
Understanding the Continuum between High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia


Abstract
1. Introduction
2. Genetic Alterations Underlie MDS Progression to AML-MRC
2.1. Chromosomal Abnormalities
2.2. Somatic Mutations
2.3. Single-Cell Analysis Reveals Biologic Mechanisms Implicated in MDS Progression to AML-MRC
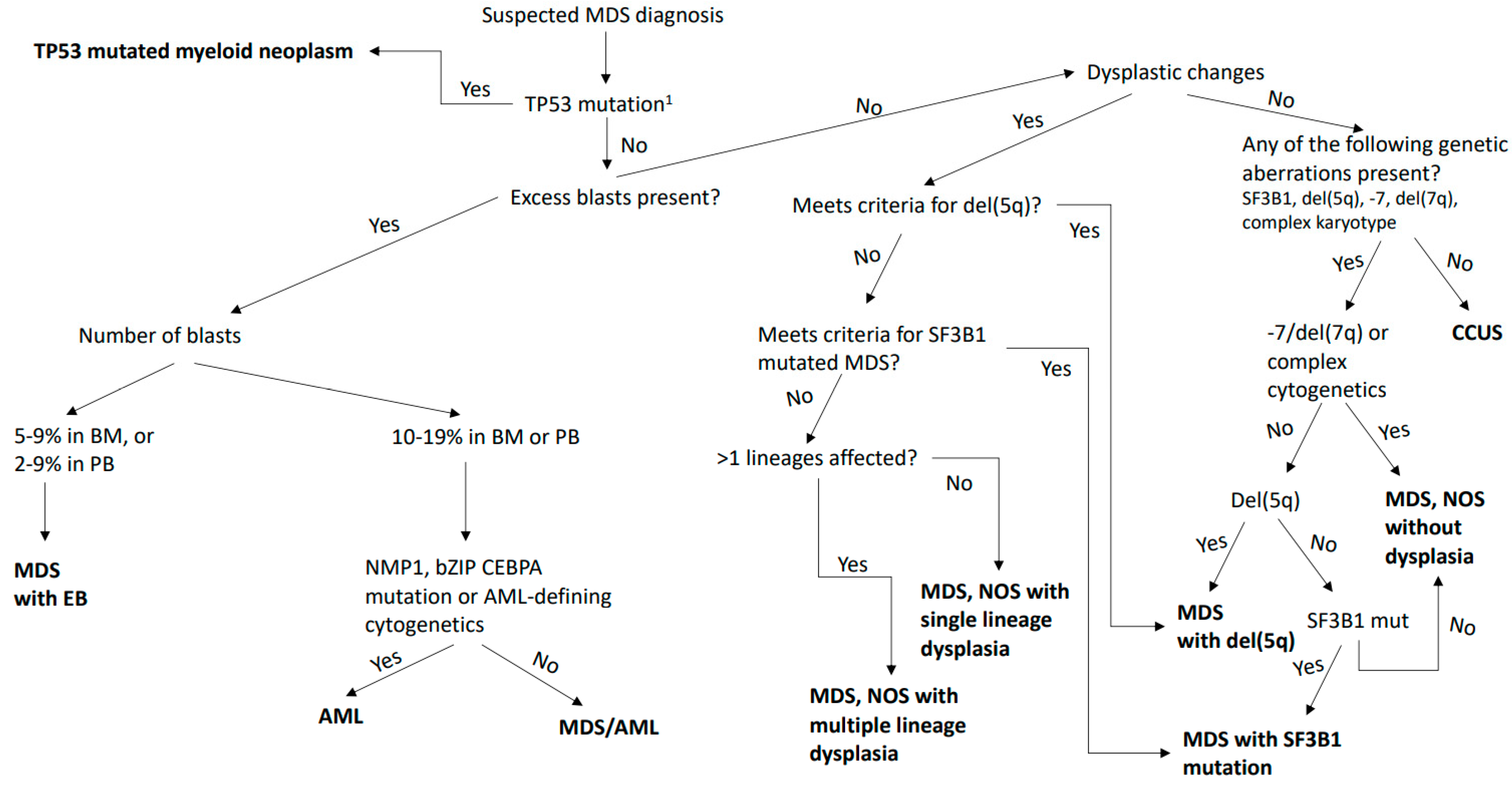
3. Changes in the Definition and Classification of MDS and AML-MRC
3.1. MDS and AML with Myelodysplasia-Related Gene Mutations as a Disease Continuum
3.2. TP53-Mutated Disease
3.3. Therapy-Related Disease
3.4. Other Changes Introduced by the ICC and the WHO
4. Risk Stratification Changes in Myeloid Neoplasms
4.1. MDS
4.2. AML
- FLT3-ITD-mutated AML irrespective of the allelic ratio (formerly FLT3-ITDhigh for biallelic mutation and FLT3-ITDlow for mono-allelic mutation) or the concurrent NMP1 mutation status is now considered intermediate risk disease. This is based on evidence of disease modifying impact of midostaurin-based therapy [128].
- The previously recognized RUNX1 and ASXL1-mutated AML categories are expanded to include the whole AML category with gene-related mutations classified under high-risk disease. This new category encompasses the nine above-mentioned gene mutations (ASXL1, RUNX1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2) associated with AML arising from MDS.
- NMP1-mutated AML with concurrent adverse-risk-associated cytogenetic abnormalities has been reclassified as a high-risk disease, based on new data that shows an association with worse outcomes [129].
- AML with in-frame monoallelic mutations in the basic leucine zipper region (bZIP) of CEBPA is now also classified under favorable-risk disease, along with biallelic mutated disease [130].
- The following cytogenetic abnormalities are added to the adverse risk group: t(3q26.2;v), involving the MECOM gene, and t(8;16)(p11.2;p13.3), associated with KAT6A::CREBBP gene fusion (39, 40). Last, hyperploidy with multiple trisomies or polysomies is no longer considered a complex karyotype and is therefore not classified as an adverse risk [14].
