

Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression

Abstract
1. Introduction
1.1. Macrophages Play a Significant Role in T2DM Progression
1.2. Treating Patients with T2DM
1.3. Metformin and Empagliflozin Can Affect Macrophages Responses
2. Results
2.1. Metformin Promotes Tnfa and Il1b Inflammatory Gene Expression in Macrophages
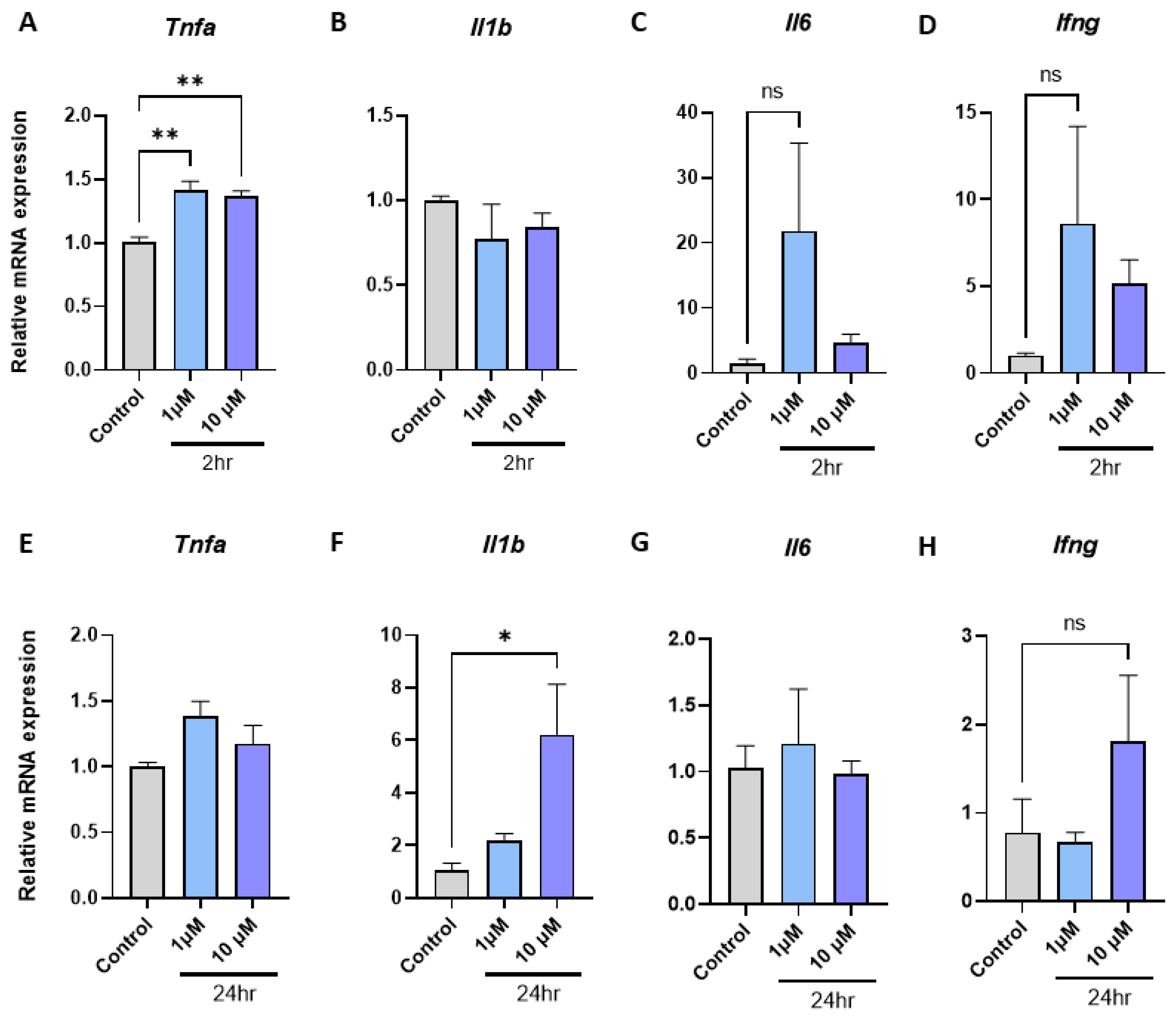
2.2. Empagliflozin Promotes Tnfa, Il1b, Il6, and Ifng Inflammatory Gene Expression in Macrophages
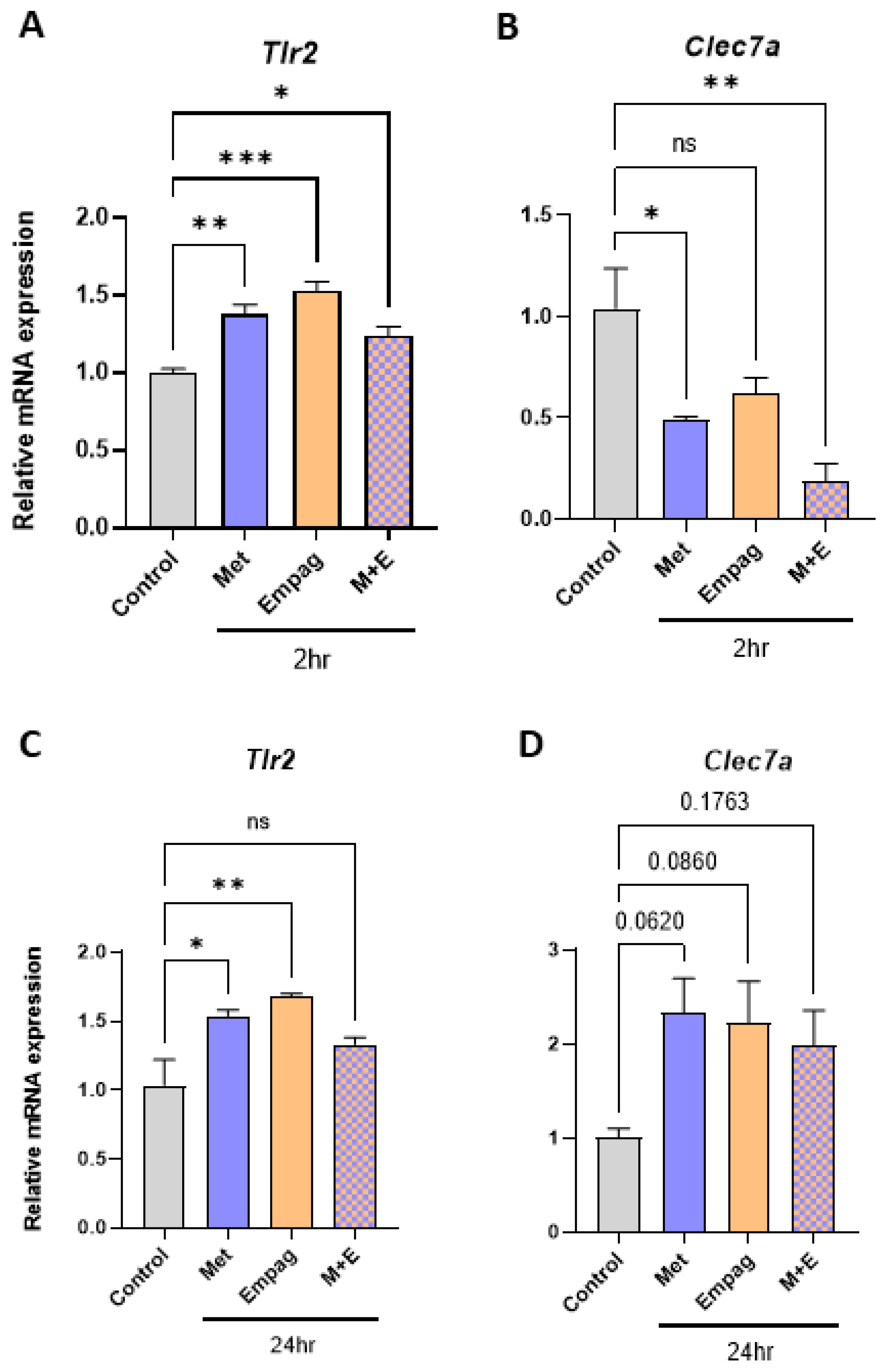
2.3. Metformin and Empagliflozin in Combination have Contrasting Effects on Macrophage Inflammatory Gene Expression
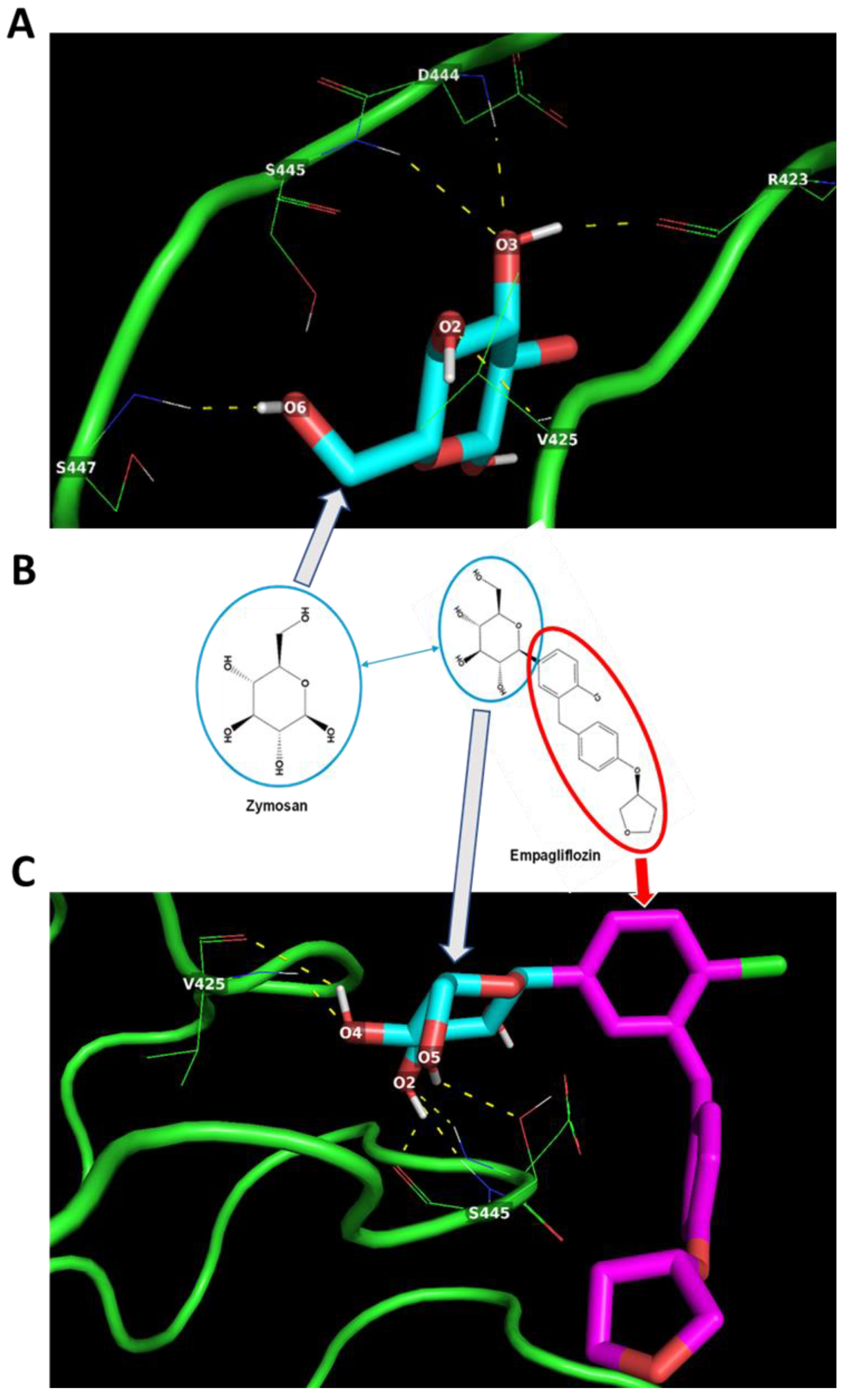
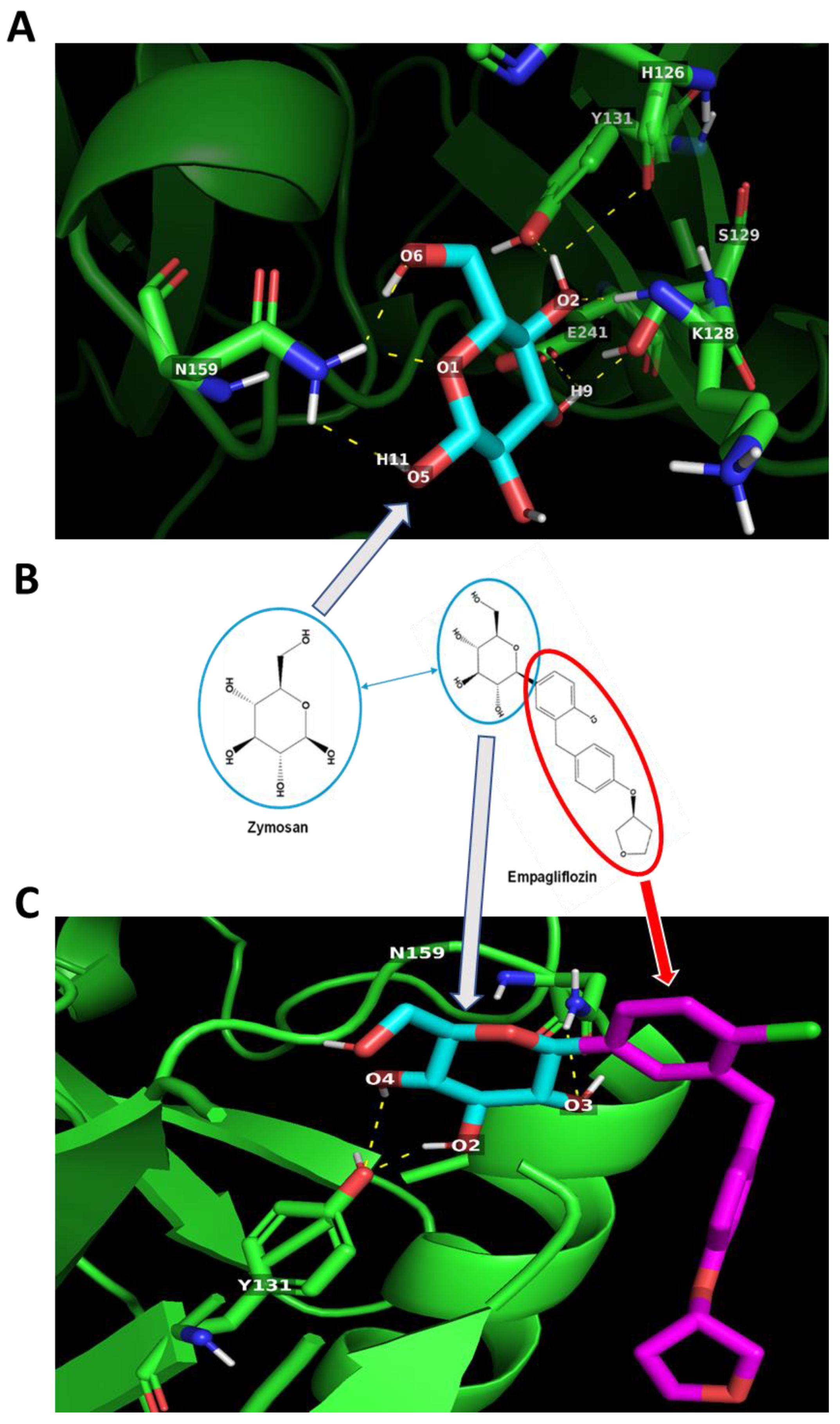
2.4. In Silico Docking of Empagliflozin with TLR2 and DECTIN1
2.5. Metformin and Empagliflozin can Interact with Tlr2 and Clec7a and Modulate Their Expression
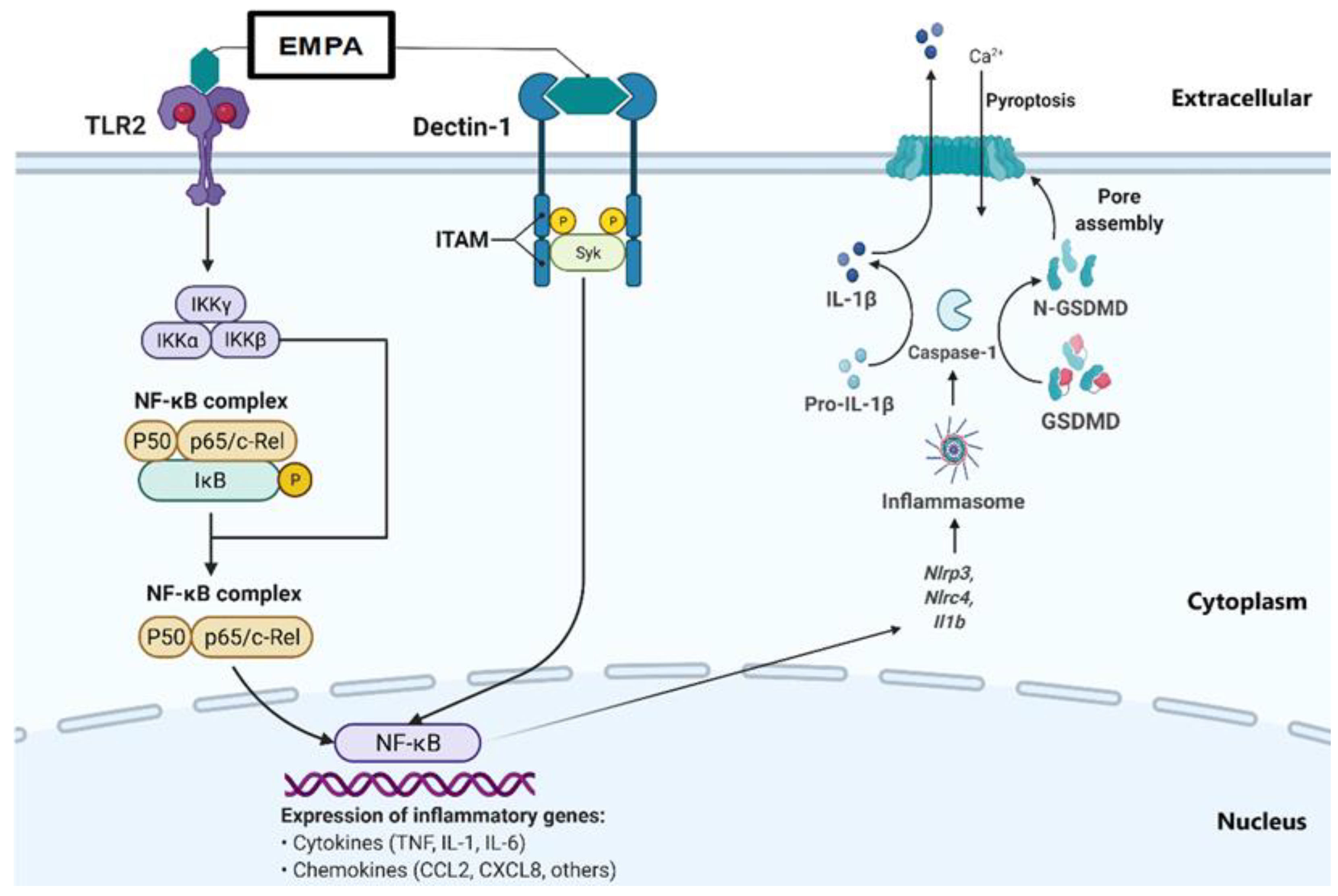
3. Discussion
4. Materials and Methods
4.1. Animal Work and Cell Culture
4.2. Gene Expression Analysis
4.3. In Silico Molecular Docking Simulation
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siu, A.L. Screening for Abnormal Blood Glucose and Type 2 Diabetes Mellitus: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2015, 163, 861. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; IDF: Brussels, Belgium, 2017; pp. 1–150. ISBN 978-2-930229-87-4. [Google Scholar]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2019. Diabetes Care 2018, 42, S13–S28. [Google Scholar] [CrossRef]
- American Diabetes Association. 15. Diabetes Care in the Hospital: Standards of Medical Care in Diabetes—2020. Diabetes Care 2019, 43, S193–S202. [Google Scholar] [CrossRef]
- Cade, W.T. Diabetes-Related Microvascular and Macrovascular Diseases in the Physical Therapy Setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study Group. Tight Blood Pressure Control and Risk of Macrovascular and Microvascular Complications in Type 2 Diabetes: UKPDS 38. BMJ 1998, 317, 703–713. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study Group. Intensive Blood-Glucose Control with Sulphonylureas or Insulin Compared with Conventional Treatment and Risk of Complications in Patients with Type 2 Diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Miller, M.E.; Williamson, J.D.; Gerstein, H.C.; Byington, R.P.; Cushman, W.C.; Ginsberg, H.N.; Ambrosius, W.T.; Lovato, L.; Applegate, W.B. Effects of Randomization to Intensive Glucose Control on Adverse Events, Cardiovascular Disease, and Mortality in Older versus Younger Adults in the ACCORD Trial. Diabetes Care 2013, 37, 634–643. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Liang, C.-P.; Han, S.; Senokuchi, T.; Tall, A.R. The Macrophage at the Crossroads of Insulin Resistance and Atherosclerosis. Circ. Res. 2007, 100, 1546–1555. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose Tissue Macrophages Promote Myelopoiesis and Monocytosis in Obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef]
- Woollard, K.J.; Geissmann, F. Monocytes in Atherosclerosis: Subsets and Functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of Blood Vessels and Tissues by a Population of Monocytes with Patrolling Behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed]
- De Kleer, I.; Willems, F.; Lambrecht, B.; Goriely, S. Ontogeny of Myeloid Cells. Front. Immunol. 2014, 5, 423. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 Modulates Inflammatory and Metabolic Effects of High-Fat Feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S.; et al. Hyperglycemia Promotes Myelopoiesis and Impairs the Resolution of Atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a Link between Obesity, Metabolic Syndrome and Type 2 Diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Lachmandas, E.; Vrieling, F.; Wilson, L.G.; Joosten, S.A.; Netea, M.G.; Ottenhoff, T.H.; van Crevel, R. The Effect of Hyperglycaemia on in Vitro Cytokine Production and Macrophage Infection with Mycobacterium Tuberculosis. PLoS ONE 2015, 10, e0117941. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 Inflamasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals That Form Early in Disease. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Hu, G.; Su, Y.; Kang, B.H.; Fan, Z.; Dong, T.; Brown, D.R.; Cheah, J.; Wittrup, K.D.; Chen, J. High-Throughput Phenotypic Screen and Transcriptional Analysis Identify New Compounds and Targets for Macrophage Reprogramming. Nat. Commun. 2021, 12, 773. [Google Scholar] [CrossRef]
- Morris, D.L. Minireview: Emerging Concepts in Islet Macrophage Biology in Type 2 Diabetes. Mol. Endocrinol. 2015, 29, 946–962. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Biessels, G.J.; Braithwaite, S.S.; Casanueva, F.F.; Draznin, B.; Halter, J.B.; Hirsch, I.B.; McDonnell, M.E.; Molitch, M.E.; Murad, M.H.; et al. Treatment of Diabetes in Older Adults: An Endocrine Society* Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2019, 104, 1520–1574. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 5. Lifestyle Management: Standards of Medical Care in Diabetes—2019. Diabetes Care 2018, 42, S46–S60. [Google Scholar] [CrossRef]
- Maggi, D.; Montecucco, F.; Adami, G.; Cordera, R. Glycosylated Haemoglobin (A1c) Best Values for Type 2 Diabetes in the Battlefield Much Ado about Nothing? (Apparently). Diabetol. Metab. Syndr. 2019, 11, 48. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care 2018, 42, S90–S102. [Google Scholar] [CrossRef]
- Chaplin, S. NICE Guidance on Managing Type 2 Diabetes in Adults. Prescriber 2016, 27, 22–29. [Google Scholar] [CrossRef]
- Espinoza, P.; Varela, C.A.; Vargas, I.E.; Ortega, G.; Silva, P.A.; Boehmer, K.B.; Montori, V.M. The Burden of Treatment in People Living with Type 2 Diabetes: A Qualitative Study of Patients and Their Primary Care Clinicians. PLoS ONE 2020, 15, e0241485. [Google Scholar] [CrossRef]
- Boye, K.S.; Mody, R.; Lage, M.J.; Douglas, S.; Patel, H. Chronic Medication Burden and Complexity for US Patients with Type 2 Diabetes Treated with Glucose-Lowering Agents. Diabetes Ther. 2020, 11, 1513–1525. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef]
- Rakel, D. Integrative Medicine, 4th ed.; Elsevier: Philadelphia, PA, USA, 2018; Chapter 33; pp. 334–346. ISBN 9780323358682. [Google Scholar]
