
Frontline Sodium Channel-Blocking Antiseizure Medicine Use Promotes Future Onset of Drug-Resistant Chronic Seizures

Abstract
1. Introduction
2. Results
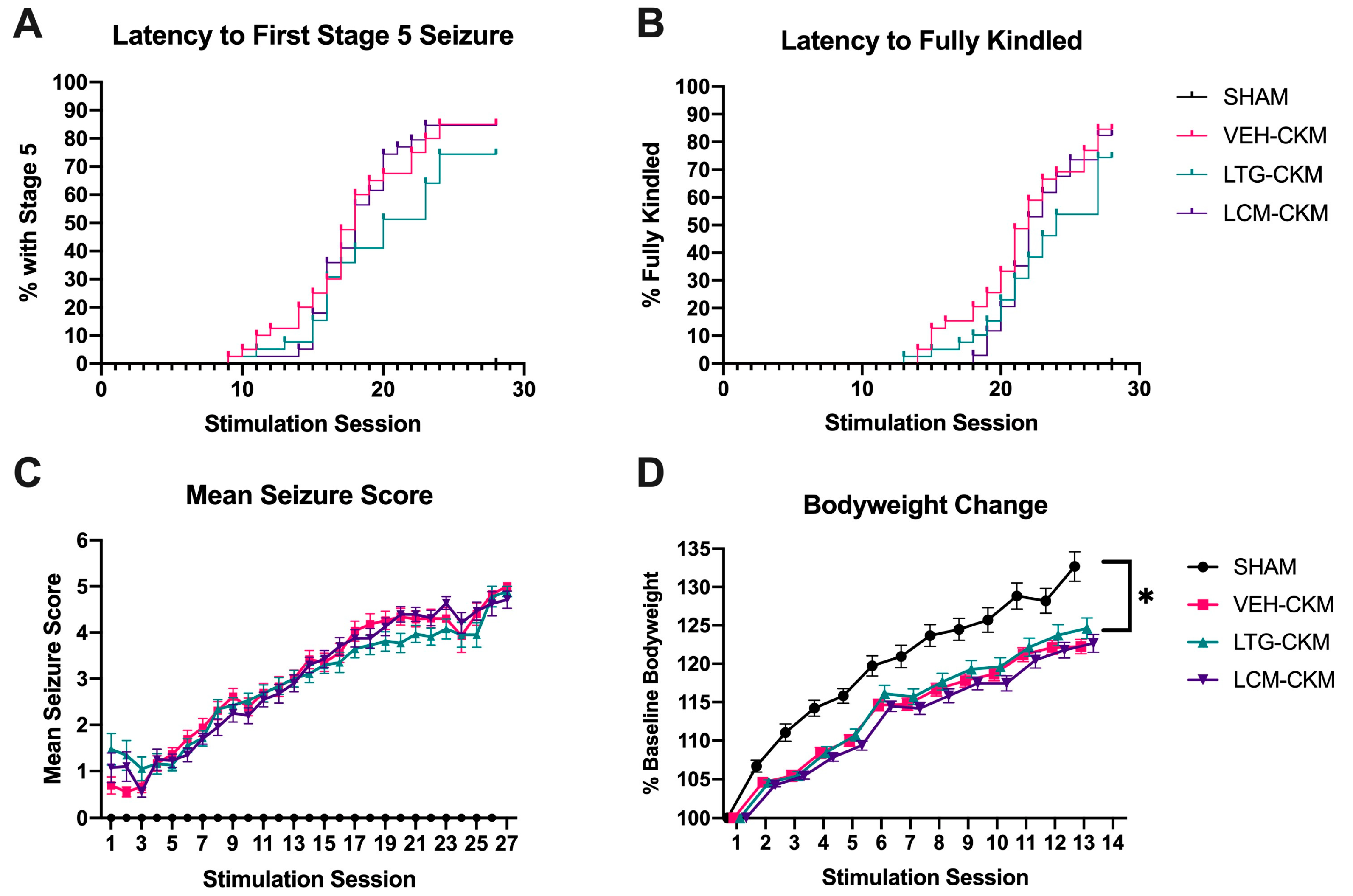
2.1. Administration of Neither LTG nor LCM at Anticonvulsant Doses Delays Kindling Acquistion
2.2. Administration of Either LTG or LCM at Anticonvulsant Doses during Kindling Reduces Subsequent Kindled Seizure Cross-Sensitivity
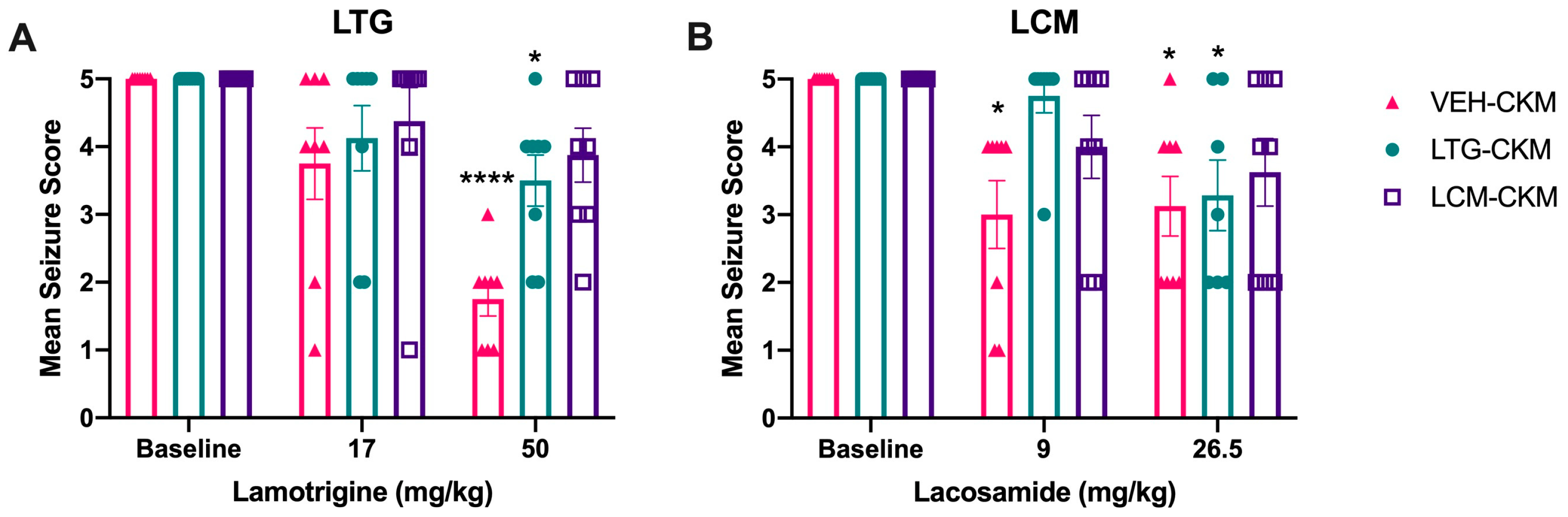
2.3. Administration of Either LTG or LCM at Anticonvulsant Doses during Kindling Leads to Significant Drug Resistance across a Range of ASMs with Diverse Mechanims
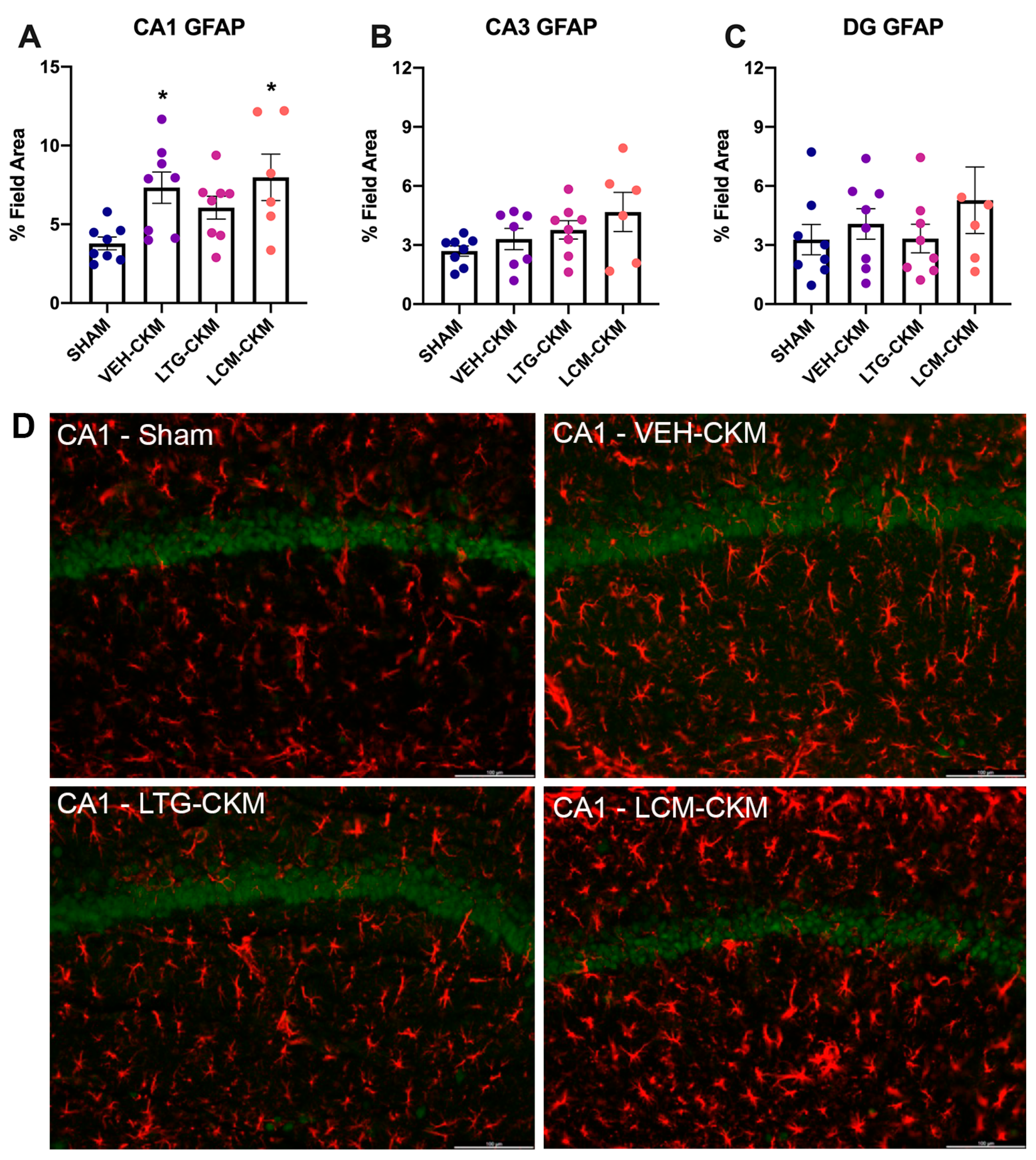
2.4. LTG Administration during Corneal Kindling Blunts Chronic Seizure-Induced Increases in Reactive Gliosis in Area CA1 of Dorsal Hippocampus
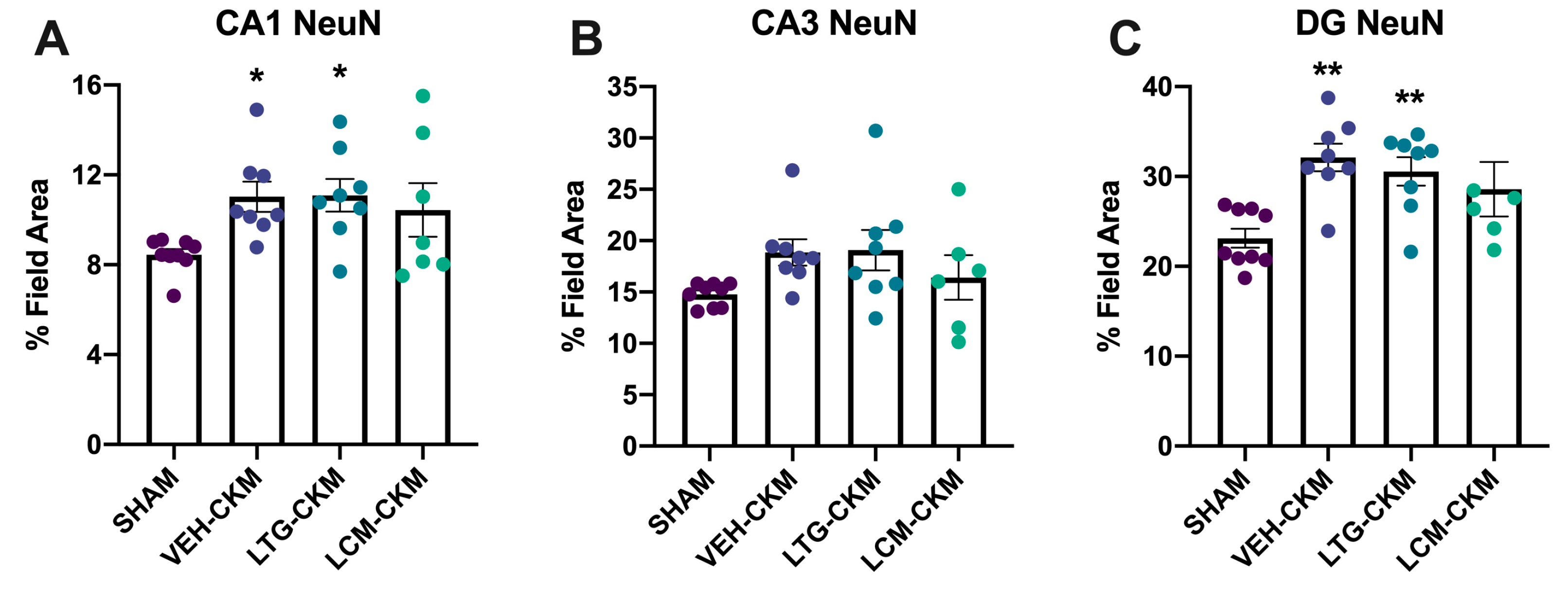
2.5. LCM Administration during Corneal Kindling Blunts Chronic Seizure-Induced Increases in Neuronal Density in Dorsal Hippocampus
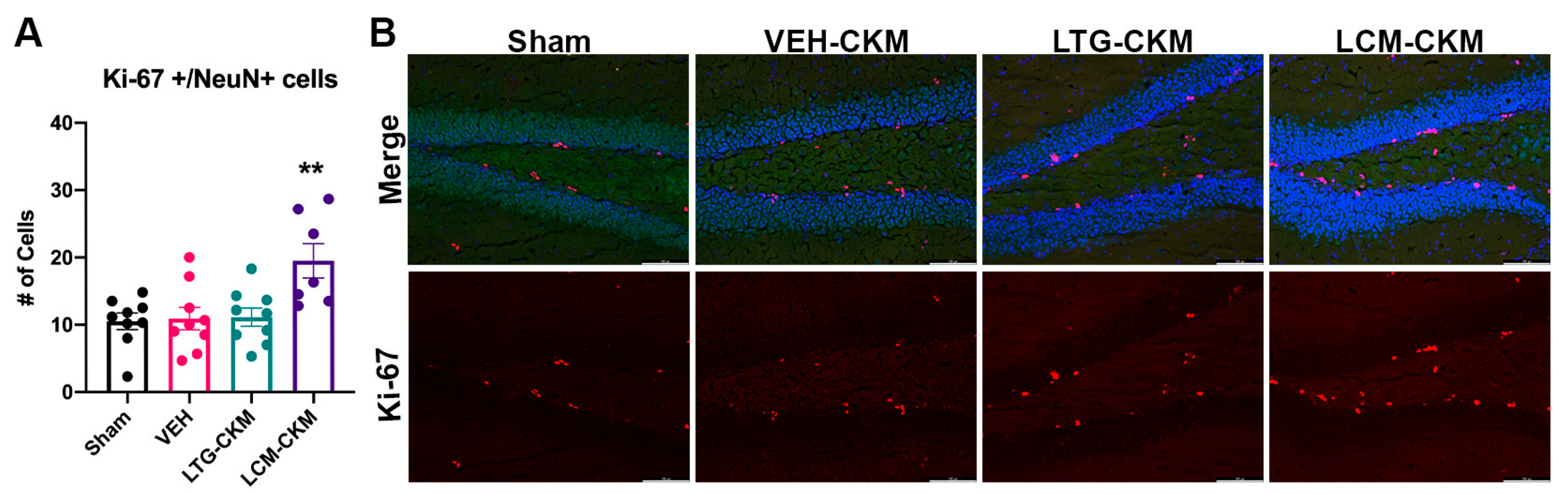
2.6. LCM Administration during Corneal Kindling Is Associated with Increased Neurogenesis in Dentate Gyrus
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Matagne, A.; Klitgaard, H. Validation of corneally kindled mice: A sensitive screening model for partial epilepsy in man. Epilepsy Res. 1998, 31, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, H.; Matagne, A.; Gobert, J.; Wulfert, E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur. J. Pharm. 1998, 353, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.L.; Johnson, K.; Billingsley, P.; Huff, J.; Handy, L.J.; Khaleel, R.; Lu, Z.; Mau, M.J.; Pruess, T.H.; Rueda, C.; et al. Validation of a Preclinical Drug Screening Platform for Pharmacoresistant Epilepsy. Neurochem. Res. 2017, 42, 1904–1918. [Google Scholar] [CrossRef]
