
Culture Conditions for Human Induced Pluripotent Stem Cell-Derived Schwann Cells: A Two-Centre Study

 , , and
, , and
Abstract
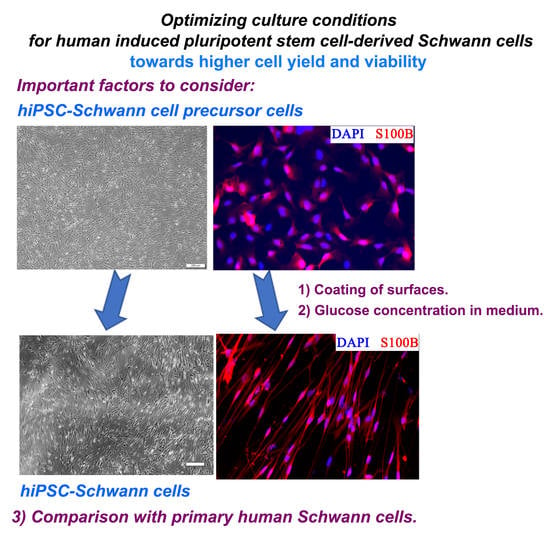
1. Introduction
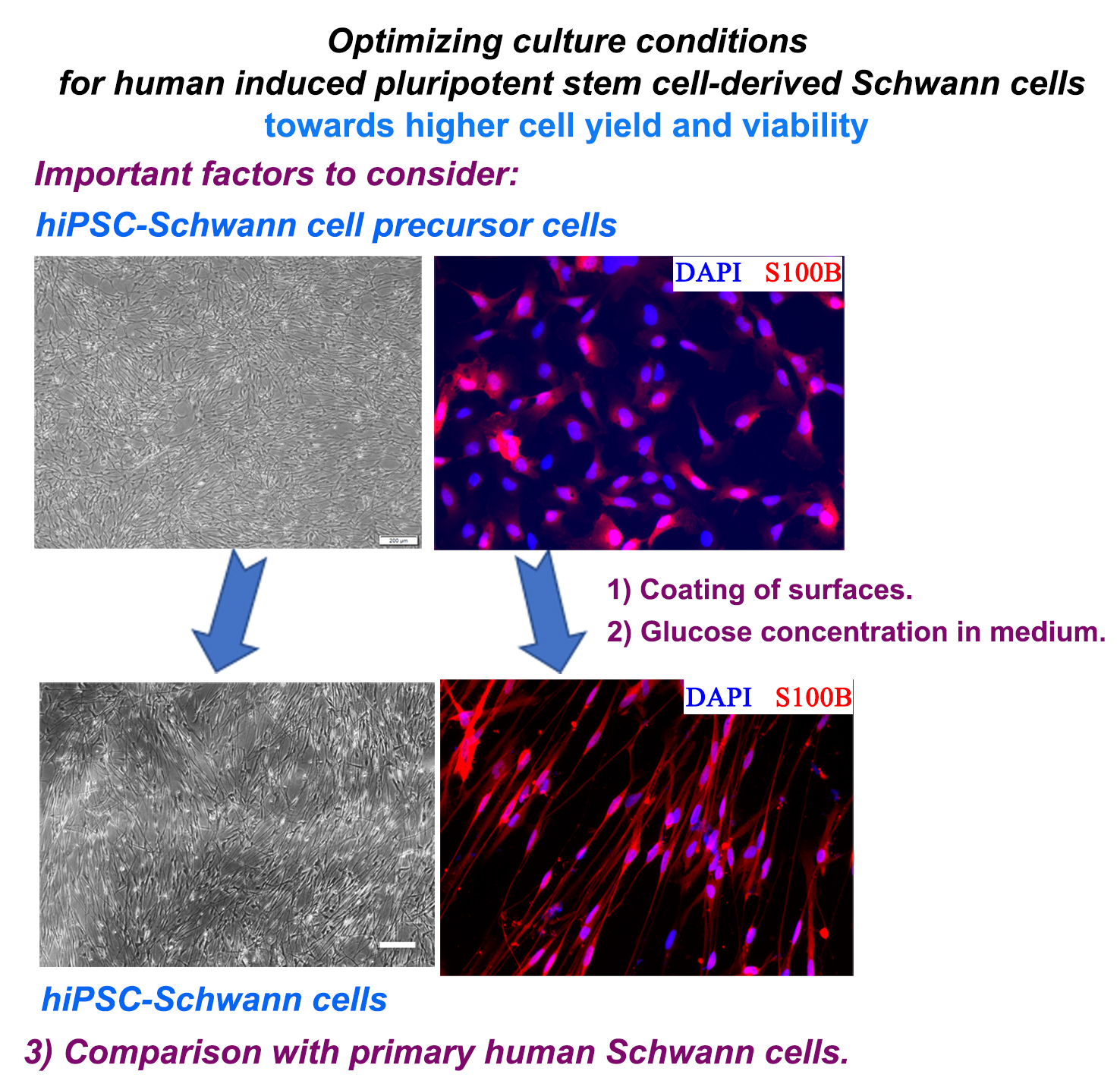
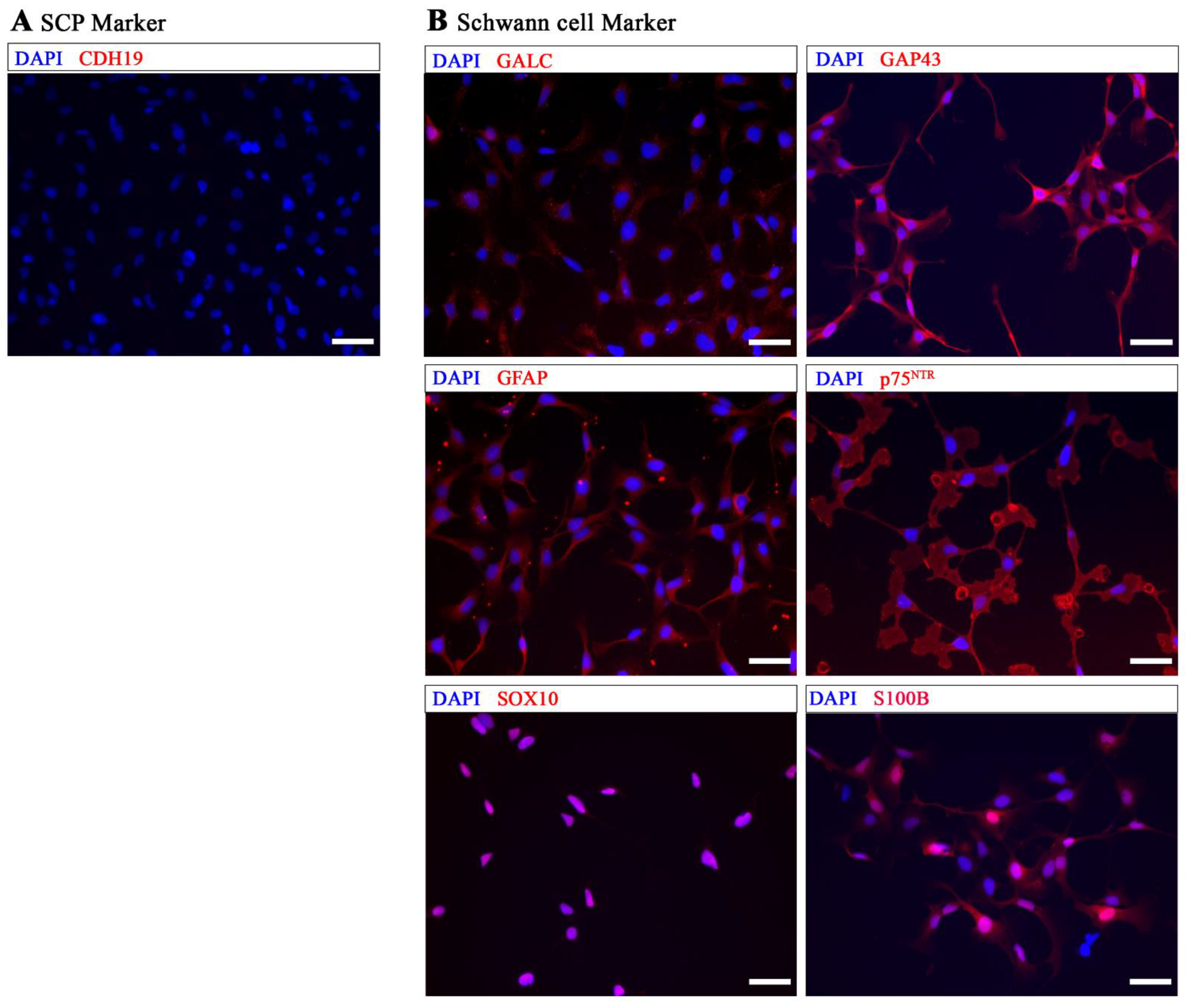
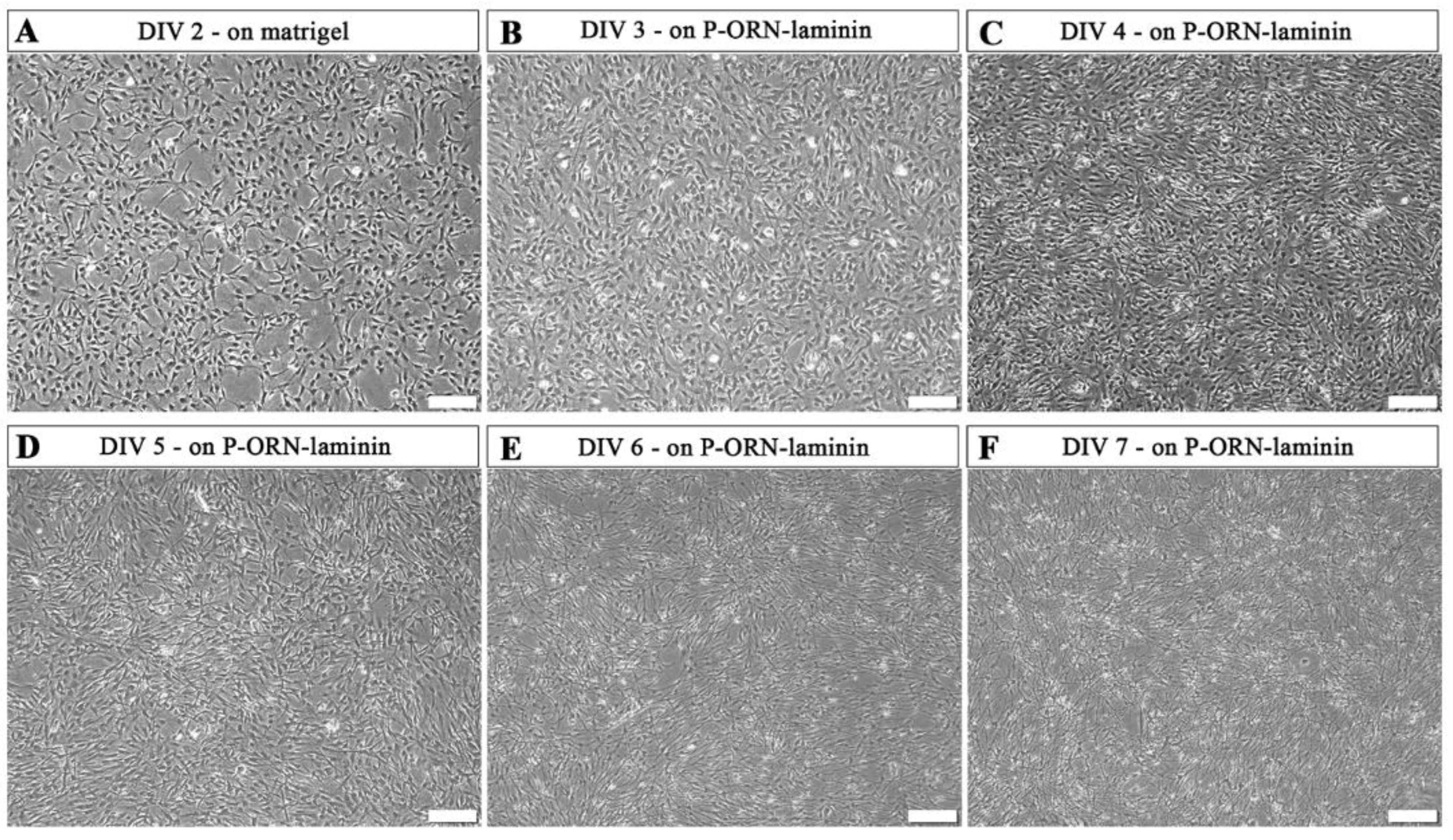
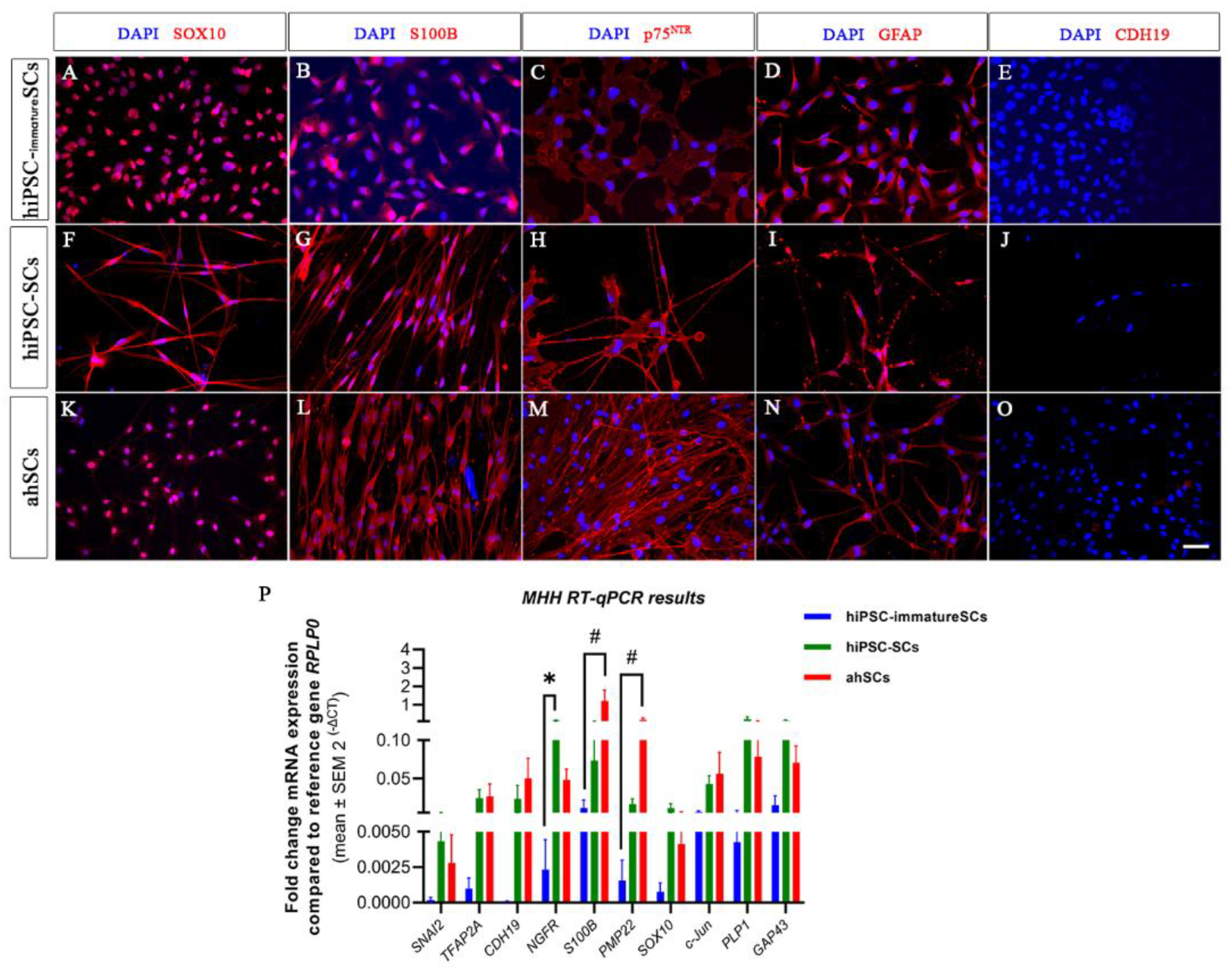
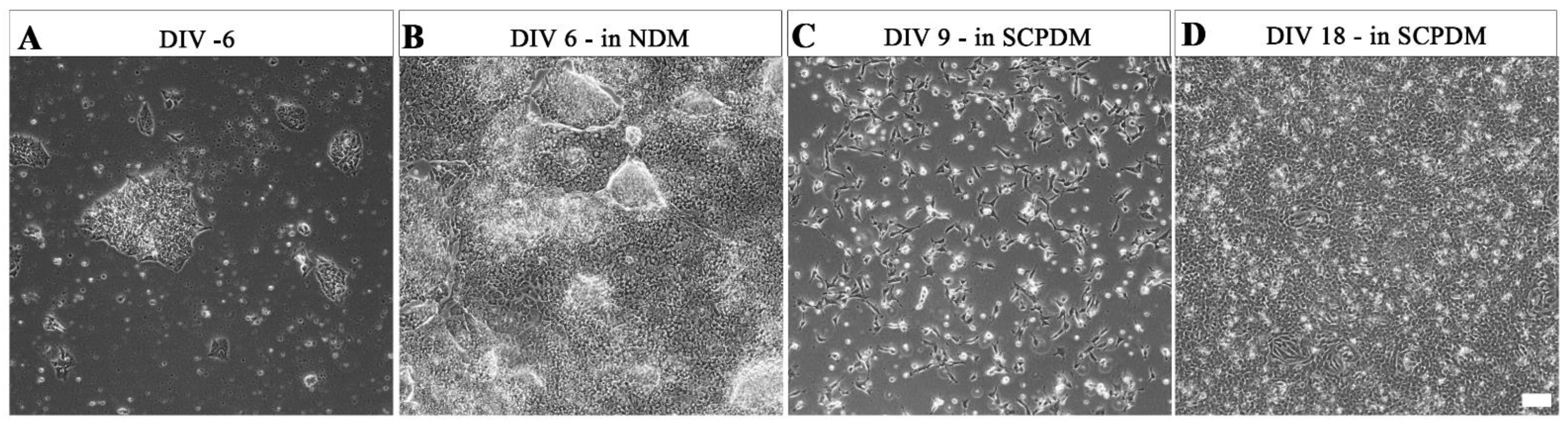
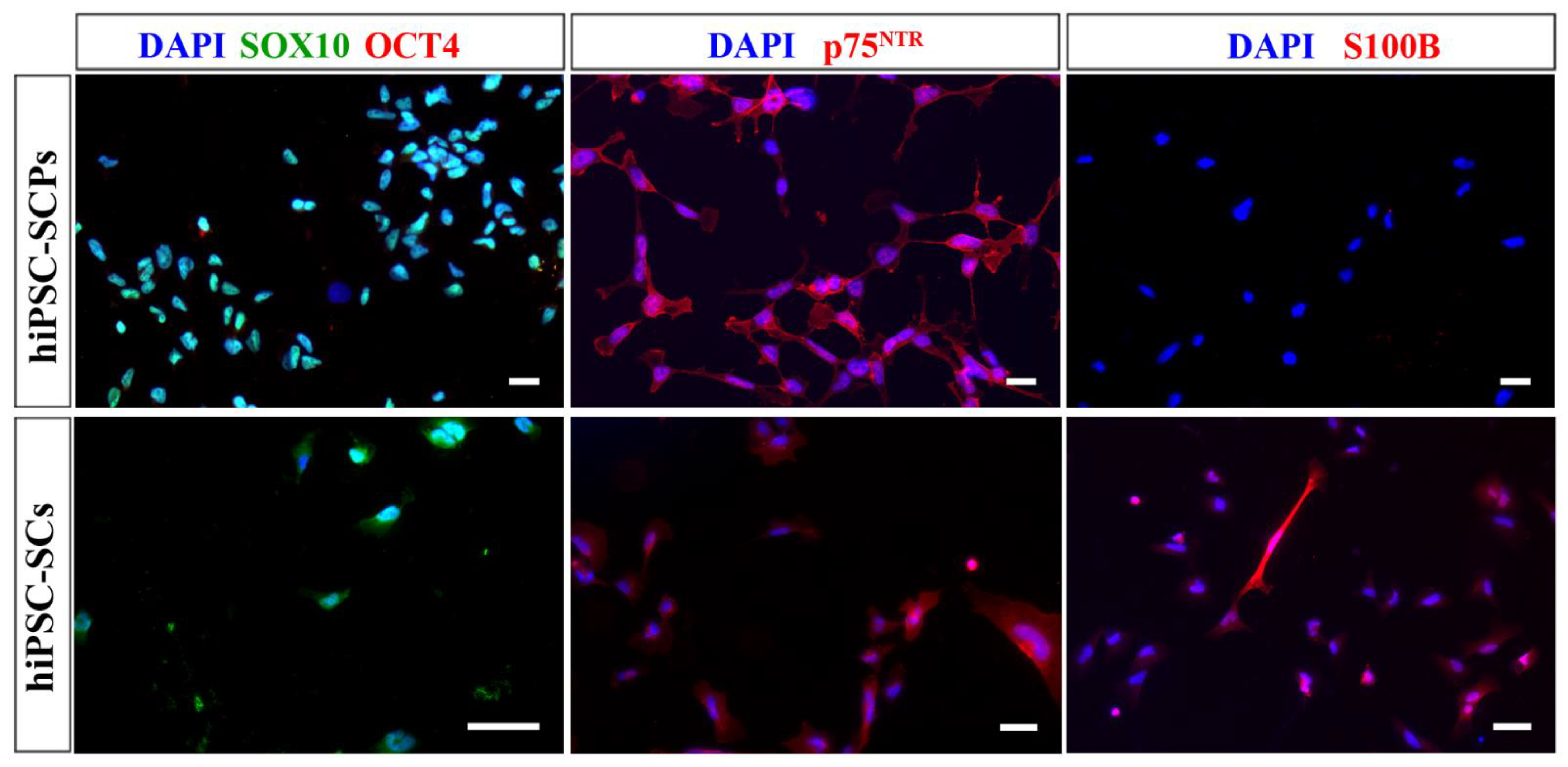
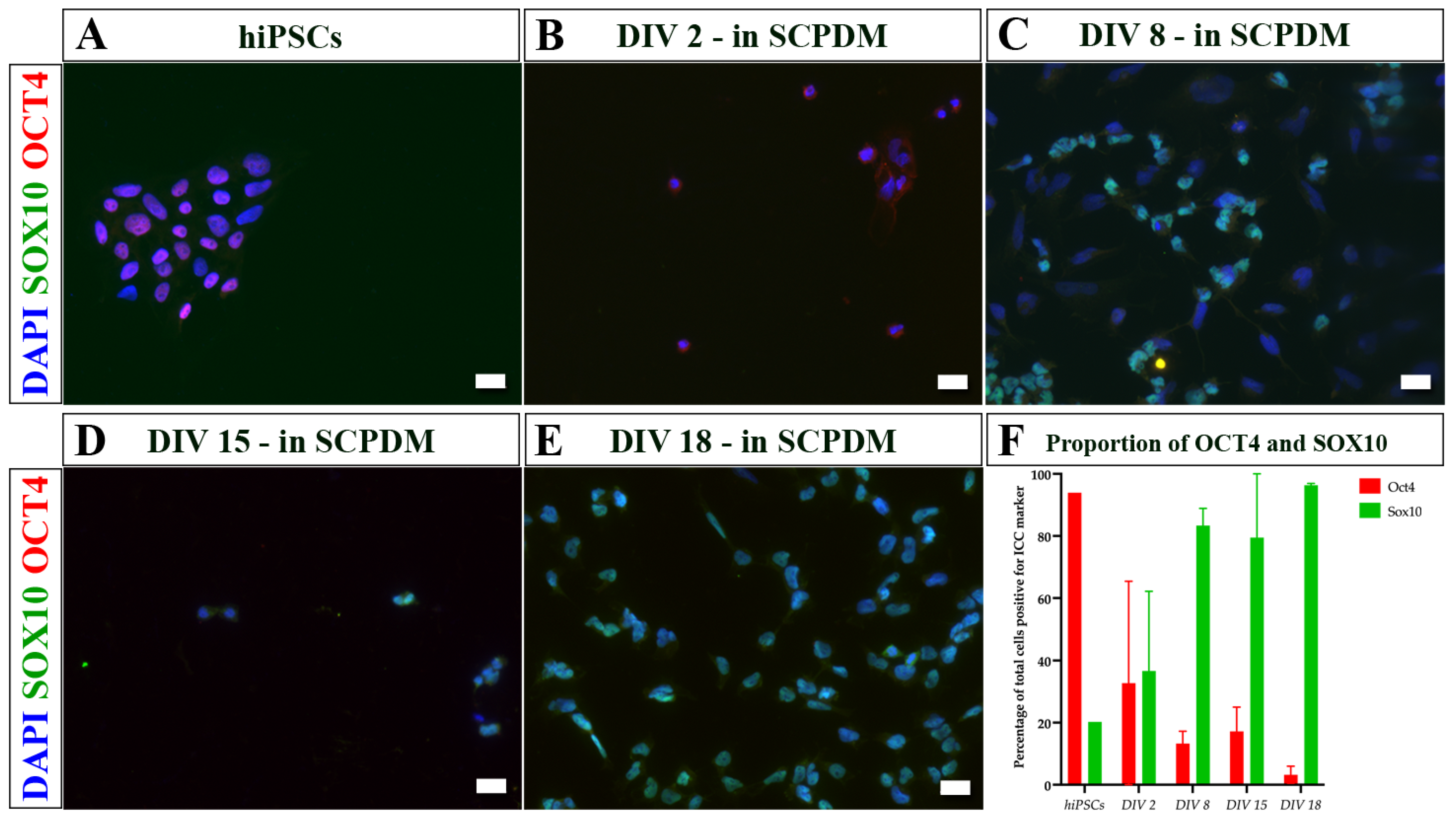
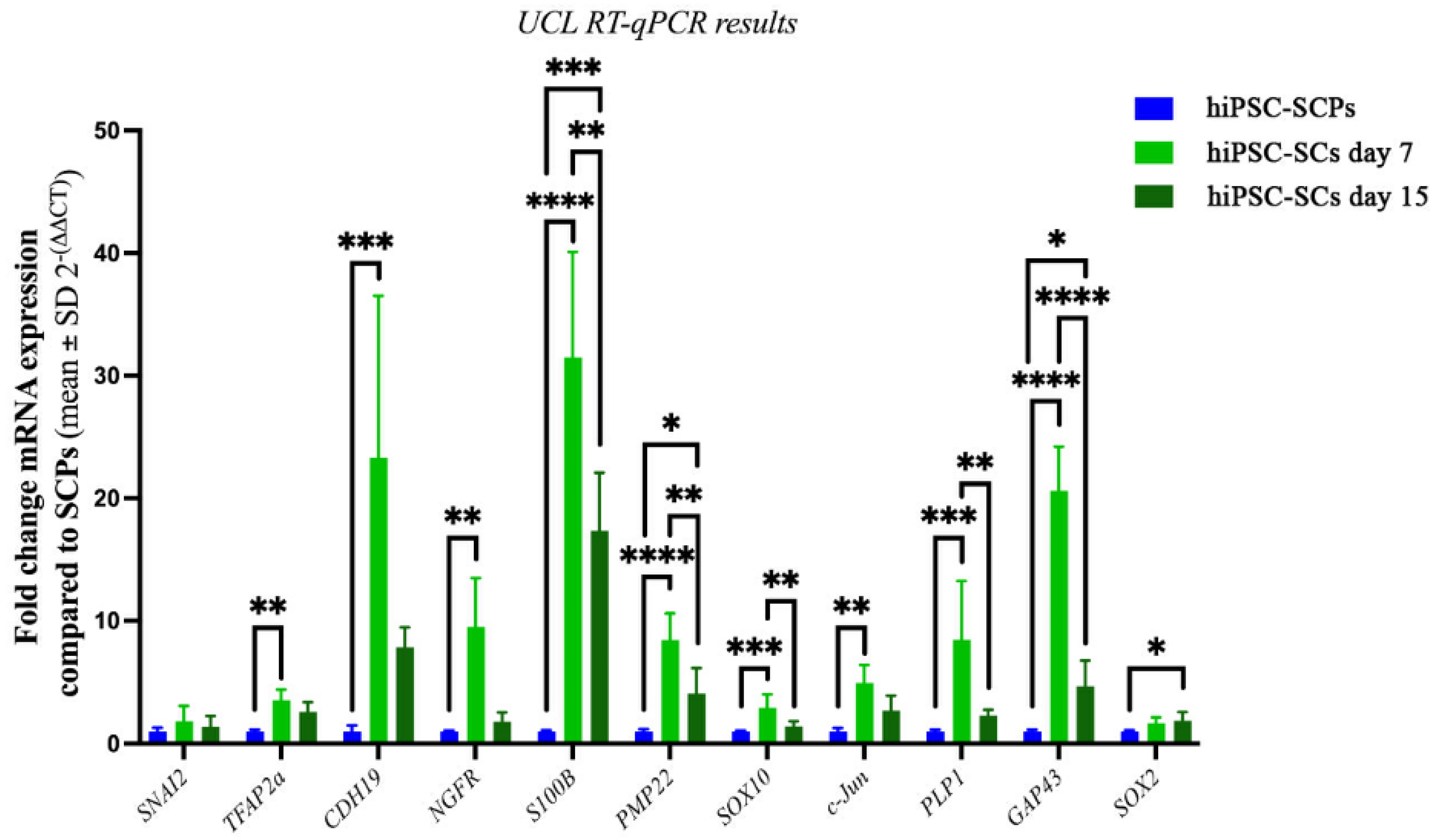
2. Results
3. Discussion
4. Materials and Methods
4.1. Culturing of Human Induced Pluripotent Stem Cells
4.1.1. By the Hannover Medical School Team-MHH
4.1.2. By the University College London Team-UCL
4.2. Culturing of Primary Adult Human Schwann Cells—Both MHH and UCL
4.3. Differentiation from hiPSCs to hiPSC-SCPs or hiPSC-immatureSCs
4.3.1. By the Hannover Medical School Team—MHH
4.3.2. By the University College London Team—UCL
4.4. Differentiation from hiPSC-SCPs or hiPSC-immatureSCs to hiPSC-SCs
4.4.1. By the Hannover Medical School Team—MHH
4.4.2. By the University College London Team—UCL
4.5. Immunocytochemistry
4.5.1. By the Hannover Medical School Team—MHH
4.5.2. By the University College London Team—UCL
4.6. RNA Extraction and Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (Real-Time RT-qPCR)
4.6.1. By the Hannover Medical School Team—MHH
4.6.2. By the University College Team—UCL
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Powell, R.; Phillips, J.B. Engineered Tissues Made from Human iPSC-Derived Schwann Cells for Investigating Peripheral Nerve Regeneration In Vitro. In In Vitro Models for Stem Cell Therapy: Methods and Protocols; Stock, P., Christ, B., Eds.; Springer US: New York, NY, USA, 2021; Volume 2269, pp. 245–254. [Google Scholar]
- Varadarajan, S.G.; Hunyara, J.L.; Hamilton, N.R.; Kolodkin, A.L.; Huberman, A.D. Central nervous system regeneration. Cell 2022, 185, 77–94. [Google Scholar] [CrossRef]