5. Current Management of Patients with High-Risk MDS and AML-MRC
5.1. Current Standard Approaches
5.1.1. AlloBMT
5.1.2. HMAs
5.1.3. Venetoclax
5.1.4. Chemotherapy
5.2. Specific Molecular Subtypes
5.2.1. TP53 Mutated High-Risk MDS and AML-MRC
5.2.2. IDH1/2 Mutated High-Risk MDS and AML-MRC
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nimer, S.D. Myelodysplastic syndromes. Blood 2008, 111, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Karantanos, T.; DeZern, A.E. Biology and clinical management of hypoplastic MDS: MDS as a bone marrow failure syndrome. Best Pract. Res. Clin. Haematol. 2021, 34, 101280. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.J.; Walter, M.J. Genetics of progression from MDS to secondary leukemia. Blood 2020, 136, 50–60. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Granfeldt Østgård, L.S.; Medeiros, B.C.; Sengeløv, H.; Nørgaard, M.; Andersen, M.K.; Dufva, I.H.; Friis, L.S.; Kjeldsen, E.; Marcher, C.W.; Preiss, B.; et al. Epidemiology and Clinical Significance of Secondary and Therapy-Related Acute Myeloid Leukemia: A National Population-Based Cohort Study. J. Clin. Oncol. 2015, 33, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Le Beau, M.M.; Espinosa, R.; Davis, E.M.; Eisenbart, J.D.; Larson, R.; Green, E.D. Cytogenetic and molecular delineation of a region of chromosome 7 commonly deleted in malignant myeloid diseases. Blood 1996, 88, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Galli’, A.; Pellagatti, A.; Karimi, M.; Sato-Otsubo, A.; Satoru, M.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood 2017, 130, 2642–2653. [Google Scholar] [CrossRef]
- Takahashi, K.; Jabbour, E.; Wang, X.; Luthra, R.; Buesoramos, C.; Patel, K.K.; Pierce, S.; Yang, H.; Wei, Y.; Daver, N.; et al. Dynamic acquisition of FLT3 or RAS alterations drive a subset of patients with lower risk MDS to secondary AML. Leukemia 2013, 27, 2081–2083. [Google Scholar] [CrossRef]
- Guess, T.; Potts, C.R.; Bhat, P.; Cartailler, J.A.; Brooks, A.; Holt, C.; Yenamandra, A.; Wheeler, F.C.; Savona, M.R.; Cartailler, J.-P.; et al. Distinct Patterns of Clonal Evolution Drive Myelodysplastic Syndrome Progression to Secondary Acute Myeloid Leukemia. Blood Cancer Discov. 2022, 3, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Ganan-Gomez, I.; Yang, H.; Ma, F.; Montalban-Bravo, G.; Thongon, N.; Marchica, V.; Richard-Carpentier, G.; Chien, K.; Manyam, G.; Wang, F.; et al. Stem cell architecture drives myelodysplastic syndrome progression and predicts response to venetoclax-based therapy. Nat. Med. 2022, 28, 557–567. [Google Scholar] [CrossRef]
- Chilton, L.; Hills, R.; Harrison, C.; Burnett, A.K.; Grimwade, D.; Moorman, A.V. Hyperdiploidy with 49–65 chromosomes represents a heterogeneous cytogenetic subgroup of acute myeloid leukemia with differential outcome. Leukemia 2013, 28, 321–328. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Giagounidis, A.A.; Germing, U.; Aul, C. Biological and prognostic significance of chromosome 5q deletions in myeloid malignancies. Clin. Cancer Res. 2006, 12, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Solé, F.; Espinet, B.; Sanz, G.F.; Cervera, J.; Calasanz, M.J.; Luño, E.; Prieto, F.; Granada, I.; Hernández, J.M.; Cigudosa, J.C.; et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Br. J. Haematol. 2000, 108, 346–356. [Google Scholar] [CrossRef]
- Ebert, B.L. Genetic deletions in AML and MDS. Best Pract. Res. Clin. Haematol. 2010, 23, 457–461. [Google Scholar] [CrossRef]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.; Lorenzo-Abalde, S.; Lane, A.L.; E Jolin, H.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q– syndrome. Nat. Med. 2009, 16, 59–66. [Google Scholar] [CrossRef]
- Boultwood, J. The role of haploinsufficiency of RPS14 and p53 activation in the molecular pathogenesis of the 5q- syndrome. Pediatr. Rep. 2011, 3, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, B.; Göhring, G.; Bernard, E.; Nilsson, L.; Tobiasson, M.; Jädersten, M.; Garelius, H.; Dybedal, I.; Grønbaek, K.; Ejerblad, E.; et al. “Randomized phase II study of azacitidine ± lenalidomide in higher-risk myelodysplastic syndromes and acute myeloid leukemia with a karyotype including Del(5q)”. Leukemia 2022, 36, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Pitel, B.A.; Sharma, N.; Zepeda-Mendoza, C.; Smadbeck, J.B.; Pearce, K.E.; Cook, J.M.; Vasmatzis, G.; Sachs, Z.; Kanagal-Shamanna, R.; Viswanatha, D.; et al. Myeloid malignancies with 5q and 7q deletions are associated with extreme genomic complexity, biallelic TP53 variants, and very poor prognosis. Blood Cancer J. 2021, 11, 18. [Google Scholar] [CrossRef]
- Cordoba, I.; González-Porras, J.R.; Nomdedeu, B.; Luño, E.; de Paz, R.; Such, E.; Tormo, M.; Vallespi, T.; Collado, R.; Xicoy, B.; et al. Better prognosis for patients with del(7q) than for patients with monosomy 7 in myelodysplastic syndrome. Cancer 2011, 118, 127–133. [Google Scholar] [CrossRef]
- Inaba, T.; Honda, H.; Matsui, H. The enigma of monosomy 7. Blood 2018, 131, 2891–2898. [Google Scholar] [CrossRef]
- Nagamachi, A.; Matsui, H.; Asou, H.; Ozaki, Y.; Aki, D.; Kanai, A.; Takubo, K.; Suda, T.; Nakamura, T.; Wolff, L.; et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell 2013, 24, 305–317. [Google Scholar] [CrossRef]
- Wong, C.C.; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium; Martincorena, I.; Rust, A.; Rashid, M.; Alifrangis, C.; Alexandrov, L.B.; Tiffen, J.C.; Kober, C.; Green, A.; et al. Inactivating CUX1 mutations promote tumorigenesis. Nat. Genet. 2013, 46, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.A.; Hao, D.; Tomczak, K.; Barrodia, P.; Im, J.S.; Reville, P.K.; Alaniz, Z.; Wang, W.; Wang, R.; Wang, F.; et al. Single cell T cell landscape and T cell receptor repertoire profiling of AML in context of PD-1 blockade therapy. Nat. Commun. 2021, 12, 6071. [Google Scholar] [CrossRef]