- Kehne, J.H.; Klein, B.D.; Raeissi, S.; Sharma, S. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem. Res. 2017, 42, 1894–1903. [Google Scholar] [CrossRef]
- Barker-Haliski, M.L.; Vanegas, F.; Mau, M.J.; Underwood, T.K.; White, H.S. Acute cognitive impact of antiseizure drugs in naive rodents and corneal-kindled mice. Epilepsia 2016, 57, 1386–1397. [Google Scholar] [CrossRef]
- Remigio, G.J.; Loewen, J.L.; Heuston, S.; Helgeson, C.; White, H.S.; Wilcox, K.S.; West, P.J. Corneal kindled C57BL/6 mice exhibit saturated dentate gyrus long-term potentiation and associated memory deficits in the absence of overt neuron loss. Neurobiol. Dis. 2017, 105, 221–234. [Google Scholar] [CrossRef]
- Koneval, Z.; Knox, K.M.; White, H.S.; Barker-Haliski, M. Lamotrigine-resistant corneal-kindled mice: A model of pharmacoresistant partial epilepsy for moderate-throughput drug discovery. Epilepsia 2018, 59, 1245–1256. [Google Scholar] [CrossRef]
- Loewen, J.L.; Barker-Haliski, M.L.; Dahle, E.J.; White, H.S.; Wilcox, K.S. Neuronal Injury, Gliosis, and Glial Proliferation in Two Models of Temporal Lobe Epilepsy. J. Neuropathol. Exp. Neurol. 2016, 75, 366–378. [Google Scholar] [CrossRef]
- Srivastava, A.K.; White, H.S. Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy Res. 2013, 104, 26–34. [Google Scholar] [CrossRef]
- Metcalf, C.S.; Huff, J.; Thomson, K.E.; Johnson, K.; Edwards, S.F.; Wilcox, K.S. Evaluation of antiseizure drug efficacy and tolerability in the rat lamotrigine-resistant amygdala kindling model. Epilepsia Open 2019, 4, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Sutula, T.P.; Kotloski, R.J. Kindling: A Model and Phenomenon of Epilepsy. In Models of Seizure and Epilepsy, 2nd ed.; Pitkanen, A., Buckmaster, P.S., Galanopoulou, A.S., Moshe, S.L., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 813–825. [Google Scholar]