- Cattin, A.-L.; Lloyd, A.C. The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol. 2016, 39, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Stierli, S.; Imperatore, V.; Lloyd, A.C. Schwann cell plasticity-roles in tissue homeostasis, regeneration, and disease. Glia 2019, 67, 2203–2215. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Stierli, S.; Napoli, I.; White, I.J.; Cattin, A.-L.; Cabrejos, A.M.; Calavia, N.G.; Malong, L.; Ribeiro, S.; Nihouarn, J.; Williams, R.; et al. The regulation of the homeostasis and regeneration of peripheral nerve is distinct from the CNS and independent of a stem cell population. Development 2018, 145, dev170316. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. c-Jun Reprograms Schwann Cells of Injured Nerves to Generate a Repair Cell Essential for Regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef]
- Wilcox, M.; Gregory, H.; Powell, R.; Quick, T.J.; Phillips, J.B. Strategies for Peripheral Nerve Repair. Curr. Tissue Microenviron. Rep. 2020, 1, 49–59. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Chapter 39—Signaling Pathways that Regulate Glial Development and Early Migration—Schwann Cells. In Patterning and Cell Type Specification in the Developing CNS and PNS, 2nd ed.; Academic Press, ELSEVIER: Amsterdam, The Netherlands, 2020; pp. 953–975. [Google Scholar]
- Wagstaff, L.J.; A Gomez-Sanchez, J.A.; Fazal, S.V.; Otto, G.W.; Kilpatrick, A.M.; Michael, K.; Wong, L.Y.N.; Ma, K.H.; Turmaine, M.; Svaren, J.; et al. Failures of nerve regeneration caused by aging or chronic denervation are rescued by restoring Schwann cell c-Jun. eLife 2021, 10, e62232. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Belfiore, L.; Chu, T.-H.; Fleming, T.; Midha, R.; Biernaskie, J.; Schuurmans, C. Insights Into the Role and Potential of Schwann Cells for Peripheral Nerve Repair From Studies of Development and Injury. Front. Mol. Neurosci. 2021, 13, 608442. [Google Scholar] [CrossRef]
- López-Cebral, R.; Silva-Correia, J.S.; Reis, R.L.; Silva, T.H.; Oliveira, J.M. Peripheral Nerve Injury: Current Challenges, Conventional Treatment Approaches, and New Trends in Biomaterials-Based Regenerative Strategies. ACS Biomater. Sci. Eng. 2017, 3, 3098–3122. [Google Scholar] [CrossRef]