- Waller, D.; Sampson, A.P. Medical Pharmacology & Therapeutics, 5th ed.; Elsevier: Edinburgh, Scottish, 2018; Chapter 40; pp. 459–473. ISBN 9780702071676. [Google Scholar]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the Promise of Insulin Sensitization in Type 2 Diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef]
- Douros, A.; Dell’Aniello, S.; Yu, O.H.Y.; Filion, K.B.; Azoulay, L.; Suissa, S. Sulfonylureas as Second Line Drugs in Type 2 Diabetes and the Risk of Cardiovascular and Hypoglycaemic Events: Population Based Cohort Study. BMJ 2018, 362, k2693. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.-T.; Chang, K.-C.; Li, C.-Y.; Wu, J.-S. Risks of Cardiovascular Diseases Associated with Dipeptidyl Peptidase-4 Inhibitors and Other Antidiabetic Drugs in Patients with Type 2 Diabetes: A Nation-Wide Longitudinal Study. Cardiovasc. Diabetol. 2016, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Ceriello, A.; Baeres, F.M.M.; Fioretto, P.; Garber, A.; Stough, W.G.; George, J.T.; Grant, P.J.; Khunti, K.; Langkilde, A.M.; et al. Addressing Cardiovascular Risk in Type 2 Diabetes Mellitus: A Report from the European Society of Cardiology Cardiovascular Roundtable. Eur. Heart J. 2018, 40, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kerins, D.M.; Walther, T. Cardiovascular Safety of Anti-Diabetic Drugs. Eur. Heart J. Cardiovasc. Pharmacother. 2015, 2, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Paquot, N.; Scheen, A.J. Anti-Inflammatory Agents to Treat or Prevent Type 2 Diabetes, Metabolic Syndrome and Cardiovascular Disease. Expert Opin. Investig. Drugs 2014, 24, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Ehses, J.A.; Donath, M.Y.; Mandrup-Poulsen, T. Sustained Effects of Interleukin-1 Receptor Antagonist Treatment in Type 2 Diabetes. Diabetes Care 2009, 32, 1663–1668. [Google Scholar] [CrossRef]