- Sutula, T.P. Secondary epileptogenesis, kindling, and intractable epilepsy: A reappraisal from the perspective of neural plasticity. Int. Rev. Neurobiol. 2001, 45, 355–386. [Google Scholar] [PubMed]
- Pawluski, J.L.; Kuchenbuch, M.; Hadjadj, S.; Dieuset, G.; Costet, N.; Vercueil, L.; Biraben, A.; Martin, B. Long-term negative impact of an inappropriate first antiepileptic medication on the efficacy of a second antiepileptic medication in mice. Epilepsia 2018, 59, e109–e113. [Google Scholar] [CrossRef] [PubMed]
- Jandova, K.; Pasler, D.; Antonio, L.L.; Raue, C.; Ji, S.; Njunting, M.; Kann, O.; Kovacs, R.; Meencke, H.J.; Cavalheiro, E.A.; et al. Carbamazepine-resistance in the epileptic dentate gyrus of human hippocampal slices. Brain A J. Neurol. 2006, 129, 3290–3306. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; NC3Rs Reporting Guidelines Working Group. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. J. Gene Med. 2010, 12, 561–563. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef]
- Brandt, C.; Heile, A.; Potschka, H.; Stoehr, T.; Loscher, W. Effects of the novel antiepileptic drug lacosamide on the development of amygdala kindling in rats. Epilepsia 2006, 47, 1803–1809. [Google Scholar] [CrossRef]
- Stohr, T.; Kupferberg, H.J.; Stables, J.P.; Choi, D.; Harris, R.H.; Kohn, H.; Walton, N.; White, H.S. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res. 2007, 74, 147–154. [Google Scholar] [CrossRef]
- Koneval, Z.; Knox, K.; Memon, A.; Zierath, D.K.; White, H.S.; Barker-Haliski, M. Antiseizure drug efficacy and tolerability in established and novel drug discovery seizure models in outbred versus inbred mice. Epilepsia 2020, 61, 2022–2034. [Google Scholar] [CrossRef]
- Florek-Luszczki, M.; Zagaja, M.; Luszczki, J.J. Influence of WIN 55,212-2 on the anticonvulsant and acute neurotoxic potential of clobazam and lacosamide in the maximal electroshock-induced seizure model and chimney test in mice. Epilepsy Res. 2014, 108, 1728–1733. [Google Scholar] [CrossRef]