- Schanz, J.; Steidl, C.; Fonatsch, C.; Pfeilstöcker, M.; Nösslinger, T.; Tuechler, H.; Valent, P.; Hildebrandt, B.; Giagounidis, A.; Aul, C.; et al. Coalesced multicentric analysis of 2,351 patients with myelodysplastic syndromes indicates an underestimation of poor-risk cytogenetics of myelodysplastic syndromes in the international prognostic scoring system. J. Clin. Oncol. 2011, 29, 1963–1970. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef]
- Saez, B.; Walter, M.J.; Graubert, T.A. Splicing factor gene mutations in hematologic malignancies. Blood 2017, 129, 1260–1269. [Google Scholar] [CrossRef]
- Sperling, A.; Gibson, C.J.; Ebert, A.S.S.C.J.G.B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2016, 17, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Dalton, W.B.; Helmenstine, E.; Walsh, N.; Gondek, L.P.; Kelkar, D.S.; Read, A.; Natrajan, R.; Christenson, E.S.; Roman, B.; Das, S.; et al. Hotspot SF3B1 mutations induce metabolic reprogramming and vulnerability to serine deprivation. J. Clin. Investig. 2019, 129, 4708–4723. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. SomaticSF3B1Mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef]
- Dalton, W.B.; Helmenstine, E.; Pieterse, L.; Li, B.; Gocke, C.D.; Donaldson, J.; Xiao, Z.; Gondek, L.P.; Ghiaur, G.; Gojo, I.; et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv. 2020, 4, 1192–1196. [Google Scholar] [CrossRef]
- Hou, H.-A.; Tsai, C.-H.; Lin, C.-C.; Chou, W.-C.; Kuo, Y.-Y.; Liu, C.-Y.; Tseng, M.-H.; Peng, Y.-L.; Liu, M.-C.; Liu, C.-W.; et al. Incorporation of mutations in five genes in the revised International Prognostic Scoring System can improve risk stratification in the patients with myelodysplastic syndrome. Blood Cancer J. 2018, 8, 39. [Google Scholar] [CrossRef]
- Zheng, X.; Zhan, Z.; Naren, D.; Li, J.; Yan, T.; Gong, Y. Prognostic value of SRSF2 mutations in patients with de novo myelodysplastic syndromes: A meta-analysis. PLoS ONE 2017, 12, e0185053. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Kon, A.; Yamazaki, S.; Nannya, Y.; Kataoka, K.; Ota, Y.; Nakagawa, M.M.; Yoshida, K.; Shiozawa, Y.; Morita, M.; Yoshizato, T.; et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 2018, 131, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.; Tang, J.-L.; Lin, C.-T.; Kuo, Y.-Y.; Li, L.-Y.; Tseng, M.-H.; Huang, C.-F.; Lai, Y.-J.; Lee, F.-Y.; Liu, M.-C.; et al. Clinical implications of U2AF1 mutation in patients with myelodysplastic syndrome and its stability during disease progression. Am. J. Hematol. 2013, 88, E277–E282. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Ou, J.; Chamberlain, L.; Simone, T.M.; Yang, H.; Virbasius, C.-M.; Ali, A.M.; Zhu, L.J.; Mukherjee, S.; Raza, A.; et al. U2AF35(S34F) Promotes Transformation by Directing Aberrant ATG7 Pre-mRNA 3′ End Formation. Mol. Cell 2016, 62, 479–490. [Google Scholar] [CrossRef]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nature 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- Wang, X.; Song, X.; Yan, X. Effect of RNA splicing machinery gene mutations on prognosis of patients with MDS: A meta-analysis. Medicine 2019, 98, e15743. [Google Scholar] [CrossRef]
- Haering, C.H.; Löwe, J.; Hochwagen, A.; Nasmyth, K. Molecular Architecture of SMC Proteins and the Yeast Cohesin Complex. Mol. Cell 2002, 9, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Gondek, L.P.; Varadhan, R.; Moliterno, A.R.; DeZern, A.E.; Jones, R.J.; Jain, T. Gender-related differences in the outcomes and genomic landscape of patients with myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes. Br. J. Haematol. 2021, 193, 1142–1150. [Google Scholar] [CrossRef]
- Viny, A.D.; Levine, R.L. Cohesin mutations in myeloid malignancies made simple. Curr. Opin. Hematol. 2018, 25, 61–66. [Google Scholar] [CrossRef]
- Patel, J.L.; Schumacher, J.A.; Frizzell, K.; Sorrells, S.; Shen, W.; Clayton, A.; Jattani, R.; Kelley, T.W. Coexisting and cooperating mutations in NPM1-mutated acute myeloid leukemia. Leuk. Res. 2017, 56, 7–12. [Google Scholar] [CrossRef]
- Dolnik, A.; Engelmann, J.C.; Scharfenberger-Schmeer, M.; Mauch, J.; Kelkenberg-Schade, S.; Haldemann, B.; Fries, T.; Krönke, J.; Kühn, M.W.M.; Paschka, P.; et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 2012, 120, e83–e92. [Google Scholar] [CrossRef]
- Christofides, A.; Karantanos, T.; Bardhan, K.; Boussiotis, V.A. Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget 2016, 7, 85624–85640. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 mutations and impact on clinical outcome: An analysis in 1,604 patients with newly diagnosed acute myeloid leukemia. Haematologica 2019, 105, e228–e231. [Google Scholar] [CrossRef]
- Triviai, I.; Zeschke, S.; Rentel, J.; Spanakis, M.; Scherer, T.; Gabdoulline, R.; Panagiota, V.; Thol, F.; Heuser, M.; Stocking, C.; et al. ASXL1/EZH2 mutations promote clonal expansion of neoplastic HSC and impair erythropoiesis in PMF. Leukemia 2018, 33, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef] [PubMed]
- Grubach, L.; Juhl-Christensen, C.; Rethmeier, A.; Olesen, L.H.; Aggerholm, A.; Hokland, P.; Østergaard, M. Gene expression profiling of Polycomb, Hox and Meis genes in patients with acute myeloid leukaemia. Eur. J. Haematol. 2008, 81, 112–122. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyagi, S.; Sashida, G.; Chiba, T.; Yuan, J.; Mochizuki-Kashio, M.; Suzuki, Y.; Sugano, S.; Nakaseko, C.; Yokote, K.; et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 2012, 120, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Tsai, H.-L.; Gondek, L.P.; DeZern, A.E.; Ghiaur, G.; Dalton, W.B.; Gojo, I.; Prince, G.T.; Webster, J.; Ambinder, A.; et al. Genomic landscape of myelodysplastic/myeloproliferative neoplasm can predict response to hypomethylating agent therapy. Leuk. Lymphoma 2022, 63, 1942–1948. [Google Scholar] [CrossRef] [PubMed]
