
Genome-Wide Mining of the Tandem Duplicated Type III Polyketide Synthases and Their Expression, Structure Analysis of Senna tora


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
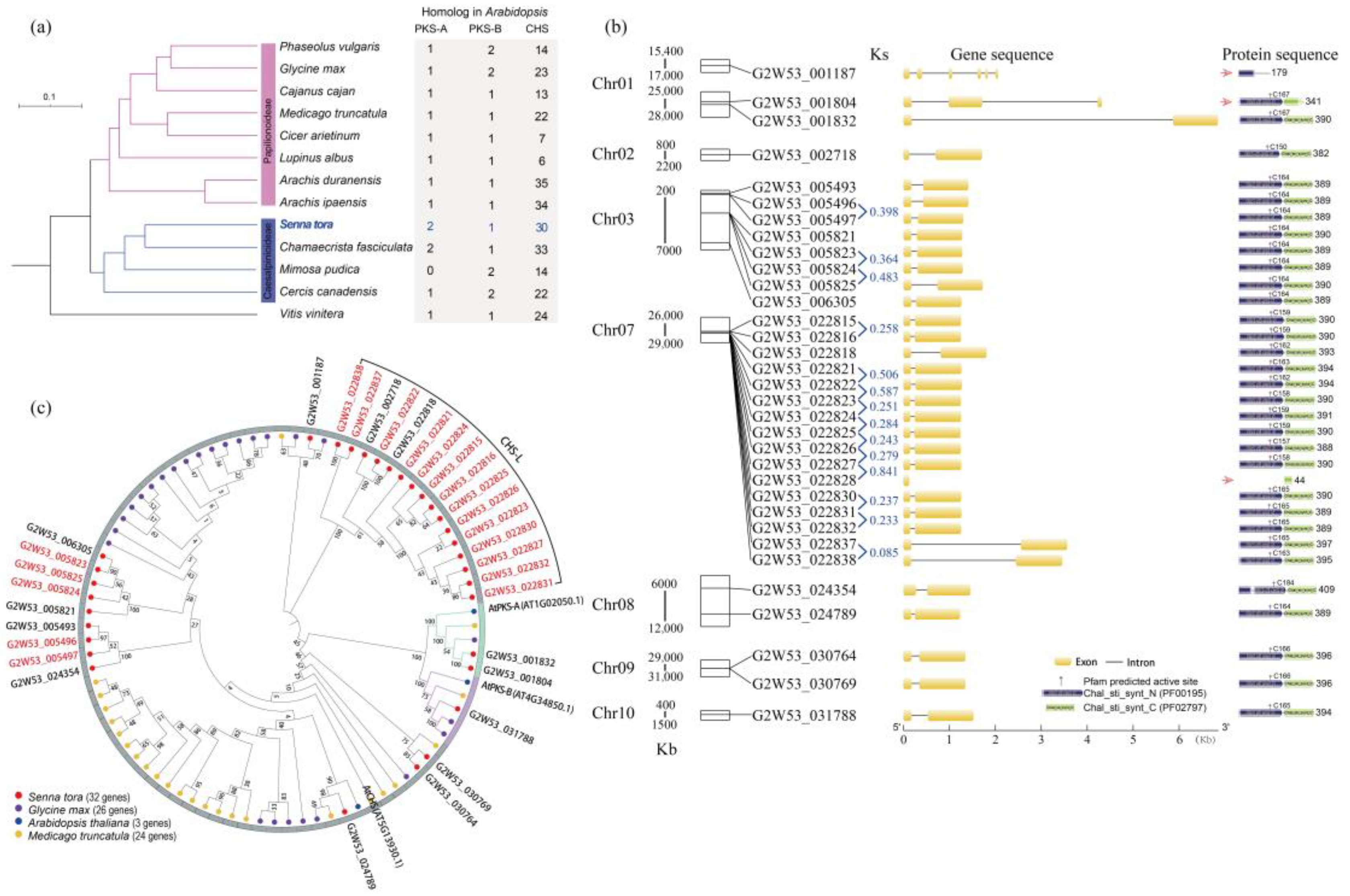
2.1. Genomic Distribution, Ks and KEGG Enrichment Analysis of the Tandem Duplicated Genes
2.2. The Identification, Distribution, and Protein Phylogenetic Tree Analysis of the Type III Polyketide Synthases
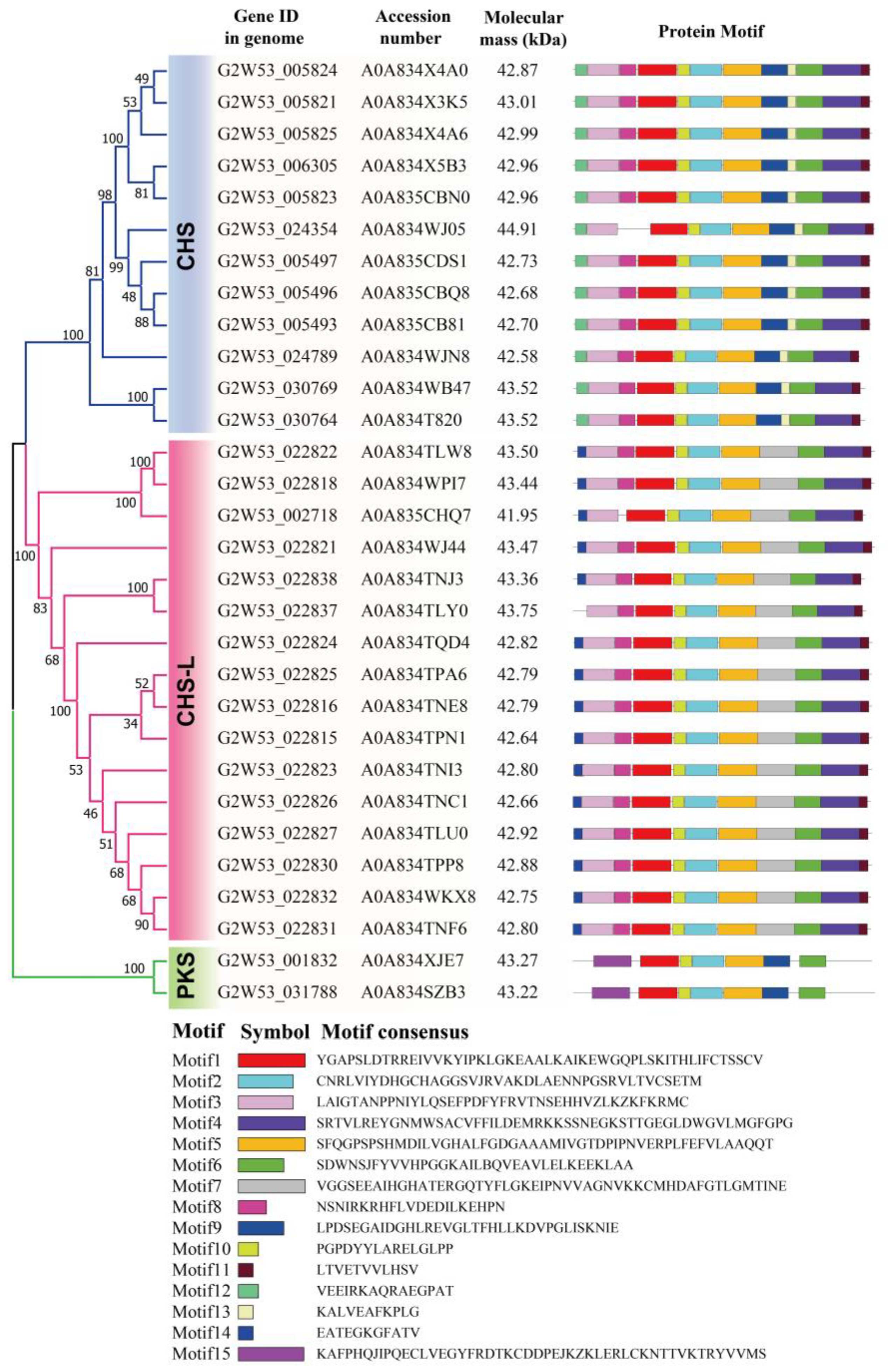
2.3. Motif Analysis of the Type III Polyketide Synthases
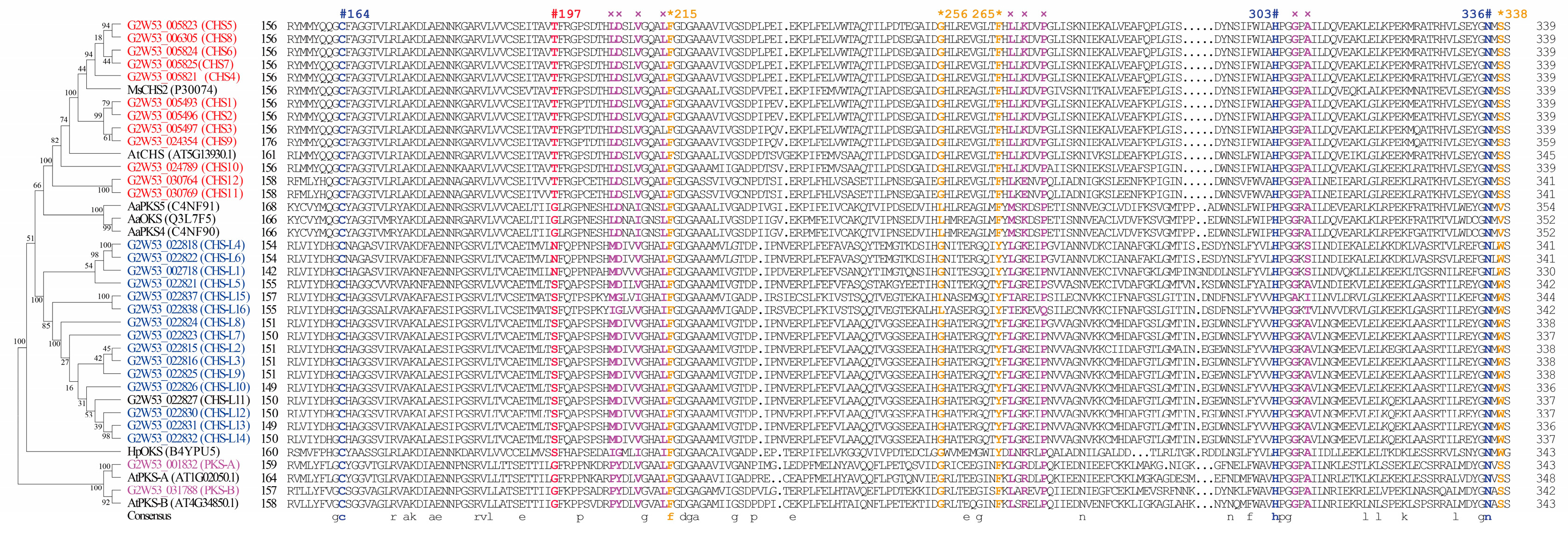
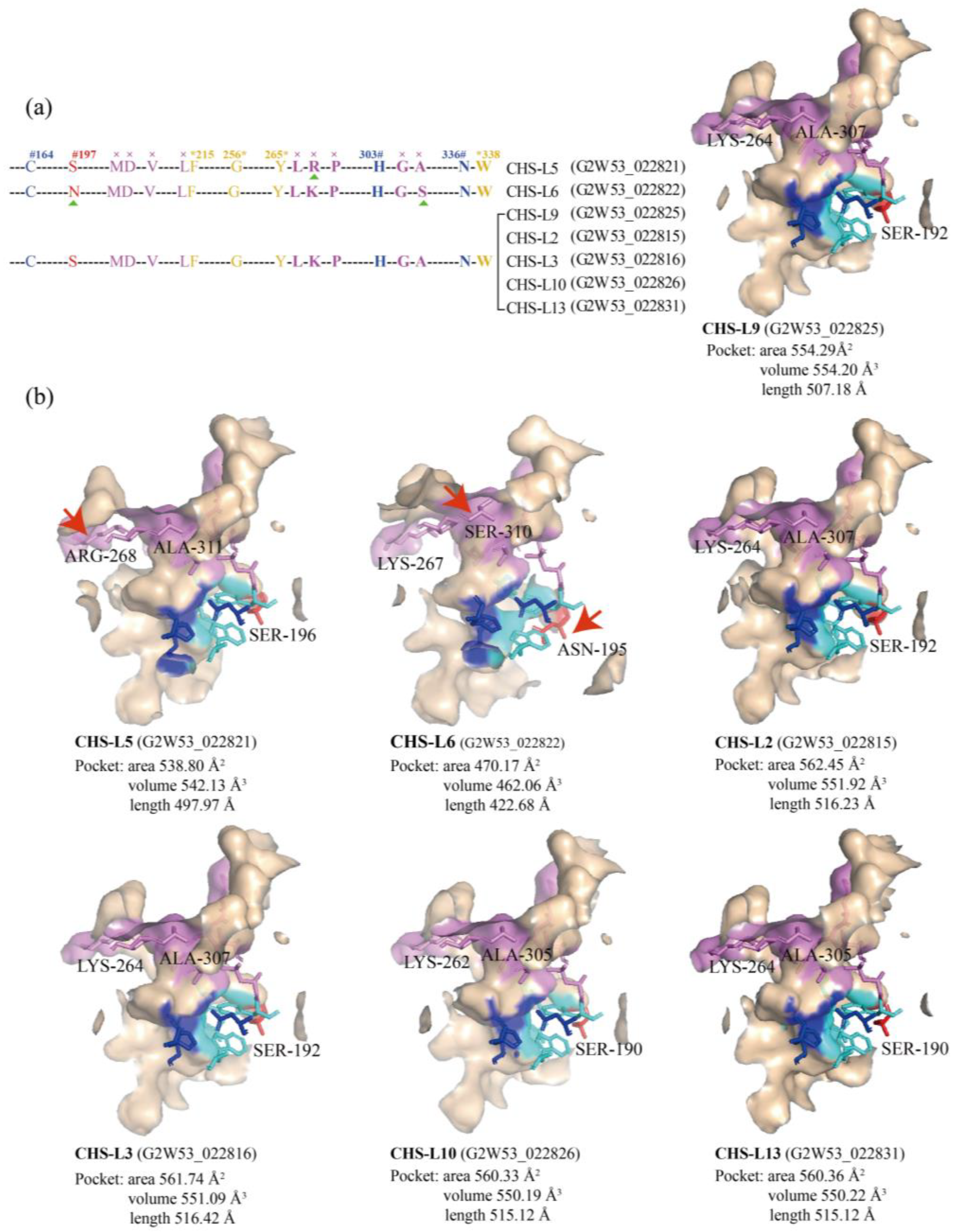
2.4. Protein Active-Site Analysis of the Type III Polyketide Synthases
2.5. Transcriptome Expression Analysis of the Type III Polyketide Synthases
2.6. Quantitative Real-Time PCR of the Chalcone Synthases-like Genes
2.7. Structure Analysis of the Chalcone Synthases-like Genes
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Genome-Wide Identification and Analysis of the Tandem Duplicated Genes
4.3. Identification of the Type III Polyketide Synthase Genes
4.4. Characteristics of the Type III Polyketide Synthase Genes
4.5. Transcriptome Expression Analysis of the Type III Polyketide Synthase Genes
4.6. Quantitative Real-Time PCR of the Chalcone Synthase-Like Genes
4.7. Three-Dimensional Structure Analysis of the Chalcone Synthase-Like Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, Y.; Zheng, H.; Yan, Z.; Liao, D.; Li, C.; Zhou, J.; Liao, H. Full-length transcriptome survey and expression analysis of Cassia obtusifolia to discover putative genes related to aurantio-obtusin biosynthesis, seed formation and development, and stress response. Int. J. Mol. Sci. 2018, 19, 2476. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Wang, L.M.; Zhang, Y.C.; Kang, L.P.; Yu, H.S.; Fan, G.W.; Han, L.F. New anthraquinone and eurotinone analogue from the seeds of Senna obtusifolia and their inhibitory effects on human organic anion transporters 1 and 3. Nat. Prod. Res. 2018, 33, 3409–3416. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.D.; Waminal, N.E.; Nguyen, T.H.; Pellerin, R.J.; Kim, H.H. Omparative FISH analysis of Senna tora tandem repeats revealed insights into the chromosome dynamics in Senna. Genes Genom. 2021, 43, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K. Editorial: The potential medicinal uses of Cassia tora Linn leaf and seed extracts. Rev. Recent Clin. Trials 2018, 13, 3–4. [Google Scholar] [CrossRef]
- Kumar, R.S.; Narasingappa, R.B.; Joshi, C.G.; Girish, T.K.; Prasada Rao, U.J.; Danagoudar, A. Evaluation of Cassia tora Linn. against oxidative stress-induced DNA and cell membrane damage. J. Pharm. Bioallied Sci. 2017, 9, 33–43. [Google Scholar] [CrossRef]
- Zaman, W.; Ye, J.; Saqib, S.; Liu, Y.; Shan, Z.; Hao, D.; Chen, Z.; Xiao, P. Predicting potential medicinal plants with phylogenetic topology: Inspiration from the research of traditional Chinese medicine. J. Ethnopharmacol. 2021, 281, 114515. [Google Scholar] [CrossRef]
- Larsen, J.S.; Pearson, L.A.; Neilan, B.A. Genome mining and evolutionary analysis reveal diverse type III polyketide synthase pathways in Cyanobacteria. Genome Biol. Evol. 2021, 13, evab056. [Google Scholar] [CrossRef]
- Mizuuchi, Y.; Shimokawa, Y.; Wanibuchi, K.; Noguchi, H.; Abe, I. Structure function analysis of novel type III polyketide synthases from Arabidopsis thaliana. Biol. Pharm. Bull. 2008, 31, 2205–2210. [Google Scholar] [CrossRef]