- Rowley, N.M.; White, H.S. Comparative anticonvulsant efficacy in the corneal kindled mouse model of partial epilepsy: Correlation with other seizure and epilepsy models. Epilepsy Res. 2010, 92, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Knox, K.M.; Beckman, M.; Smith, C.L.; Jayadev, S.; Barker-Haliski, M. Chronic seizures induce sex-specific cognitive deficits with loss of presenilin 2 function. Exp. Neurol. 2023, 361, 114321. [Google Scholar] [CrossRef] [PubMed]
- Kee, N.; Sivalingam, S.; Boonstra, R.; Wojtowicz, J.M. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J. Neurosci. Methods 2002, 115, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Brodie, M.J.; Liew, D.; Kwan, P. Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol. 2018, 75, 279–286. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Epilepsy after the first drug fails: Substitution or add-on? Seizure 2000, 9, 464–468. [Google Scholar] [CrossRef]
- Kumar, S.; Goel, R.K. Pharmacokinetic, pharmacodynamic, and neurochemical investigations of lamotrigine-pentylenetetrazole kindled mice to ascertain it as a reliable model for clinical drug-resistant epilepsy. Anim. Model Exp. Med. 2020, 3, 245–255. [Google Scholar] [CrossRef]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain A J. Neurol. 2006, 129, 18–35. [Google Scholar] [CrossRef]
- Brandt, C.; Bethmann, K.; Gastens, A.M.; Loscher, W. The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol. Dis. 2006, 24, 202–211. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Johnson, M.R. Intrinsic severity as a determinant of antiepileptic drug refractoriness. Epilepsy Currents/American Epilepsy Society 2008, 8, 127–130. [Google Scholar] [CrossRef]
- Thomson, K.E.; Modi, A.; Glauser, T.A.; White, H.S. The Impact of Nonadherence to Antiseizure Drugs on Seizure Outcomes in an Animal Model of Epilepsy. Epilepsia 2017, 58, 1054–1062. [Google Scholar] [CrossRef]