- Hood, B.; Levene, H.B.; Levi, A.D. Transplantation of autologous Schwann cells for the repair of segmental peripheral nerve defects. Neurosurg. Focus 2009, 26, E4. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, Y.A.; Almeida, V.W.; Gupta, R.; Levi, A.D. Transplantation of Schwann cells in a collagen tube for the repair of large, segmental peripheral nerve defects in rats. J. Neurosurg. 2013, 119, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Powell, R.; Phillips, J.B.; Haastert-Talini, K. Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering. Cells 2020, 9, 2497. [Google Scholar] [CrossRef] [PubMed]
- Monje, P.V. The properties of human Schwann cells: Lessons from in vitro culture and transplantation studies. Glia 2019, 68, 797–810. [Google Scholar] [CrossRef]
- Vijayavenkataraman, S. Nerve guide conduits for peripheral nerve injury repair: A review on design, materials and fabrication methods. Acta Biomater. 2020, 106, 54–69. [Google Scholar] [CrossRef]
- Jessen, K.R.; Arthur-Farraj, P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef]
- Kubiak, C.A.; Grochmal, J.; Kung, T.A.; Cederna, P.S.; Midha, R.; Kemp, S.W. Stem-cell-based therapies to enhance peripheral nerve regeneration. Muscle Nerve 2020, 61, 449–459. [Google Scholar] [CrossRef]
- Monje, P.V. Schwann Cell Cultures: Biology, Technology and Therapeutics. Cells 2020, 9, 1848. [Google Scholar] [CrossRef]
- Sugai, K.; Sumida, M.; Shofuda, T.; Yamaguchi, R.; Tamura, T.; Kohzuki, T.; Abe, T.; Shibata, R.; Kamata, Y.; Ito, S.; et al. First-in-human clinical trial of transplantation of iPSC-derived NS/PCs in subacute complete spinal cord injury: Study protocol. Regen. Ther. 2021, 18, 321–333. [Google Scholar] [CrossRef]
- Jessen, K.; Brennan, A.; Morgan, L.; Mirsky, R.; Kent, A.; Hashimoto, Y.; Gavrilovic, J. The schwann cell precursor and its fate: A study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron 1994, 12, 509–527. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Adameyko, I. Schwann cell precursor: A neural crest cell in disguise? Dev. Biol. 2018, 444 (Suppl. S1), S25–S35. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Lee, J.; Lee, D.Y.; Kim, Y.-D.; Kim, J.Y.; Lim, H.J.; Lim, S.; Cho, Y.S. Schwann Cell Precursors from Human Pluripotent Stem Cells as a Potential Therapeutic Target for Myelin Repair. Stem Cell Rep. 2017, 8, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Hörner, S.J.; Couturier, N.; Bruch, R.; Koch, P.; Hafner, M.; Rudolf, R. hiPSC-Derived Schwann Cells Influence Myogenic Differentiation in Neuromuscular Cocultures. Cells 2021, 10, 3292. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kim, J.Y.; Song, C.L.; Jeong, J.E.; Cho, Y.S. Directly induced human Schwann cell precursors as a valuable source of Schwann cells. Stem Cell Res. Ther. 2020, 11, 257. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-W.; Huang, W.-C.; Qiu, X.; da Silva, F.F.F.; Wang, A.; Patel, S.; Nesti, L.J.; Poo, M.-M.; Li, S. The Differentiation Stage of Transplanted Stem Cells Modulates Nerve Regeneration. Sci. Rep. 2017, 7, 17401. [Google Scholar] [CrossRef] [PubMed]
- Hörner, S.J.; Couturier, N.; Gueiber, D.C.; Hafner, M.; Rudolf, R. Development and In Vitro Differentiation of Schwann Cells. Cells 2022, 11, 3753. [Google Scholar] [CrossRef]
- Muangsanit, P.; Day, A.; Dimiou, S.; Ataç, A.F.; Kayal, C.; Park, H.; Nazhat, S.N.; Phillips, J.B. Rapidly formed stable and aligned dense collagen gels seeded with Schwann cells support peripheral nerve regeneration. J. Neural Eng. 2020, 17, 46036. [Google Scholar] [CrossRef]
- Skubis, A.; Gola, J.; Sikora, B.; Hybiak, J.; Paul-Samojedny, M.; Mazurek, U.; Łos, M.J. Impact of Antibiotics on the Proliferation and Differentiation of Human Adipose-Derived Mesenchymal Stem Cells. Int. J. Mol. Sci. 2017, 18, 2522. [Google Scholar] [CrossRef]
- Ryu, A.H.; Eckalbar, W.L.; Kreimer, A.; Yosef, N.; Ahituv, N. Use antibiotics in cell culture with caution: Genome-wide identification of antibiotic-induced changes in gene expression and regulation. Sci. Rep. 2017, 7, 7533. [Google Scholar] [CrossRef]
- Cohen, S.; Samadikuchaksaraei, A.; Polak, J.M.; Bishop, A.E. Antibiotics Reduce the Growth Rate and Differentiation of Embryonic Stem Cell Cultures. Tissue Eng. 2006, 12, 2025–2030. [Google Scholar] [CrossRef] [PubMed]
- Duman, M.; Vaquié, A.; Nocera, G.; Heller, M.; Stumpe, M.; Sankar, D.S.; Dengjel, J.; Meijer, D.; Yamaguchi, T.; Matthias, P.; et al. EEF1A1 deacetylation enables transcriptional activation of remyelination. Nat. Commun. 2020, 11, 3420. [Google Scholar] [CrossRef]
- Chu, T.; Baral, K.; Labit, E.; Rosin, N.; Sinha, S.; Umansky, D.; Alzahrani, S.; Arora, R.; Cao, L.; Rancourt, D.; et al. Comparison of human skin- and nerve-derived Schwann cells reveals many similarities and subtle genomic and functional differences. Glia 2022, 70, 2131–2156. [Google Scholar] [CrossRef] [PubMed]