- Austin, M.B.; Bowman, M.E.; Ferrer, J.L.; Schröder, J.; Noel, J.P. An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 2004, 11, 1179–1194. [Google Scholar] [CrossRef]
- Kong, X.; Khan, A.; Li, Z.; You, J.; Munsif, F.; Kang, H.; Zhou, R. Identification of chalcone synthase genes and their expression patterns reveal pollen abortion in cotton. Saudi J. Biol. Sci. 2020, 27, 3691–3699. [Google Scholar] [CrossRef]
- Zhou, Q.; Bräuer, A.; Adihou, H.; Schmalhofer, M.; Saura, P.; Grammbitter, G.L.C.; Kaila, V.R.I.; Groll, M.; Bode, H.B. Molecular mechanism of polyketide shortening in anthraquinone biosynthesis of Photorhabdus luminescens. Chem. Sci. 2019, 10, 6341–6349. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Lee, W.H.; Lee, C.M.; Sim, J.S.; Won, S.Y.; Han, S.R.; Kwon, S.J.; Kim, J.S.; Kim, C.K.; Oh, T.J. De novo transcriptome sequence of Senna tora provides insights into anthraquinone biosynthesis. PLoS ONE 2020, 15, e0225564. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, Y.; Shi, S.P.; Wanibuchi, K.; Kojima, A.; Morita, H.; Noguchi, H.; Abe, I. Novel type III polyketide synthases from Aloe arborescens. FEBS J. 2009, 276, 2391–2401. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, I.A.M.; Beuerle, T.; Ernst, L.; Abdel-Baky, A.M.; Desoky, E.E.D.K.; Ahmed, A.S.; Beerhues, L. In vitro formation of the anthranoid scaffold by cell-free extracts from yeast-extract-treated Cassia bicapsularis cell cultures. Phytochemistry 2013, 88, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Pandey, R.P.; Lee, C.M.; Sim, J.S.; Jeong, J.T.; Choi, B.S.; Jung, M.; Ginzburg, D.; Zhai, K.; Won, S.Y.; et al. Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 2020, 11, 5875. [Google Scholar] [CrossRef] [PubMed]