- Kuo, C.C. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol. Pharmacol. 1998, 54, 712–721. [Google Scholar] [PubMed]
- Lipkind, G.M.; Fozzard, H.A. Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol. Pharmacol. 2010, 78, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Hebeisen, S.; Pires, N.; Loureiro, A.I.; Bonifacio, M.J.; Palma, N.; Whyment, A.; Spanswick, D.; Soares-da-Silva, P. Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: A comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Biagini, G.; Perucca, E.; Avoli, M. Lacosamide: A new approach to target voltage-gated sodium currents in epileptic disorders. CNS Drugs 2009, 23, 555–568. [Google Scholar] [CrossRef]
- Beyreuther, B.K.; Freitag, J.; Heers, C.; Krebsfanger, N.; Scharfenecker, U.; Stohr, T. Lacosamide: A review of preclinical properties. CNS Drug Rev. 2007, 13, 21–42. [Google Scholar] [CrossRef]
- Remy, S.; Urban, B.W.; Elger, C.E.; Beck, H. Anticonvulsant pharmacology of voltage-gated Na+ channels in hippocampal neurons of control and chronically epileptic rats. Eur. J. Neurosci. 2003, 17, 2648–2658. [Google Scholar] [CrossRef]
- Catterall, W.A.; Kalume, F.; Oakley, J.C. NaV1.1 channels and epilepsy. J. Physiol. 2010, 588, 1849–1859. [Google Scholar] [CrossRef]
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Miyazaki, H.; Hoshi, N.; Smith, B.J.; Nukina, N.; Goldin, A.L.; Chandy, K.G. Modulation of voltage-gated K+ channels by the sodium channel beta1 subunit. Proc. Natl. Acad. Sci. USA 2012, 109, 18577–18582. [Google Scholar] [CrossRef]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R.; et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef]
