

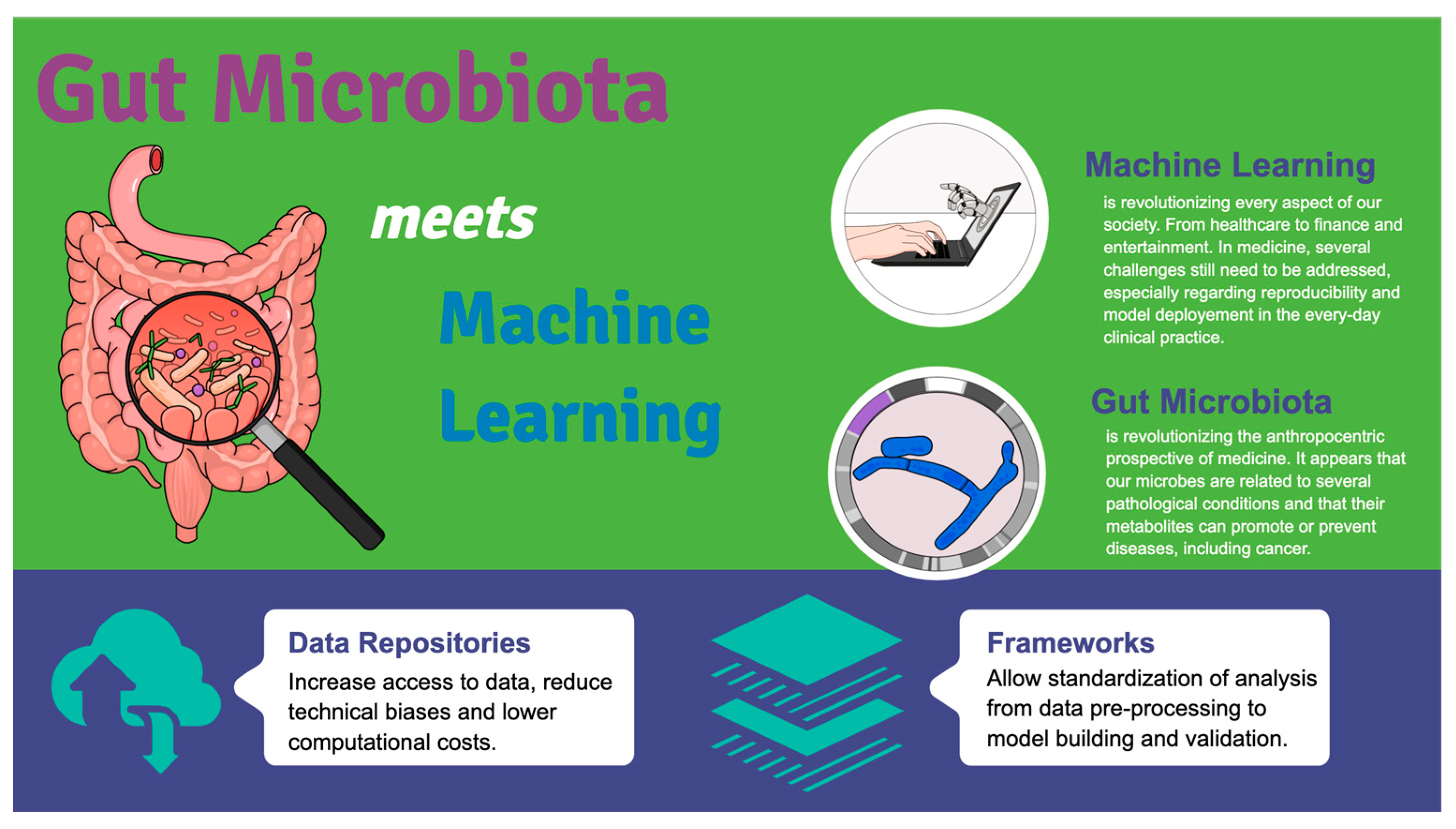
Gut Microbes Meet Machine Learning: The Next Step towards Advancing Our Understanding of the Gut Microbiome in Health and Disease


{kind=link}
Abstract
1. The Human Microbiome
2. Machine Learning and Gut Microbiome
2.1. Challenges in Current Application
2.2. The Importance of Data Repositories and Data Preprocessing
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Chassaing, B.; Etienne-Mesmin, L.; Gewirtz, A.T. Microbiota-liver axis in hepatic disease. Hepatology 2013, 59, 328–339. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A story of liver and gut microbes: How does the intestinal flora affect liver disease? A review of the literature. Am. J. Physiol. Liver Physiol. 2020, 318, G889–G906. [Google Scholar] [CrossRef]
- Giuffrè, M.; Moretti, R.; Campisciano, G.; Da Silveira, A.B.M.; Monda, V.M.; Comar, M.; Di Bella, S.; Antonello, R.M.; Luzzati, R.; Crocè, L.S. You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS. J. Clin. Med. 2020, 9, 3705. [Google Scholar] [CrossRef]
- Giuffrè, M.; Gazzin, S.; Zoratti, C.; Llido, J.P.; Lanza, G.; Tiribelli, C.; Moretti, R. Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation. Int. J. Mol. Sci. 2022, 23, 15564. [Google Scholar] [CrossRef]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinform. 2016, 12, 5–6. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]
- Ghannam, R.B.; Techtmann, S.M. Machine learning applications in microbial ecology, human microbiome studies, and environmental monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef]
- Goodswen, S.J.; Barratt, J.L.N.; Kennedy, P.J.; Kaufer, A.; Calarco, L.; Ellis, J.T. Machine learning and applications in microbiology. FEMS Microbiol. Rev. 2021, 45, fuab015. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Turukalo, T.L.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Henn, J.; Buness, A.; Schmid, M.; Kalff, J.C.; Matthaei, H. Machine learning to guide clinical decision-making in abdominal surgery—A systematic literature review. Langenbeck’s Arch. Surg. 2021, 407, 51–61. [Google Scholar] [CrossRef]
- Meskó, B.; Görög, M. A short guide for medical professionals in the era of artificial intelligence. NPJ Digit. Med. 2020, 3, 126. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Fidelle, M.; Iebba, V.; Alla, L.; Pasolli, E.; Segata, N.; Desnoyer, A.; Pietrantonio, F.; Ferrere, G.; et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur. Urol. 2020, 78, 195–206. [Google Scholar] [CrossRef]