- Rinke, J.; Müller, J.P.; Blaess, M.F.; A Chase, A.; Meggendorfer, M.; Schäfer, V.; Winkelmann, N.; Haferlach, C.; Cross, N.C.P.; A Hochhaus, A.; et al. Molecular characterization of EZH2 mutant patients with myelodysplastic/myeloproliferative neoplasms. Leukemia 2017, 31, 1936–1943. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.; Thakker, N.; Corcoran, C.M.; Donnai, D.; Perveen, R.; Schneider, A.; Hadley, D.W.; Tifft, C.; Zhang, L.; Wilkie, A.O.M.; et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat. Genet. 2004, 36, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Abuhadra, N.; Mukherjee, S.; Al-Issa, K.; Adema, V.; Hirsch, C.M.; Advani, A.; Przychodzen, B.; Makhoul, A.; Awada, H.; Maciejewski, J.P.; et al. BCOR and BCORL1 mutations in myelodysplastic syndromes (MDS): Clonal architecture and impact on outcomes. Leuk. Lymphoma 2019, 60, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Gearhart, M.D.; Gery, S.; Shojaee, S.; Yang, H.; Sun, H.; Lin, D.-C.; Bai, J.-W.; Mead, M.; Zhao, Z.; et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia 2016, 30, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Nazha, A.; Narkhede, M.; Radivoyevitch, T.; Seastone, D.J.; Patel, B.J.; Gerds, A.T.; Mukherjee, S.; Kalaycio, M.; Advani, A.; Przychodzen, B.; et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016, 30, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Nazha, A.; Al-Issa, K.; Hamilton, B.K.; Radivoyevitch, T.; Gerds, A.; Mukherjee, S.; Adema, V.; Zarzour, A.; Abuhadra, N.; Patel, B.J.; et al. Adding molecular data to prognostic models can improve predictive power in treated patients with myelodysplastic syndromes. Leukemia 2017, 31, 2848–2850. [Google Scholar] [CrossRef]
- Ichikawa, M.; Yoshimi, A.; Nakagawa, M.; Nishimoto, N.; Watanabe-Okochi, N.; Kurokawa, M. A role for RUNX1 in hematopoiesis and myeloid leukemia. Int. J. Hematol. 2013, 97, 726–734. [Google Scholar] [CrossRef]
- Song, W.-J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 1999, 23, 166–175. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lin, L.-I.; Tang, J.-L.; Ko, B.-S.; Tsay, W.; Chou, W.-C.; Yao, M.; Wu, S.-J.; Tseng, M.-H.; Tien, H.-F. RUNX1 gene mutation in primary myelodysplastic syndrome—The mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. Br. J. Haematol. 2007, 139, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhou, F.; Cai, X.; Chao, H.; Zhang, R.; Chen, S. Mutational landscape of patients with acute myeloid leukemia or myelodysplastic syndromes in the context of RUNX1 mutation. Hematology 2020, 25, 211–218. [Google Scholar] [CrossRef]
- Kaisrlikova, M.; Vesela, J.; Kundrat, D.; Votavova, H.; Merkerova, M.D.; Krejcik, Z.; Divoky, V.; Jedlicka, M.; Fric, J.; Klema, J.; et al. RUNX1 mutations contribute to the progression of MDS due to disruption of antitumor cellular defense: A study on patients with lower-risk MDS. Leukemia 2022, 36, 1898–1906. [Google Scholar] [CrossRef]
- Wu, P.; Weng, J.; Li, M.; Lu, Z.; Deng, C.; Sun, Q.; Xu, R.; Geng, S.; Du, X. Co-occurrence of RUNX1 and ASXL1 mutations underlie poor response and outcome for MDS patients treated with HMAs. Am. J. Transl. Res. 2019, 11, 3651–3658. [Google Scholar]
- Nakagawa, T.; Saitoh, S.; Imoto, S.; Itoh, M.; Tsutsumi, M.; Hikiji, K.; Nakamura, H.; Matozaki, S.; Ogawa, R.; Nakao, Y.; et al. Multiple point mutation of N-ras and K-ras oncogenes in myelodysplastic syndrome and acute myelogenous leukemia. Oncology 1992, 49, 114–122. [Google Scholar] [CrossRef]
- Paquette, R.L.; Landaw, E.M.; Pierre, R.V.; Kahan, J.; Lübbert, M.; Lazcano, O.; Isaac, G.; McCormick, F.; Koeffler, H.P. N-ras mutations are associated with poor prognosis and increased risk of leukemia in myelodysplastic syndrome. Blood 1993, 82, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Al-Kali, A.; Quintás-Cardama, A.; Luthra, R.; Bueso-Ramos, C.; Pierce, S.; Kadia, T.; Borthakur, G.; Estrov, Z.; Jabbour, E.; Faderl, S.; et al. Prognostic impact of RAS mutations in patients with myelodysplastic syndrome. Am. J. Hematol. 2013, 88, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Padua, R.; Guinn, B.-A.; Al-Sabah, A.; Smith, M.; Taylor, C.; Pettersson, T.; Ridge, S.; Carter, G.; White, D.; Oscier, D.; et al. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: A 10-year follow-up. Leukemia 1998, 12, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, J.; Liu, Y.; Sidik, H.; Young, K.H.; Lodish, H.F.; Fleming, M.D. Oncogenic Kras-induced leukemogeneis: Hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood 2009, 113, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.; Subrahmanyam, R.; Ren, R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood 2006, 108, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Haferlach, T.; Kern, W.; Haferlach, C.; Schnittger, S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica 2007, 92, 744–752. [Google Scholar] [CrossRef]
- Daver, N.; Strati, P.; Jabbour, E.; Kadia, T.; Luthra, R.; Wang, S.; Patel, K.; Ravandi, F.; Cortes, J.; Dong, X.Q.; et al. FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am. J. Hematol. 2012, 88, 56–59. [Google Scholar] [CrossRef]
- Levis, M.; Small, D. FLT3: ITDoes matter in leukemia. Leukemia 2003, 17, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Patel, K.P.; Thompson, P.A.; DiNardo, C.; Takahashi, K.; Cabrero, M.; Borthakur, G.; Cortes, J.; Konopleva, M.; Kadia, T.; et al. Detectable FLT3-ITD or RAS mutation at the time of transformation from MDS to AML predicts for very poor outcomes. Leuk. Res. 2015, 39, 1367–1374. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Maiti, A.; Kadia, T.M.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Muto, T.; Walker, C.S.; Choi, K.; Hueneman, K.; Smith, M.A.; Gul, Z.; Garcia-Manero, G.; Ma, A.; Zheng, Y.; Starczynowski, D.T. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol. 2020, 21, 535–545. [Google Scholar] [CrossRef]