- Yang, T.; Chun, Y.W.; Stroud, D.M.; Mosley, J.D.; Knollmann, B.C.; Hong, C.; Roden, D.M. Screening for acute IKr block is insufficient to detect torsades de pointes liability: Role of late sodium current. Circulation 2014, 130, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Luna-Tortos, C.; Fedrowitz, M.; Loscher, W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology 2008, 55, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhang, Y.P.; Hou, Z.; Huang, C.; Zhu, H.; Zhang, C.Q.; Yin, Q. Novel Insights into NeuN: From Neuronal Marker to Splicing Regulator. Mol. Neurobiol. 2016, 53, 1637–1647. [Google Scholar] [CrossRef]
- Parent, J.M.; Lowenstein, D.H. Seizure-induced neurogenesis: Are more new neurons good for an adult brain? Prog. Brain Res. 2002, 135, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.L.; Donermeyer, D.L.; Allen, P.M. A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nat. Immunol. 2012, 13, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.F.; Chen, Y.Z.; Nishimura, Y.; Nishi, K. An amiloride-sensitive and voltage-dependent Na+ channel in an HLA-DR-restricted human T cell clone. J. Immunol. 2000, 165, 83–90. [Google Scholar] [CrossRef]
- Fraser, S.P.; Diss, J.K.; Lloyd, L.J.; Pani, F.; Chioni, A.M.; George, A.J.; Djamgoz, M.B. T-lymphocyte invasiveness: Control by voltage-gated Na+ channel activity. FEBS Lett. 2004, 569, 191–194. [Google Scholar] [CrossRef]
- Faught, E.; Helmers, S.; Thurman, D.; Kim, H.; Kalilani, L. Patient characteristics and treatment patterns in patients with newly diagnosed epilepsy: A US database analysis. Epilepsy Behav. EB 2018, 85, 37–44. [Google Scholar] [CrossRef]
- Nicholas, J.M.; Ridsdale, L.; Richardson, M.P.; Ashworth, M.; Gulliford, M.C. Trends in antiepileptic drug utilisation in UK primary care 1993-2008: Cohort study using the General Practice Research Database. Seizure 2012, 21, 466–470. [Google Scholar] [CrossRef]