- Rayner, M.L.; Day, A.G.; Bhangra, K.S.; Sinden, J.; Phillips, J.B. Engineered neural tissue made using clinical-grade human neural stem cells supports regeneration in a long gap peripheral nerve injury model. Acta Biomater. 2021, 135, 203–213. [Google Scholar] [CrossRef]
- O’Rourke, C.; Day, A.G.E.; Murray-Dunning, C.; Thanabalasundaram, L.; Cowan, J.; Stevanato, L.; Grace, N.; Cameron, G.; Drake, R.A.L.; Sinden, J.; et al. An allogeneic ‘off the shelf’ therapeutic strategy for peripheral nerve tissue engineering using clinical grade human neural stem cells. Sci. Rep. 2018, 8, 2951. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Bunting, S.C.; Davies, H.A.; Loughlin, A.J.; Golding, J.P.; Phillips, J.B. Engineered neural tissue for peripheral nerve repair. Biomaterials 2013, 34, 7335–7343. [Google Scholar] [CrossRef] [PubMed]
- Casella, G.T.; Wieser, R.; Bunge, R.P.; Margitich, I.S.; Katz, J.; Olson, L.; Wood, P.M. Density dependent regulation of human Schwann cell proliferation. Glia 2000, 30, 165–177. [Google Scholar] [CrossRef]
- Haastert, K.; Mauritz, C.; Chaturvedi, S.; Grothe, C. Human and rat adult Schwann cell cultures: Fast and efficient enrichment and highly effective non-viral transfection protocol. Nat. Protoc. 2007, 2, 99–104. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Li, Y.; Tennekoon, G.I.; Birnbaum, M.; Marchionni, M.A.; Rutkowski, J.L. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell. Neurosci. 2001, 17, 761–767. [Google Scholar] [CrossRef]
- Fregien, N.L.; White, L.A.; Bunge, M.B.; Wood, P.M. Forskolin increases neuregulin receptors in human Schwann cells without increasing receptor mRNA. Glia 2004, 49, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, F.R.; Salomonis, N.; Sheehan, A.; Huang, M.; Park, J.S.; Spindler, M.J.; Lizarraga, P.; A Weiss, W.; So, P.-L.; Conklin, B.R. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cells 2013, 2, 119–131. [Google Scholar] [PubMed]