- Kong, T.; Laranjeira, A.B.A.; Yang, K.; Fisher, D.A.C.; Yu, L.; De La Frégonnière, L.P.; Wang, A.Z.; Ruzinova, M.B.; Fowles, J.S.; Fulbright, M.C.; et al. DUSP6 mediates resistance to JAK2 inhibition and drives leukemic progression. Nat. Cancer 2022, 4, 108–127. [Google Scholar] [CrossRef]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2018, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Ghannam, J.; Nosiri, C.; Gui, G.; Goswami, M.; Calvo, K.R.; Lindblad, K.E.; Oetjen, K.A.; Wilkerson, M.D.; Soltis, A.R.; et al. Personalized Single-Cell Proteogenomics to Distinguish Acute Myeloid Leukemia from Nonmalignant Clonal Hematopoiesis. Blood Cancer Discov. 2021, 2, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Sampson, A.M.; Ishikawa, C.; Hueneman, K.M.; Choi, K.; Pujato, M.A.; Chutipongtanate, S.; Wyder, M.; Haffey, W.D.; O’Brien, E.; et al. Blocking UBE2N abrogates oncogenic immune signaling in acute myeloid leukemia. Sci. Transl. Med. 2022, 14. [Google Scholar] [CrossRef]
- Agarwal, P.; Li, H.; Choi, K.; Hueneman, K.; He, J.; Welner, R.S.; Starczynowski, D.T.; Bhatia, R. TNF-α-induced alterations in stromal progenitors enhance leukemic stem cell growth via CXCR2 signaling. Cell Rep. 2021, 36, 109386. [Google Scholar] [CrossRef] [PubMed]
- Schinke, C.; Giricz, O.; Carolina, S.; Shastri, A.; Gordon, S.; Barreyro, L.; Bhagat, T.; Bhattacharyya, S.; Ramachandra, N.; Bartenstein, M.; et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015, 125, 3144–3152. [Google Scholar] [CrossRef]
- Karantanos, T.; Teodorescu, P.; Perkins, B.; Christodoulou, I.; Esteb, C.; Varadhan, R.; Helmenstine, E.; Rajkhowa, T.; Paun, B.C.; Bonifant, C.; et al. The role of the atypical chemokine receptor CCRL2 in myelodysplastic syndrome and secondary acute myeloid leukemia. Sci. Adv. 2022, 8, eabl8952. [Google Scholar] [CrossRef]
- Karantanos, T.; Teodorescu, P.; Arvanitis, M.; Perkins, B.; Jain, T.; DeZern, A.E.; Dalton, W.B.; Christodoulou, I.; Paun, B.C.; Varadhan, R.; et al. CCRL2 affects the sensitivity of myelodysplastic syndrome and secondary acute myeloid leukemia cells to azacytidine. Haematologica 2022. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Othus, M.; Wood, B.L.; Walter, R.B.; Becker, P.S.; Percival, M.-E.; Abkowitz, J.L.; Appelbaum, F.R.; Estey, E.H. Comparison of myeloid blast counts and variant allele frequencies of gene mutations in myelodysplastic syndrome with excess blasts and secondary acute myeloid leukemia. Leuk. Lymphoma 2020, 62, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Garcia-Manero, G.; Pierce, S.; Nazha, A.; Bueso-Ramos, C.; Jabbour, E.; Ravandi, F.; Cortes, J.; Kantarjian, H. Interactions and relevance of blast percentage and treatment strategy among younger and older patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Am. J. Hematol. 2016, 91, 227–232. [Google Scholar] [CrossRef]
- Fang, H.; He, R.; Chiu, A.; Viswanatha, D.S.; Ketterling, R.P.; Patnaik, M.S.; Reichard, K.K. Genetic Factors in Acute Myeloid Leukemia with Myelodysplasia-Related Changes. Am. J. Clin. Pathol. 2020, 153, 656–663. [Google Scholar] [CrossRef]
- Malcovati, L.; Galli’, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; Kooy, M.V.M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef]
- Short, N.J.; Montalban-Bravo, G.; Hwang, H.; Ning, J.; Franquiz, M.J.; Kanagal-Shamanna, R.; Patel, K.P.; DiNardo, C.D.; Ravandi, F.; Garcia-Manero, G.; et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020, 4, 5681–5689. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Siddon, A.J.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Cin, P.D.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. [Google Scholar] [CrossRef]
- Ertz-Archambault, N.; Kosiorek, H.; Taylor, G.E.; Kelemen, K.; Dueck, A.; Castro, J.; Marino, R.; Gauthier, S.; Finn, L.; Sproat, L.Z.; et al. Association of Therapy for Autoimmune Disease with Myelodysplastic Syndromes and Acute Myeloid Leukemia. JAMA Oncol. 2017, 3, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, J.; Cotta, C.V.; Medeiros, L.J.; Lin, P.; Singh, S.; Sharma, S.; Jain, M.; Chauhan, R. Myelodysplastic syndrome with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) has a high risk for progression to acute myeloid leukemia. Am. J. Clin. Pathol. 2011, 136, 282–288. [Google Scholar] [CrossRef]
- Fang, H.; Yabe, M.; Zhang, X.; Kim, Y.; Wu, X.; Wei, P.; Chi, S.; Zheng, L.; Garcia-Manero, G.; Shao, L.; et al. Myelodysplastic syndrome with t(6;9)(p22;q34.1)/DEK-NUP214 better classified as acute myeloid leukemia? A multicenter study of 107 cases. Mod. Pathol. 2021, 34, 1143–1152. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Sasaki, K.; Patel, K.; Gañán-Gómez, I.; Jabbour, E.; Kadia, T.; Ravandi, F.; Dinardo, C.; Borthakur, G.; et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv. 2019, 3, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Zarka, J.; Sasaki, K.; Qiao, W.; Pak, D.; Ning, J.; Short, N.J.; Haddad, F.; Tang, Z.; Patel, K.P.; et al. Predictors of outcomes in adults with acute myeloid leukemia and KMT2A rearrangements. Blood Cancer J. 2021, 11, 162. [Google Scholar] [CrossRef]
- Taube, F.; Georgi, J.A.; Kramer, M.; Stasik, S.; Middeke, J.M.; Röllig, C.; Krug, U.; Krämer, A.; Scholl, S.; Hochhaus, A.; et al. CEBPA mutations in 4708 patients with acute myeloid leukemia: Differential impact of bZIP and TAD mutations on outcome. Blood 2022, 139, 87–103. [Google Scholar] [CrossRef]
- Wakita, S.; Sakaguchi, M.; Oh, I.; Kako, S.; Toya, T.; Najima, Y.; Doki, N.; Kanda, J.; Kuroda, J.; Mori, S.; et al. Prognostic impact of CEBPA bZIP domain mutation in acute myeloid leukemia. Blood Adv. 2022, 6, 238–247. [Google Scholar] [CrossRef]