- Meeker, S.; Beckman, M.; Knox, K.M.; Treuting, P.M.; Barker-Haliski, M. Repeated Intraperitoneal Administration of Low-Concentration Methylcellulose Leads to Systemic Histologic Lesions Without Loss of Preclinical Phenotype. J. Pharmacol. Exp. Ther. 2019, 371, 25–35. [Google Scholar] [CrossRef]
- White, H.S.; Woodhead, J.H.; Franklin, M.R. General principles: Experimental selection, quantification, and evaluation of antiepileptic drugs. In Antiepileptic Drugs, 4th ed.; Levy, R.H.M., Meldrum, B.S., Eds.; Raven Press: New York, NY, USA, 1995; pp. 99–110. [Google Scholar]
- White, H.S.; Johnson, M.; Wolf, H.H.; Kupferberg, H.J. The early identification of anticonvulsant activity: Role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital. J. Neurol. Sci. 1995, 16, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Finney, D.J. Probit Analysis. A Statistical Treatment of the Sigmoid Response Curve; University Press: Cambridge, UK, 1952. [Google Scholar]
- Cho, C.; Zeigler, M.; Mizuno, S.; Morrison, R.S.; Totah, R.A.; Barker-Haliski, M. Reductions in Hydrogen Sulfide and Changes in Mitochondrial Quality Control Proteins Are Evident in the Early Phases of the Corneally Kindled Mouse Model of Epilepsy. Int. J. Mol. Sci. 2022, 23, 1434. [Google Scholar] [CrossRef]
- Barker-Haliski, M.L.; Oldenburger, K.; Keefe, K.A. Disruption of subcellular Arc/Arg 3.1 mRNA expression in striatal efferent neurons following partial monoamine loss induced by methamphetamine. J. Neurochem. 2012, 123, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Knox, K.M.; Zierath, D.K.; White, H.S.; Barker-Haliski, M. Continuous seizure emergency evoked in mice with pharmacological, electrographic, and pathological features distinct from status epilepticus. Epilepsia 2021, 62, 3076–3090. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (mg/kg, i.p.) | Protected (N/F) | Motor Impairment (T/F) |
|---|---|---|
| 2.5 | 1/8 | 0/8 |
| 3 | 1/16 | 0/16 |
| 4 | 5/15 | 0/15 |
| 5 | 8/8 | 0/8 |
| 10 | 8/8 | 0/8 |
| 15 | 8/8 | 0/8 |