- Liu, Q.; Spusta, S.C.; Mi, R.; Lassiter, R.N.; Stark, M.R.; Höke, A.; Rao, M.S.; Zeng, X. Human Neural Crest Stem Cells Derived from Human ESCs and Induced Pluripotent Stem Cells: Induction, Maintenance, and Differentiation into Functional Schwann Cells. STEM CELLS Transl. Med. 2012, 1, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, M.; Andersson, P.-H.; Åkesson, E.; Odeberg, J.; Holmberg, L.; Inzunza, J.; Falci, S.; Öhman, J.; Suuronen, R.; Skottman, H.; et al. Markers of Pluripotency and Differentiation in Human Neural Precursor Cells Derived from Embryonic Stem Cells and CNS Tissue. Cell Transplant. 2011, 20, 177–192. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R.; Lloyd, A.C. Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a020487. [Google Scholar] [CrossRef]
- Mirsky, R.; Woodhoo, A.; Parkinson, D.B.; Arthur-Farraj, P.; Bhaskaran, A.; Jessen, K.R. Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J. Peripher. Nerv. Syst. 2008, 13, 122–135. [Google Scholar] [CrossRef]
- Birchmeier, C.; Nave, K. Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia 2008, 56, 1491–1497. [Google Scholar] [CrossRef]
- Monje, P.V.; Sant, D.; Wang, G. Phenotypic and Functional Characteristics of Human Schwann Cells as Revealed by Cell-Based Assays and RNA-SEQ. Mol. Neurobiol. 2018, 55, 6637–6660. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Schwann Cell Precursors; Multipotent Glial Cells in Embryonic Nerves. Front. Mol. Neurosci. 2019, 12, 69. [Google Scholar] [CrossRef]
- Clements, M.P.; Byrne, E.; Guerrero, L.F.C.; Cattin, A.-L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 2017, 96, 98–114.e7. [Google Scholar] [CrossRef] [PubMed]
- Bhatheja, K.; Field, J. Schwann cells: Origins and role in axonal maintenance and regeneration. Int. J. Biochem. Cell Biol. 2006, 38, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.; Kumar, R.; Sinha, S.; Shah, P.; Stykel, M.; Shapira, Y.; Midha, R.; Biernaskie, J. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. Eneuro 2017, 4, ENEURO.0307-16.2017. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.; Pereira, J.A.; Gerber, J.; Tan, G.; Dimitrieva, S.; Yángüez, E.; Suter, U. Transcriptional profiling of mouse peripheral nerves to the single-cell level to build a sciatic nerve ATlas (SNAT). Elife 2021, 10, e58591. [Google Scholar] [CrossRef]