- Forghieri, F.; Nasillo, V.; Paolini, A.; Bettelli, F.; Pioli, V.; Giusti, D.; Gilioli, A.; Colasante, C.; Acquaviva, G.; Riva, G.; et al. NPM1-Mutated Myeloid Neoplasms with <20% Blasts: A Really Distinct Clinico-Pathologic Entity? Int. J. Mol. Sci. 2020, 21, 8975. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Évid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Short, N.J.; Zhou, S.; Fu, C.; Berry, D.A.; Walter, R.B.; Freeman, S.D.; Hourigan, C.S.; Huang, X.; Gonzalez, G.N.; Hwang, H.; et al. Association of Measurable Residual Disease with Survival Outcomes in Patients with Acute Myeloid Leukemia: A Systematic Review and Meta-analysis. JAMA Oncol. 2020, 6, 1890. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Wang, S.A.; Jorgensen, J.; Kadia, T.M.; Daver, N.G.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; Borthakur, G.; et al. Prognostic value of measurable residual disease after venetoclax and decitabine in acute myeloid leukemia. Blood Adv. 2021, 5, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Simoes, C.; Paiva, B.; Martínez-Cuadrón, D.; Bergua, J.-M.; Vives, S.; Algarra, L.; Tormo, M.; Martinez, P.; Serrano, J.; Herrera, P.; et al. Measurable residual disease in elderly acute myeloid leukemia: Results from the PETHEMA-FLUGAZA phase 3 clinical trial. Blood Adv. 2021, 5, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Döhner, K.; Thiede, C.; Jahn, N.; Panina, E.; Gambietz, A.; Larson, R.A.; Prior, T.W.; Marcucci, G.; Jones, D.; Krauter, J.; et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood 2020, 135, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Röllig, C.; Montesinos, P.; Martínez-Cuadrón, D.; Barragan, E.; García, R.; Botella, C.; Martínez, P.; Ravandi, F.; Kadia, T.; et al. Chromosomal Abnormalities and Prognosis in NPM1-Mutated Acute Myeloid Leukemia: A Pooled Analysis of Individual Patient Data from Nine International Cohorts. J. Clin. Oncol. 2019, 37, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.; Nomdedeu, J.F. CEBPA bZip mutations: Just a single shot. Blood 2021, 138, 1091–1092. [Google Scholar] [CrossRef]
- Madanat, Y.F.; Xie, Z.; Zeidan, A.M. Advances in myelodysplastic syndromes: Promising novel agents and combination strategies. Expert Rev. Hematol. 2023, 16, 51–63. [Google Scholar] [CrossRef]
- Estey, E.H.; Hasserjian, R.P.; Döhner, H. Distinguishing AML from MDS: A fixed blast percentage may no longer be optimal. Blood 2022, 139, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, S.; Ziagkos, D.; de Wreede, L.C.; van Biezen, A.; Finke, J.; Platzbecker, U.; Niederwieser, D.; Einsele, H.; Bethge, W.; Schleuning, M.; et al. Allogeneic Stem Cell Transplantation for Patients Age ≥ 70 Years with Myelodysplastic Syndrome: A Retrospective Study of the MDS Subcommittee of the Chronic Malignancies Working Party of the EBMT. Biol. Blood Marrow Transplant. 2016, 23, 44–52. [Google Scholar] [CrossRef]
- Atallah, E.; Logan, B.; Chen, M.; Cutler, C.; Deeg, J.; Jacoby, M.; Champlin, R.; Nishihori, T.; Confer, D.; Gajewski, J.; et al. Comparison of Patient Age Groups in Transplantation for Myelodysplastic Syndrome: The Medicare Coverage with Evidence Development Study. JAMA Oncol. 2020, 6, 486. [Google Scholar] [CrossRef]
- Cutler, C.S.; Lee, S.J.; Greenberg, P.; Deeg, H.J.; Pérez, W.S.; Anasetti, C.; Bolwell, B.J.; Cairo, M.S.; Gale, R.P.; Klein, J.P.; et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: Delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 2004, 104, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Franke, G.-N.; Lückemeier, P.; Platzbecker, U. Allogeneic Stem-Cell Transplantation in Patients with Myelodysplastic Syndromes and Prevention of Relapse. Clin. Lymphoma Myeloma Leuk. 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Kröger, N.; Iacobelli, S.; Franke, G.-N.; Platzbecker, U.; Uddin, R.; Hübel, K.; Scheid, C.; Weber, T.; Robin, M.; Stelljes, M.; et al. Dose-Reduced Versus Standard Conditioning Followed by Allogeneic Stem-Cell Transplantation for Patients with Myelodysplastic Syndrome: A Prospective Randomized Phase III Study of the EBMT (RICMAC Trial). J. Clin. Oncol. 2017, 35, 2157–2164. [Google Scholar] [CrossRef]
- Scott, B.L.; Pasquini, M.C.; Fei, M.; Fraser, R.; Wu, J.; Devine, S.M.; Porter, D.L.; Maziarz, R.T.; Warlick, E.; Fernandez, H.F.; et al. Myeloablative versus Reduced-Intensity Conditioning for Hematopoietic Cell Transplantation in Acute Myelogenous Leukemia and Myelodysplastic Syndromes—Long-Term Follow-Up of the BMT CTN 0901 Clinical Trial. Transplant. Cell. Ther. 2021, 27, 483.e1–483.e6. [Google Scholar] [CrossRef]
- Webster, J.A.; Yogarajah, M.; Zahurak, M.; Symons, H.; Dezern, A.E.; Gojo, I.; Prince, G.T.; Morrow, J.; Jones, R.J.; Smith, B.D.; et al. A phase II study of azacitidine in combination with granulocyte-macrophage colony-stimulating factor as maintenance treatment, after allogeneic blood or marrow transplantation in patients with poor-risk acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Leuk. Lymphoma 2021, 62, 3181–3191. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Allen, C.; Mirza, A.-S.; Grimshaw, A.A.; Giri, S.; Podoltsev, N.A.; Gowda, L.; Cho, C.; Tallman, M.S.; Zeidan, A.M.; et al. Hypomethylating Agents and FLT3 Inhibitors as Maintenance Treatment for Acute Myeloid Leukemia and Myelodysplastic Syndrome After Allogeneic Hematopoietic Stem Cell Transplantation–A Systematic Review and Meta-Analysis. Transplant. Cell. Ther. 2021, 27, 997.e1–997.e11. [Google Scholar] [CrossRef]
- Oran, B.; de Lima, M.; Garcia-Manero, G.; Thall, P.F.; Lin, R.; Popat, U.; Alousi, A.M.; Hosing, C.; Giralt, S.; Rondon, G.; et al. A phase 3 randomized study of 5-azacitidine maintenance vs observation after transplant in high-risk AML and MDS patients. Blood Adv. 2020, 4, 5580–5588. [Google Scholar] [CrossRef]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Prébet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Adès, L.; et al. Outcome of High-Risk Myelodysplastic Syndrome after Azacitidine Treatment Failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef]