- Reams, A.B.; Roth, J.R. Mechanisms of gene duplication and amplification. Cold Spring Harb. Perspect. Biol. 2015, 7, a016592. [Google Scholar] [CrossRef]
- Kono, T.J.Y.; Brohammer, A.B.; McGaugh, S.E.; Hirsch, C.N. Tandem duplicate genes in maize are abundant and date to two distinct periods of time. G3 (Bethesda) 2018, 8, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc. Biol. Sci. 2012, 279, 5048–5057. [Google Scholar] [CrossRef] [PubMed]
- Leister, D. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 2004, 20, 116–122. [Google Scholar] [CrossRef]
- Liu, C.; Wu, Y.; Liu, Y.; Yang, L.; Dong, R.; Jiang, L.; Liu, P.; Liu, G.; Wang, Z.; Luo, L. Genome-wide analysis of tandem duplicated genes and their contribution to stress resistance in pigeonpea (Cajanus cajan). Genomics 2021, 113, 728–735. [Google Scholar] [CrossRef]
- Ai, H.L.; Lv, X.; Ye, K.; Zhang, X.; Zhou, L.Y.; Li, Z.H. The complete chloroplast genome of a well-known medicinal herb, Senna tora. Mitochondrial DNA Part B 2020, 5, 1659–1660. [Google Scholar] [CrossRef]
- Kontturi, J.; Osama, R.; Deng, X.; Bashandy, H.; Albert, V.A.; Teeri, T.H. Functional characterization and expression of GASCL1 and GASCL2, two anther-specific chalcone synthase like enzymes from Gerbera hybrida. Phytochemistry 2017, 134, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhao, W.; Wang, Z.; Zhu, J.; Liu, Q. Molecular evolution and sequence divergence of plant chalcone synthase and chalcone synthase-Like genes. Genetica 2014, 142, 215–225. [Google Scholar] [CrossRef]
- Ji, Y.; Hongya, G. Duplication and divergent evolution of the CHS and CHS-like genes in the chalcone synthase (CHS) superfamily. Sci. Bull. 2006, 51, 505–509. [Google Scholar] [CrossRef]