- Wanner, I.B.; Wood, P.M. N-Cadherin Mediates Axon-Aligned Process Growth and Cell–Cell Interaction in Rat Schwann Cells. J. Neurosci. 2002, 22, 4066–4079. [Google Scholar] [CrossRef]
- Shi, H.; Gong, Y.; Qiang, L.; Li, X.; Zhang, S.; Gao, J.; Li, K.; Ji, X.; Tian, L.; Gu, X.; et al. Derivation of Schwann cell precursors from neural crest cells resident in bone marrow for cell therapy to improve peripheral nerve regeneration. Biomaterials 2016, 89, 25–37. [Google Scholar] [CrossRef]
- Shi, H.; Li, X.; Yang, J.; Zhao, Y.; Xue, C.; Wang, Y.; He, Q.; Shen, M.; Zhang, Q.; Yang, Y.; et al. Bone marrow-derived neural crest precursors improve nerve defect repair partially through secreted trophic factors. Stem Cell Res. Ther. 2019, 10, 397. [Google Scholar] [CrossRef]
- Haase, A.; Göhring, G.; Martin, U. Generation of non-transgenic iPS cells from human cord blood CD34 + cells under animal component-free conditions. Stem Cell Res. 2017, 21, 71–73. [Google Scholar] [CrossRef]
- Haastert, K.; Mauritz, C.; Matthies, C.; Grothe, C. Autologous adult human Schwann cells genetically modified to provide alternative cellular transplants in peripheral nerve regeneration. J. Neurosurg. 2006, 104, 778–786. [Google Scholar] [CrossRef]
- Gryshkov, O.; Al Halabi, F.; Kuhn, A.I.; Leal-Marin, S.; Freund, L.J.; Förthmann, M.; Meier, N.; Barker, S.A.; Haastert-Talini, K.; Glasmacher, B. PVDF and P(VDF-TrFE) Electrospun Scaffolds for Nerve Graft Engineering: A Comparative Study on Piezoelectric and Structural Properties, and In Vitro Biocompatibility. Int. J. Mol. Sci. 2021, 22, 11373. [Google Scholar] [CrossRef]
- Huang, Z.; Kankowski, S.; Ertekin, E.; Almog, M.; Nevo, Z.; Rochkind, S.; Haastert-Talini, K. Modified Hyaluronic Acid-Laminin-Hydrogel as Luminal Filler for Clinically Approved Hollow Nerve Guides in a Rat Critical Defect Size Model. Int. J. Mol. Sci. 2021, 22, 6554. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Ratzka, A.; Brinkmann, H.; Klimaschewski, L.; Grothe, C.; Claus, P. Analysis of the Fibroblast Growth Factor System Reveals Alterations in a Mouse Model of Spinal Muscular Atrophy. PLoS ONE 2012, 7, e31202. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D0-D6 | D6-D24 | D24-D26 | D26-D28 | >D30 | |
|---|---|---|---|---|---|
