Fate in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.J.; Manning, J.E.; Bandak, T.M.; Ratau, M.C.; Hanser, K.R.; Silverstein, S.C. Endothelial Cell Cytosolic Free Calcium Regulates Neutrophil Migration across Monolayers of Endothelial Cells. J. Cell Biol. 1993, 120, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Eiermann, D.F.; Johnson, C.E.; Haskill, J.S. Human Monocyte Inflammatory Mediator Gene Expression Is Selecively Regulated by Adherence Substrates. J. Immunol. 1989, 142, 1970–1976. [Google Scholar] [CrossRef]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2020, 10, 1568. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.D.; Zsengellér, Z.K.; Khankin, E.V.; Lo, A.S.; Rajakumar, A.; DuPont, J.J.; McCurley, A.; Moss, M.E.; Zhang, D.; Clark, C.D.; et al. Soluble FMS-like Tyrosine Kinase 1 Promotes Angiotensin II Sensitivity in Preeclampsia. J. Clin. Investig. 2016, 126, 2561–2574. [Google Scholar] [CrossRef] [PubMed]
- Hashambhoy, Y.L.; Chappell, J.C.; Peirce, S.M.; Bautch, V.L.; mac Gabhann, F. Computational Modeling of Interacting VEGF and Soluble VEGF Receptor Concentration Gradients. Front. Physiol. 2011, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, V.A.; Buhimschi, I.A.; Lockwood, C.J.; Paidas, M.J.; Dulay, A.T.; Ramma, W.; Abdel-Razeq, S.S.; Zhao, G.; Ahmad, S.; Ahmed, A.; et al. Heparin Elevates Circulating Soluble Fms-like Tyrosine Kinase-1 Immunoreactivity in Pregnant Women Receiving Anticoagulation Therapy. Circulation 2011, 124, 2543–2553. [Google Scholar] [CrossRef]
- Geislinger, T.M.; Franke, T. Hydrodynamic Lift of Vesicles and Red Blood Cells in Flow—From Fåhræus & Lindqvist to Microfluidic Cell Sorting. Adv. Colloid Interface Sci. 2014, 208, 161–176. [Google Scholar] [CrossRef]
- Davies, H.S.; Débarre, D.; El Amri, N.; Verdier, C.; Richter, R.P.; Bureau, L. Elastohydrodynamic Lift at a Soft Wall. Phys. Rev. Lett. 2018, 120, 198001. [Google Scholar] [CrossRef]
- Peters, W.; Kusche-Vihrog, K.; Oberleithner, H.; Schillers, H. Cystic Fibrosis Transmembrane Conductance Regulator Is Involved in Polyphenol-Induced Swelling of the Endothelial Glycocalyx. Nanomedicine 2015, 11, 1521–1530. [Google Scholar] [CrossRef][Green Version]
- Bastounis, E.E.; Yeh, Y.T.; Theriot, J.A. Subendothelial Stiffness Alters Endothelial Cell Traction Force Generation While Exerting a Minimal Effect on the Transcriptome. Sci. Rep. 2019, 9, 18209. [Google Scholar] [CrossRef]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in Vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Schulz, A.; Drost, C.C.; Hesse, B.; Beul, K.; Brand, M.; Di Marco, G.S. The Soluble Fms-like Tyrosine Kinase-1 Contributes to Structural and Functional Changes in Endothelial Cells in Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 16059. [Google Scholar] [CrossRef]
- Calzada, M.J.; Annis, D.S.; Zeng, B.; Marcinkiewicz, C.; Banas, B.; Lawler, J.; Mosher, D.F.; Roberts, D.D. Identification of Novel Β1 Integrin Binding Sites in the Type 1 and Type 2 Repeats of Thrombospondin-1. J. Biol. Chem. 2004, 279, 41734–41743. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Schofield, N.R.; Mostafavi-Pour, Z.; Green, L.J.; Garratt, A.N.; Mould, A.P.; Humphries, M.J. Dual Functionality of the Anti-Β1 Integrin Antibody, 12G10, Exemplifies Agonistic Signalling from the Ligand Binding Pocket of Integrin Adhesion Receptors. J. Biol. Chem. 2005, 280, 10234–10243. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.; Jeggle, P.; Kusche-Vihrog, K.; Oberleithner, H. Cortical Actin Nanodynamics Determines Nitric Oxide Release in Vascular Endothelium. PLoS ONE 2012, 7, 9444. [Google Scholar] [CrossRef]
- Golle, L.; Gerth, H.U.; Beul, K.; Heitplatz, B.; Barth, P.; Fobker, M.; Pavenstädt, H.; di Marco, G.S.; Brand, M. Bone Marrow-Derived Cells and Their Conditioned Medium Induce Microvascular Repair in Uremic Rats by Stimulation of Endogenous Repair Mechanisms. Sci. Rep. 2017, 7, 9444. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Dustin Schaeffer, R.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Stadtmann, A.; Block, H.; Volmering, S.; Abram, C.; Sohlbach, C.; Boras, M.; Lowell, C.A.; Zarbock, A. Cross-Talk between Shp1 and PIPKIγ Controls Leukocyte Recruitment. J. Immunol. 2015, 195, 1152–1161. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, A.; Drost, C.C.; Hesse, B.; Beul, K.; Boeckel, G.R.; Lukasz, A.; Pavenstädt, H.; Brand, M.; Di Marco, G.S. The Endothelial Glycocalyx as a Target of Excess Soluble Fms-like Tyrosine Kinase-1. Int. J. Mol. Sci. 2023, 24, 5380. https://doi.org/10.3390/ijms24065380
Schulz A, Drost CC, Hesse B, Beul K, Boeckel GR, Lukasz A, Pavenstädt H, Brand M, Di Marco GS. The Endothelial Glycocalyx as a Target of Excess Soluble Fms-like Tyrosine Kinase-1. International Journal of Molecular Sciences. 2023; 24(6):5380. https://doi.org/10.3390/ijms24065380
Chicago/Turabian StyleSchulz, Annika, Carolin C. Drost, Bettina Hesse, Katrin Beul, Göran R. Boeckel, Alexander Lukasz, Hermann Pavenstädt, Marcus Brand, and Giovana S. Di Marco. 2023. "The Endothelial Glycocalyx as a Target of Excess Soluble Fms-like Tyrosine Kinase-1" International Journal of Molecular Sciences 24, no. 6: 5380. https://doi.org/10.3390/ijms24065380
APA StyleSchulz, A., Drost, C. C., Hesse, B., Beul, K., Boeckel, G. R., Lukasz, A., Pavenstädt, H., Brand, M., & Di Marco, G. S. (2023). The Endothelial Glycocalyx as a Target of Excess Soluble Fms-like Tyrosine Kinase-1. International Journal of Molecular Sciences, 24(6), 5380. https://doi.org/10.3390/ijms24065380