- van Asseldonk, E.J.P.; van Poppel, P.C.M.; Ballak, D.B.; Stienstra, R.; Netea, M.G.; Tack, C.J. One Week Treatment with the IL-1 Receptor Antagonist Anakinra Leads to a Sustained Improvement in Insulin Sensitivity in Insulin Resistant Patients with Type 1 Diabetes Mellitus. Clin. Immunol. 2015, 160, 155–162. [Google Scholar] [CrossRef]
- Vallejo, S.; Palacios, E.; Romacho, T.; Villalobos, L.; Peiró, C.; Sánchez-Ferrer, C.F. The Interleukin-1 Receptor Antagonist Anakinra Improves Endothelial Dysfunction in Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 2014, 13, 158. [Google Scholar] [CrossRef]
- Cavelti-Weder, C.; Babians-Brunner, A.; Keller, C.; Stahel, M.A.; Kurz-Levin, M.; Zayed, H.; Solinger, A.M.; Mandrup-Poulsen, T.; Dinarello, C.A.; Donath, M.Y. Effects of Gevokizumab on Glycemia and Inflammatory Markers in Type 2 Diabetes. Diabetes Care 2012, 35, 1654–1662. [Google Scholar] [CrossRef]
- Hensen, J.; Howard, C.P.; Walter, V.; Thuren, T. Impact of Interleukin-1β Antibody (Canakinumab) on Glycaemic Indicators in Patients with Type 2 Diabetes Mellitus: Results of Secondary Endpoints from a Randomized, Placebo-Controlled Trial. Diabetes Metab. 2013, 39, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, A.; Howard, C.P.; Botha, J.; Thuren, T. Effect of Anti-IL-1β Antibody (Canakinumab) on Insulin Secretion Rates in Impaired Glucose Tolerance or Type 2 Diabetes: Results of a Randomized, Placebo-Controlled Trial. Diabetes Obes. Metab. 2012, 14, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Lancaster, J.; Abu-Raddad, E.; Polzer, J.; Miller, J.W.; Scherer, J.C.; De Gaetano, A.; Berg, J.K.; Landschulz, W.H. Double-Blind, Randomized Study Evaluating the Glycemic and Anti-Inflammatory Effects of Subcutaneous LY2189102, a Neutralizing IL-1β Antibody, in Patients with Type 2 Diabetes. Diabetes Care 2013, 36, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.; Shoelson, S.E.; Bernier, R.; Goldfine, A.B. Salsalate Improves Glycemia and Inflammatory Parameters in Obese Young Adults. Diabetes Care 2007, 31, 289–294. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Silver, R.; Aldhahi, W.; Cai, D.; Tatro, E.; Lee, J.; Shoelson, S.E. Use of Salsalate to Target Inflammation in the Treatment of Insulin Resistance and Type 2 Diabetes. Clin. Transl. Sci. 2008, 1, 36–43. [Google Scholar] [CrossRef]