| Kim et al. 2017 [25] | D0 = hiPSC | D24 + = hiPSC-SCP | D30 + = hiPSC-SC | ||
| Matrigel coating | |||||
| Neurobasal/Advanced DMEM/F12 N2, B27, SB431542, CHIR99021, BSA, β-ME | DMEM (1 g/L glucose) 1% FBS, NRG1 | ||||
| NRG1 | RA, FSK, PDGF-BB | PDGF-BB | |||
| MHH-Protocol | D0 = hiPSC | D24 + = hiPSC-immatureSC | D30 + = hiPSC-SC | ||
| Matrigel coating | P-ORN-laminin coating | ||||
| Neurobasal/Advanced DMEM/F12 N2, B27, SB431542, CHIR99021, BSA, β-ME, Y27632, Pen/Strep, Amphotericin B | Advanced DMEM/F12 (4.5 g/L glucose) 10% FBS, Pen/Strep, Amphotericin B, NGR1 | ahSC medium | |||
| NRG1 | RA, FSK, PDGF-BB | PDGF-BB | |||
| Cells dissociated when reaching 60–70% confluence | Seeding density 21,052 cells/cm2 | Seeding density 42,104 cells/cm2 | |||
| UCL-Protocol | D0 = hiPSC | D24 + = hiPSC-SCP | D30 + = hiPSC-SC | ||
| GeltrexTM matrix | |||||
| Neurobasal/Advanced DMEM/F12 N2, B27, SB431542, CHIR99021, BSA, β-ME | DMEM(1 g/L glucose) 1% FBS, NRG1 | ||||
| NRG1 | RA, FSK, PDGF-BB | PDGF-BB | |||
| Cells dissociated when reaching 90% confluence | Seeding density 14,104 cells/cm2 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Powell, R.; Kankowski, S.; Phillips, J.B.; Haastert-Talini, K. Culture Conditions for Human Induced Pluripotent Stem Cell-Derived Schwann Cells: A Two-Centre Study. Int. J. Mol. Sci. 2023, 24, 5366. https://doi.org/10.3390/ijms24065366
Huang Z, Powell R, Kankowski S, Phillips JB, Haastert-Talini K. Culture Conditions for Human Induced Pluripotent Stem Cell-Derived Schwann Cells: A Two-Centre Study. International Journal of Molecular Sciences. 2023; 24(6):5366. https://doi.org/10.3390/ijms24065366
Chicago/Turabian StyleHuang, Zhong, Rebecca Powell, Svenja Kankowski, James B. Phillips, and Kirsten Haastert-Talini. 2023. "Culture Conditions for Human Induced Pluripotent Stem Cell-Derived Schwann Cells: A Two-Centre Study" International Journal of Molecular Sciences 24, no. 6: 5366. https://doi.org/10.3390/ijms24065366
APA StyleHuang, Z., Powell, R., Kankowski, S., Phillips, J. B., & Haastert-Talini, K. (2023). Culture Conditions for Human Induced Pluripotent Stem Cell-Derived Schwann Cells: A Two-Centre Study. International Journal of Molecular Sciences, 24(6), 5366. https://doi.org/10.3390/ijms24065366