- Aminu, M.; Ahmad, N.A. Complex Chemical Data Classification and Discrimination Using Locality Preserving Partial Least Squares Discriminant Analysis. ACS Omega 2020, 5, 26601–26610. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Reiman, D.; Layden, B.T.; Dai, Y. MiMeNet: Exploring microbiome-metabolome relationships using neural networks. PLoS Comput. Biol. 2021, 17, e1009021. [Google Scholar] [CrossRef]
- Hornik, K.; Stinchcombe, M.; White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 1989, 2, 359–366. [Google Scholar] [CrossRef]
- Chen, R.Y.; Mostafa, I.; Hibberd, M.C.; Das, S.; Mahfuz, M.; Naila, N.N.; Islam, M.M.; Huq, S.; Alam, M.A.; Zaman, M.U.; et al. A Microbiota-Directed Food Intervention for Undernourished Children. N. Engl. J. Med. 2021, 384, 1517–1528. [Google Scholar] [CrossRef]
- Schloss, P.D. Identifying and Overcoming Threats to Reproducibility, Replicability, Robustness, and Generalizability in Microbiome Research. Mbio 2018, 9, e00525-18. [Google Scholar] [CrossRef]
- Poussin, C.; Sierro, N.; Boué, S.; Battey, J.; Scotti, E.; Belcastro, V.; Peitsch, M.C.; Ivanov, N.V.; Hoeng, J. Interrogating the microbiome: Experimental and computational considerations in support of study reproducibility. Drug Discov. Today 2018, 23, 1644–1657. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Smialowski, P.; Frishman, D.; Kramer, S. Pitfalls of supervised feature selection. Bioinformatics 2009, 26, 440–443. [Google Scholar] [CrossRef]
- Pasolli, E.; Schiffer, L.; Manghi, P.; Renson, A.; Obenchain, V.; Truong, D.T.; Beghini, F.; Malik, F.; Ramos, M.; Dowd, J.B.; et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 2017, 14, 1023–1024. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2019, 48, D570–D578. [Google Scholar] [CrossRef]
- Dai, D.; Zhu, J.; Sun, C.; Li, M.; Liu, J.; Wu, S.; Ning, K.; He, L.-J.; Zhao, X.-M.; Chen, W.-H. GMrepo v2: A curated human gut microbiome database with special focus on disease markers and cross-dataset comparison. Nucleic Acids Res. 2021, 50, D777–D784. [Google Scholar] [CrossRef]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vázquez-Baeza, Y.; Ackermann, G.; Dereus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, web-enabled microbiome meta-analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef]
- Mirzayi, C.; Renson, A.; Furlanello, C.; Sansone, S.-A.; Zohra, F.; Elsafoury, S.; Geistlinger, L.; Kasselman, L.J.; Eckenrode, K.; van de Wijgert, J.; et al. Reporting guidelines for human microbiome research: The STORMS checklist. Nat. Med. 2021, 27, 1885–1892. [Google Scholar] [CrossRef]
- Wirbel, J.; Zych, K.; Essex, M.; Karcher, N.; Kartal, E.; Salazar, G.; Bork, P.; Sunagawa, S.; Zeller, G. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 2021, 22, 93. [Google Scholar] [CrossRef]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet Multinomial Mixtures: Generative Models for Microbial Metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef]