- Koska, J.; Ortega, E.; Bunt, J.C.; Gasser, A.; Impson, J.; Hanson, R.L.; Forbes, J.; de Courten, B.; Krakoff, J. The Effect of Salsalate on Insulin Action and Glucose Tolerance in Obese Non-Diabetic Patients: Results of a Randomised Double-Blind Placebo-Controlled Study. Diabetologia 2008, 52, 385–393. [Google Scholar] [CrossRef]
- Goldfine, A.B. The Effects of Salsalate on Glycemic Control in Patients with Type 2 Diabetes. Ann. Intern. Med. 2010, 152, 346. [Google Scholar] [CrossRef]
- Goldfine, A.B. Salicylate (Salsalate) in Patients with Type 2 Diabetes. Ann. Intern. Med. 2013, 159, 1. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Conlin, P.R.; Halperin, F.; Koska, J.; Permana, P.; Schwenke, D.; Shoelson, S.E.; Reaven, P.D. A Randomised Trial of Salsalate for Insulin Resistance and Cardiovascular Risk Factors in Persons with Abnormal Glucose Tolerance. Diabetologia 2013, 56, 714–723. [Google Scholar] [CrossRef]
- Faghihimani, E.; Aminorroaya, A.; Rezvanian, H.; Adibi, P.; Ismail-Beigi, F.; Amini, M. Salsalate Improves Glycemic Control in Patients with Newly Diagnosed Type 2 Diabetes. Acta Diabetol. 2011, 50, 537–543. [Google Scholar] [CrossRef]
- Bernstein, L.E.; Berry, J.; Kim, S.; Canavan, B.; Grinspoon, S.K. Effects of Etanercept in Patients with the Metabolic Syndrome. Arch. Intern. Med. 2006, 166, 902. [Google Scholar] [CrossRef] [PubMed]
- Stanley, T.L.; Zanni, M.V.; Johnsen, S.; Rasheed, S.; Makimura, H.; Lee, H.; Khor, V.K.; Ahima, R.S.; Grinspoon, S.K. TNF-α Antagonism with Etanercept Decreases Glucose and Increases the Proportion of High Molecular Weight Adiponectin in Obese Subjects with Features of the Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E146–E150. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Hattori, K.; Hayashi, T. Pleiotropic Benefits of Metformin: Macrophage Targeting Its Anti-Inflammatory Mechanisms. Diabetes 2015, 64, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S. Anti-Inflammatory Effects of Empagliflozin in Patients with Type 2 Diabetes and Insulin Resistance. Diabetol. Metab. Syndr. 2018, 10, 93. [Google Scholar] [CrossRef]