- Ball, B.; Komrokji, R.S.; Adès, L.; Sekeres, M.A.; DeZern, A.E.; Pleyer, L.; Vey, N.; Almeida, A.; Germing, U.; Cluzeau, T.; et al. Evaluation of induction chemotherapies after hypomethylating agent failure in myelodysplastic syndromes and acute myeloid leukemia. Blood Adv. 2018, 2, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, Q.; Li, S.; Wang, X. Resistance to Hypomethylating Agents in Myelodysplastic Syndrome and Acute Myeloid Leukemia from Clinical Data and Molecular Mechanism. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.K.; Nguyen, A.; Shi, T.; Tang, L.; Ni, X.; Escoubet, L.; MacBeth, K.J.; DiMartino, J.; Wells, J.A. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc. Natl. Acad. Sci. USA 2019, 116, 695–700. [Google Scholar] [CrossRef]
- Bowler, E.H.; Bell, J.; Divecha, N.; Skipp, P.; Ewing, R.M. Proteomic Analysis of Azacitidine-Induced Degradation Profiles Identifies Multiple Chromatin and Epigenetic Regulators Including Uhrf1 and Dnmt1 as Sensitive to Azacitidine. J. Proteome Res. 2019, 18, 1032–1042. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Papaemmanuil, E.; Beck, D.; Deshpande, N.P.; Verma, A.; Kumari, A.; Woll, P.S.; Richards, L.A.; Knezevic, K.; Chandrakanthan, V.; et al. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep. 2017, 20, 572–585. [Google Scholar] [CrossRef]
- Valencia, A.; Masala, E.; Rossi, A.; Martino, A.; Sanna, A.; Buchi, F.; Canzian, F.; Cilloni, D.; Gaidano, V.; Voso, M.T.; et al. Expression of nucleoside-metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia 2013, 28, 621–628. [Google Scholar] [CrossRef]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.-H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by abt-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Bazinet, A.; Darbaniyan, F.; Jabbour, E.; Montalban-Bravo, G.; Ohanian, M.; Chien, K.; Kadia, T.; Takahashi, K.; Masarova, L.; Short, N.; et al. Azacitidine plus venetoclax in patients with high-risk myelodysplastic syndromes or chronic myelomonocytic leukaemia: Phase 1 results of a single-centre, dose-escalation, dose-expansion, phase 1–2 study. Lancet Haematol. 2022, 9, e756–e765. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V.; Pratz, K.; Dohner, H.; Recher, C.; Thirman, M.J.; Dinardo, C.D.; Fenaux, P.; Schuh, A.C.; Wei, A.H.; Pigneux, A.; et al. Venetoclax and azacitidine combination in chemotherapy ineligible untreated patients with therapy-related myeloid neoplasms, antecedent myelodysplastic syndromes, or myelodysplastic/myeloproliferative neoplasms. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 4–8 June 2021. [Google Scholar]
- Guo, Y.; Liu, B.; Deng, L.; Qiao, Y.; Jian, J. The efficacy and adverse events of venetoclax in combination with hypomethylating agents treatment for patients with acute myeloid leukemia and myelodysplastic syndrome: A systematic review and meta-analysis. Hematology 2020, 25, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Borate, U.; Pollyea, D.A.; Brunner, A.M.; Roncolato, F.; Garcia, J.S.; Filshie, R.; Odenike, O.; Watson, A.M.; Krishnadasan, R.; et al. A phase 1b study of venetoclax and azacitidine combination in patients with relapsed or refractory myelodysplastic syndromes. Am. J. Hematol. 2022, 98, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Derkach, A.; Gowda, L.; Menghrajani, K.; DeWolf, S.; Ruiz, J.D.; Ponce, D.M.; Shaffer, B.C.; Tamari, R.; Young, J.W.; et al. Venetoclax-based combinations in AML and high-risk MDS prior to and following allogeneic hematopoietic cell transplant. Leuk. Lymphoma 2021, 62, 3394–3401. [Google Scholar] [CrossRef]
- Yang, T.-T.; Song, X.-L.; Zhao, Y.-M.; Ye, B.-D.; Luo, Y.; Xiao, H.-W.; Chen, Y.; Fu, H.-R.; Yu, J.; Liu, L.-Z.; et al. Outcome after allogeneic hematopoietic stem cell transplantation following Venetoclax-based therapy among AML and MDS patients. Ann. Hematol. 2022, 101, 2731–2741. [Google Scholar] [CrossRef]
- Bello, C.; Yu, D.; Komrokji, R.S.; Zhu, W.; Wetzstein, G.A.; List, A.F.; Lancet, J.E. Outcomes after induction chemotherapy in patients with acute myeloid leukemia arising from myelodysplastic syndrome. Cancer 2010, 117, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients with Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- E Lancet, J.; Uy, G.L.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; A Strickland, S.; Hogge, D.; Solomon, S.R.; Bixby, D.L.; et al. CPX-351 versus 7+3 cytarabine and daunorubicin chemotherapy in older adults with newly diagnosed high-risk or secondary acute myeloid leukaemia: 5-year results of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2021, 8, e481–e491. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Ball, S.; Loghavi, S.; Zeidan, A.M. TP53-altered higher-risk myelodysplastic syndromes/neoplasms and acute myeloid leukemia: A distinct genetic entity with unique unmet needs. Leuk. Lymphoma 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Bykov, V.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef]
- Ali, D.; Jönsson-Videsäter, K.; Deneberg, S.; Bengtzén, S.; Nahi, H.; Paul, C.; Lehmann, S. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur. J. Haematol. 2010, 86, 206–215. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019, 105, 1539–1551. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Gallacher, P.; Wennborg, A.; Hickman, D.K.; et al. Phase I and Expansion Study of Eprenetapopt (APR-246) in Combination with Venetoclax (VEN) and Azacitidine (AZA) in TP53-Mutant Acute Myeloid Leukemia (AML). Blood 2021, 138, 3409. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.-P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2020, 9, 1380. [Google Scholar] [CrossRef]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.A.; Asch, A.S.; Mannis, G.N.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in frontline TP53m AML patients: Phase 1b results. J. Clin. Oncol. 2022, 40, 7020. [Google Scholar] [CrossRef]