- Abe, I.; Oguro, S.; Utsumi, Y.; Sano, Y.; Noguchi, H. Engineered biosynthesis of plant polyketides: Chain length control in an octaketide-producing plant type III polyketide synthase. J. Am. Chem. Soc. 2005, 127, 12709–12716. [Google Scholar] [CrossRef]
- Deng, X.; Bashandy, H.; Ainasoja, M.; Kontturi, J.; Pietiäinen, M.; Laitinen, R.A.E.; Albert, V.A.; Valkonen, J.P.T.; Elomaa, P.; Teeri, T.H. Functional diversification of duplicated chalcone synthase genes in anthocyanin biosynthesis of Gerbera hybrida. New Phytol. 2014, 201, 1469–1483. [Google Scholar] [CrossRef]
- Abe, I.; Morita, H. Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat. Prod. Rep. 2010, 27, 809–838. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 9. [Google Scholar] [CrossRef]
- Hu, L.; He, H.; Zhu, C.; Peng, X.; Fu, J.; He, X.; Chen, X.; Ouyang, L.; Bian, J.; Liu, S. Genome-wide identification and phylogenetic analysis of the chalcone synthase gene family in rice. J. Plant Res. 2017, 130, 95–105. [Google Scholar] [CrossRef]
- Xiao, J.; Xie, X.; Li, C.; Xing, G.; Cheng, K.; Li, H.; Liu, N.; Tan, J.; Zheng, W. Identification of SPX family genes in the maize genome and their expression under different phosphate regimes. Plant Physiol. Biochem. 2021, 168, 211–220. [Google Scholar] [CrossRef]
- Sgamma, T.; Pape, J.; Massiah, A.; Jackson, S. Selection of reference genes for diurnal and developmental time-course real-time PCR expression analyses in lettuce. Plant Methods 2016, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cui, H.M.; Huang, T.H.; Liu, T.K.; Hou, X.L.; Li, Y. Identification and validation of reference genes for RT-qPCR analysisin non-heading chinese cabbage flowers. Front. Plant Sci. 2016, 7, 811. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zaman, W.; Lu, J.; Niu, Q.; Zhang, X.; Ayaz, A.; Saqib, S.; Yang, B.; Zhang, J.; Zhao, H.; et al. Natural lupeol level variation among castor accessions and the upregulation of lupeol synthesis in response to light. Ind. Crops Prod. 2023, 192, 116090. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Go, M.K.; Wongsantichon, J.; Cheung, V.W.N.; Chow, J.Y.; Robinson, R.C.; Yew, W.S. Synthetic polyketide enzymology: Platform for biosynthesis of antimicrobial polyketides. ACS Catal. 2015, 5, 4033–4042. [Google Scholar] [CrossRef]
- Jez, J.M.; Bowman, M.E.; Noel, J.P. Structure-guided programming of polyketide chain-length determination in chalcone synthase. Biochemistry 2001, 40, 14829–14838. [Google Scholar] [CrossRef]
- Griesmann, M.; Chang, Y.; Liu, X.; Song, Y.; Haberer, G.; Crook, M.B.; Billault-Penneteau, B.; Lauressergues, D.; Keller, J.; Imanishi, L.; et al. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 2018, 361, 144. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.-A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 2014, 15, 312. [Google Scholar] [CrossRef]
- Parween, S.; Nawaz, K.; Roy, R.; Pole, A.K.; Suresh, B.V.; Misra, G.; Jain, M.; Yadav, G.; Parida, S.K.; Tyagi, A.K.; et al. An advanced draft genome assembly of a desi type chickpea (Cicer arietinum L.). Sci. Rep. 2015, 5, 12806. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, H.; Pandey, M.K.; Yang, Q.; Wang, X.; Garg, V.; Li, H.; Chi, X.; Doddamani, D.; Hong, Y.; et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. USA 2016, 113, 6785–6790. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, H.; Hong, Y.; Zhang, G.; Wen, S.; Li, X.; Zhou, G.; Li, S.; Liu, H.; Liu, H.; et al. Genome sequencing and analysis of the peanut B-genome progenitor (Arachis ipaensis). Front. Plant Sci. 2018, 9, 604. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Q.; Yuan, W.; Xu, F.; Aslam, M.M.; Miao, R.; Li, Y.; Wang, Q.; Li, X.; Zhang, X.; et al. The genome evolution and low-phosphorus adaptation in white lupin. Nat. Commun. 2020, 11, 1069. [Google Scholar] [CrossRef]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Z.; Zhao, X.; Zhou, C.; Fang, T.; Liu, G.; Luo, J. Genome-Wide Mining of the Tandem Duplicated Type III Polyketide Synthases and Their Expression, Structure Analysis of Senna tora. Int. J. Mol. Sci. 2023, 24, 4837. https://doi.org/10.3390/ijms24054837
Cai Z, Zhao X, Zhou C, Fang T, Liu G, Luo J. Genome-Wide Mining of the Tandem Duplicated Type III Polyketide Synthases and Their Expression, Structure Analysis of Senna tora. International Journal of Molecular Sciences. 2023; 24(5):4837. https://doi.org/10.3390/ijms24054837
Chicago/Turabian StyleCai, Zeping, Xingkun Zhao, Chaoye Zhou, Ting Fang, Guodao Liu, and Jiajia Luo. 2023. "Genome-Wide Mining of the Tandem Duplicated Type III Polyketide Synthases and Their Expression, Structure Analysis of Senna tora" International Journal of Molecular Sciences 24, no. 5: 4837. https://doi.org/10.3390/ijms24054837
APA StyleCai, Z., Zhao, X., Zhou, C., Fang, T., Liu, G., & Luo, J. (2023). Genome-Wide Mining of the Tandem Duplicated Type III Polyketide Synthases and Their Expression, Structure Analysis of Senna tora. International Journal of Molecular Sciences, 24(5), 4837. https://doi.org/10.3390/ijms24054837