| ASM | Acute Low and High Doses Tested (mg/kg, i.p.) | VEH-Kindled Mice (N Protected/F Tested) | LTG-Kindled Mice (N Protected/F Tested) | LCM-Kindled Mice (N Protected/F Tested) | |||
|---|---|---|---|---|---|---|---|
| Low Dose | High Dose | Low Dose | High Dose | Low Dose | High Dose | ||
| LTG | 17; 50 | 2/8 | 7/8 | 2/8 | 2/8 | 1/8 | 1/8 |
| LCM | 9; 26.5 | 3/8 | 4/8 | 0/8 | 3/8 | 2/8 | 3/8 |
| LEV | 60; 180 | 7/8 | 7/8 | 6/8 | 7/8 | 6/8 | 5/8 |
| GBP | 75; 300 | 2/8 | 6/8 | 3/8 | 5/8 | 3/8 | 5/8 |
| PER | 1; 4 | 6/8 | 8/8 | 3/8 | 7/8 | 1/8 | 6/8 |
| VPA | 75; 300 | 4/8 | 7/8 | 3/8 | 5/8 | 2/8 | 5/8 |
| PB | 15; 45 | 7/8 | 7/8 | 3/8 | 4/8 | 2/8 | 4/8 |
| CBZ | 10; 40 | 5/8 | 7/8 | 1/8 | 3/8 | 1/8 | 2/8 |
| TPM | 8; 20 | 1/8 | 0/8 | 1/8 | 0/8 | 1/7 | 2/8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zierath, D.; Mizuno, S.; Barker-Haliski, M. Frontline Sodium Channel-Blocking Antiseizure Medicine Use Promotes Future Onset of Drug-Resistant Chronic Seizures. Int. J. Mol. Sci. 2023, 24, 4848. https://doi.org/10.3390/ijms24054848
Zierath D, Mizuno S, Barker-Haliski M. Frontline Sodium Channel-Blocking Antiseizure Medicine Use Promotes Future Onset of Drug-Resistant Chronic Seizures. International Journal of Molecular Sciences. 2023; 24(5):4848. https://doi.org/10.3390/ijms24054848
Chicago/Turabian StyleZierath, Dannielle, Stephanie Mizuno, and Melissa Barker-Haliski. 2023. "Frontline Sodium Channel-Blocking Antiseizure Medicine Use Promotes Future Onset of Drug-Resistant Chronic Seizures" International Journal of Molecular Sciences 24, no. 5: 4848. https://doi.org/10.3390/ijms24054848
APA StyleZierath, D., Mizuno, S., & Barker-Haliski, M. (2023). Frontline Sodium Channel-Blocking Antiseizure Medicine Use Promotes Future Onset of Drug-Resistant Chronic Seizures. International Journal of Molecular Sciences, 24(5), 4848. https://doi.org/10.3390/ijms24054848