- Naval, G.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (pts) with Newly Diagnosed Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. In Proceedings of the ASH Annual Meeting, Atlanta, GA, USA, 11–14 December 2021. [Google Scholar]
- Jin, J.; Hu, C.; Yu, M.; Chen, F.; Ye, L.; Yin, X.; Zhuang, Z.; Tong, H. Prognostic value of isocitrate dehydrogenase mutations in myelodysplastic syndromes: A retrospective cohort study and meta-analysis. PLoS ONE 2014, 9, e100206. [Google Scholar] [CrossRef]
- Patnaik, M.M.; A Hanson, C.; Hodnefield, J.M.; Lasho, T.L.; Finke, C.M.; A Knudson, R.; Ketterling, R.; Pardanani, A.; Tefferi, A. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: A Mayo Clinic Study of 277 patients. Leukemia 2011, 26, 101–105. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Frankfurt, O.; Schuh, A.C.; Döhner, H.; Martinelli, G.; Patel, P.A.; Raffoux, E.; et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination with Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2021, 39, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; A Pollyea, D.; Roboz, G.J.; Collins, R.; A Sekeres, M.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Venugopal, S.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Montalban-Bravo, G.; Wang, X.; E Carraway, H.; Sekeres, M.A.; Sukkur, A.; et al. Targeted therapy with the mutant IDH2 inhibitor enasidenib for high-risk IDH2-mutant myelodysplastic syndrome. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Fathi, A.T.; Kim, H.T.; Soiffer, R.J.; Levis, M.J.; Li, S.; Kim, A.S.; Mims, A.S.; DeFilipp, Z.; El-Jawahri, A.; McAfee, S.L.; et al. Enasidenib as maintenance following allogeneic hematopoietic cell transplantation for IDH2-mutated myeloid malignancies. Blood Adv. 2022, 6, 5857–5865. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Genetic Alteration | Incidence in MDS/AML | Possible Biologic Mechanism | Clinical Significance | References |
|---|---|---|---|---|
| Chromosomal abnormalities | ||||
| Deletion of 5q | 10–15% | Defective ribosomal biogenesis leads to degradation of MDM2 | Overall good prognosis | [16,17,18,19,20,21] |
| Deletion of chromosome 7/7q | ~20% | Loss of chromosome 7 is linked with compromised expression of interferon gamma pathway genes. | Overall poor prognosis | [23,24,25,26,27,28] |
| Complex karyotype (CK) | 30% | Mutations in TP53 and frequent LOH of this gene | Poor prognosis/survival | [29,30,31,32,33,34,35] |
| Somatic mutations | ||||
| SF3B1 | 10–15% | Alterations in the spliceosome that frequently lead to non-sense-mediated decay (NMD) | Overall good prognosis but specific variants may have different impact | [39,40,41,42] |
| SRSF2 | 10–15% | Alteration of splicing and DNA damage accumulation. | Poor prognosis | [43,44,45,46] |
| U2AF1 | 5–15% | Suppression of ATG7 levels is associated with increased oxidative stress and chromosomal instability | Poor prognosis/survival | [47,48,49] |
| ZRSR2 | 5% | Retention of U12-dependent introns causing aberrant splicing | Negative impact on survival less strong compared to SRSF2 and U2AF1 | [31,50,51] |
| STAG2 | ~10% | Dysplasia caused by abnormal hematopoietic stem cell maturation along with additional oncogenic mutations | Poor prognosis | [53,54,55,56,57] |
| EZH2 | 4–5% | Gain of function mutations lead to improper control of cell proliferation | Poor prognosis/survival | [58,59,60,63,65,93] |
| BCOR | <5% | Loss of function mutations causing increased myeloid cell proliferation | Neutral impact on survival | [69,70,71,72] |
| RUNX1 | 10–15% | Mutated gene transcripts block gene activation responsible proper hematopoiesis | Poor prognosis/survival | [67,74,76,77,78,79] |
| NRAS/KRAS | 4–5% | RAS gene mutations activate RAS/Raf/MEK and PI3K/AKT pathways | Poor prognosis/survival | [80,81,82,83,84,85] |
| FLT3-ITD | 0.6–6% | Mutations cause constitutive auto-phosphorylation of the FLT3 receptor leading to activation of downstream pathways | Poor prognosis/survival | [11,88,89] |
| TP53 | 5–10% | Mutations lead to impaired DNA damage repairs, LOH is critical for the prognosis | Poor prognosis/survival | [90,91,92] |
| Description | Clonality | Cytopenia | Dysplasia | Increased Blasts | Genetic Testing | |
|---|---|---|---|---|---|---|
| CH | Lineage derived from a single clone | Yes | No | No | No | General term to define somatic mutations or cytogenetic abnormalities or copy number mutations 1 |
| CHIP | Lineage derived from a single clone | Yes | No | No | No | Somatic mutation in a myeloid neoplasm driver gene with VAF ≥ 2%, or Non-MDS defining clonal cytogenetic abnormality in the absence of a myeloid neoplasm or cytopenia |
| CCUS | Lineage derived from a single clone + Cytopenia | Yes | Yes | No | No | Somatic mutation in a myeloid neoplasm driver gene with VAF ≥ 2% 2 |
| VEXAS syndrome | CH + anemia | Yes | Yes | No | No | Somatic mutation in UBA1 gene |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavras, P.D.; Sinanidis, I.; Tsakiroglou, P.; Karantanos, T. Understanding the Continuum between High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. Int. J. Mol. Sci. 2023, 24, 5018. https://doi.org/10.3390/ijms24055018
Zavras PD, Sinanidis I, Tsakiroglou P, Karantanos T. Understanding the Continuum between High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2023; 24(5):5018. https://doi.org/10.3390/ijms24055018
Chicago/Turabian StyleZavras, Phaedon D., Ilias Sinanidis, Panagiotis Tsakiroglou, and Theodoros Karantanos. 2023. "Understanding the Continuum between High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia" International Journal of Molecular Sciences 24, no. 5: 5018. https://doi.org/10.3390/ijms24055018
APA StyleZavras, P. D., Sinanidis, I., Tsakiroglou, P., & Karantanos, T. (2023). Understanding the Continuum between High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. International Journal of Molecular Sciences, 24(5), 5018. https://doi.org/10.3390/ijms24055018