- Topçuoğlu, B.D.; Lesniak, N.A.; Ruffin, M.T.; Wiens, J.; Schloss, P.D. A Framework for Effective Application of Machine Learning to Microbiome-Based Classification Problems. Mbio 2020, 11, e00434-20. [Google Scholar] [CrossRef]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef]
- Wilkinson, J.E.; Franzosa, E.A.; Everett, C.; Li, C.; Bae, S.; Berzansky, I.; Bhosle, A.; Bjørnevik, K.; Brennan, C.A.; Cao, Y.G.; et al. A framework for microbiome science in public health. Nat. Med. 2021, 27, 766–774. [Google Scholar] [CrossRef]
- Sanna, S.; Van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vila, A.V.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.M.A.E.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Garay, J.A.R.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Liu, X.; Tong, X.; Zou, Y.; Lin, X.; Zhao, H.; Tian, L.; Jie, Z.; Wang, Q.; Zhang, Z.; Lu, H.; et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat. Genet. 2022, 54, 52–61. [Google Scholar] [CrossRef]
- Pryszlak, A.; Wenzel, T.; Seitz, K.W.; Hildebrand, F.; Kartal, E.; Cosenza, M.R.; Benes, V.; Bork, P.; Merten, C.A. Enrichment of gut microbiome strains for cultivation-free genome sequencing using droplet microfluidics. Cell Rep. Methods 2021, 2, 100137. [Google Scholar] [CrossRef]
- Jin, W.-B.; Li, T.-T.; Huo, D.; Qu, S.; Li, X.V.; Arifuzzaman, M.; Lima, S.F.; Shi, H.-Q.; Wang, A.; Putzel, G.G.; et al. Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell 2022, 185, 547–562.e22. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuffrè, M.; Moretti, R.; Tiribelli, C. Gut Microbes Meet Machine Learning: The Next Step towards Advancing Our Understanding of the Gut Microbiome in Health and Disease. Int. J. Mol. Sci. 2023, 24, 5229. https://doi.org/10.3390/ijms24065229
Giuffrè M, Moretti R, Tiribelli C. Gut Microbes Meet Machine Learning: The Next Step towards Advancing Our Understanding of the Gut Microbiome in Health and Disease. International Journal of Molecular Sciences. 2023; 24(6):5229. https://doi.org/10.3390/ijms24065229
Chicago/Turabian StyleGiuffrè, Mauro, Rita Moretti, and Claudio Tiribelli. 2023. "Gut Microbes Meet Machine Learning: The Next Step towards Advancing Our Understanding of the Gut Microbiome in Health and Disease" International Journal of Molecular Sciences 24, no. 6: 5229. https://doi.org/10.3390/ijms24065229
APA StyleGiuffrè, M., Moretti, R., & Tiribelli, C. (2023). Gut Microbes Meet Machine Learning: The Next Step towards Advancing Our Understanding of the Gut Microbiome in Health and Disease. International Journal of Molecular Sciences, 24(6), 5229. https://doi.org/10.3390/ijms24065229