- Xu, L.; Ota, T. Emerging Roles of SGLT2 Inhibitors in Obesity and Insulin Resistance: Focus on Fat Browning and Macrophage Polarization. Adipocyte 2017, 7, 121–128. [Google Scholar] [CrossRef]
- Bastard, J.-P. Elevated Levels of Interleukin 6 Are Reduced in Serum and Subcutaneous Adipose Tissue of Obese Women after Weight Loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef]
- Wang, J.; Sun, X.; Ma, Q.; Fu, G.; Cong, L.; Zhang, H.; Fan, D.; Feng, J.; Lu, S.; Liu, J.; et al. Metformin’s Antitumour and Anti-Angiogenic Activities Are Mediated by Skewing Macrophage Polarization. J. Cell. Mol. Med. 2018, 22, 3825–3836. [Google Scholar] [CrossRef]
- Woo, S.-L.; Xu, H.; Li, H.; Zhao, Y.; Hu, X.; Zhao, J.; Guo, X.; Guo, T.; Botchlett, R.; Qi, T.; et al. Metformin Ameliorates Hepatic Steatosis and Inflammation without Altering Adipose Phenotype in Diet-Induced Obesity. PLoS ONE 2014, 9, e91111. [Google Scholar] [CrossRef]
- Stavri, S.; Trusca, V.G.; Simionescu, M.; Gafencu, A.V. Metformin Reduces the Endotoxin-Induced Down-Regulation of Apolipoprotein E Gene Expression in Macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 435–440. [Google Scholar] [CrossRef]
- Isoda, K.; Young, J.L.; Zirlik, A.; MacFarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schönbeck, U.; Libby, P. Metformin Inhibits Proinflammatory Responses and Nuclear Factor-ΚB in Human Vascular Wall Cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 611–617. [Google Scholar] [CrossRef]
- Pham, S.V.; Chilton, R. EMPA-REG OUTCOME: The Cardiologist’s Point of View. Am. J. Med. 2017, 130, S57–S62. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-Induced Obese Mice. EbioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Chen, G.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Sakai, Y.; Kaneko, S.; Ota, T. Empagliflozin Reverses Obesity and Insulin Resistance through Fat Browning and Alternative Macrophage Activation in Mice Fed a High-Fat Diet. BMJ Open Diabetes Res. Care 2019, 7, e000783. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 Inhibition Modulates NLRP3 Inflammasome Activity via Ketones and Insulin in Diabetes with Cardiovascular Disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, W.; Ni, X.; Little, P.J.; Xu, S.; Tang, L.; Weng, J. Metformin, Macrophage Dysfunction and Atherosclerosis. Front. Immunol. 2021, 12, 682853. [Google Scholar] [CrossRef]
- Vuong, L.D.; Nguyen, Q.N.; Truong, V.-L. Anti-Inflammatory and Anti-Oxidant Effects of Combination between Sulforaphane and Acetaminophen in LPS-Stimulated RAW 264.7 Macrophage Cells. Immunopharmacol. Immunotoxicol. 2019, 41, 413–419. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—Mode of Action and Clinical Implications for Diabetes and Cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef]
- Boehringer Ingelheim International GmbH. Summary of Product Characteristics; Boehringer Ingelheim International GmbH: Ingelheim am Rhein, Germany, 2019; pp. 1–53. Available online: https://www.ema.europa.eu/en/documents/product-information/jardiance-epar-product-information_en.pdf (accessed on 21 July 2022).
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef]
- InvivoGen. Dectin-1: A Major Receptor in Antifungal Immunity|Review|InvivoGen. Available online: https://www.invivogen.com/review-dectin1 (accessed on 6 November 2019).
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Sato, M.; Sano, H.; Iwaki, D.; Kudo, K.; Konishi, M.; Takahashi, H.; Takahashi, T.; Imaizumi, H.; Asai, Y.; Kuroki, Y. Direct Binding of Toll-like Receptor 2 to Zymosan, and Zymosan-Induced NF-ΚB Activation and TNF-α Secretion Are Down-Regulated by Lung Collectin Surfactant Protein A. J. Immunol. 2003, 171, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S. Yeast Zymosan, a Stimulus for TLR2 and Dectin-1, Induces Regulatory Antigen-Presenting Cells and Immunological Tolerance. J. Clin. Investig. 2006, 116, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, M.A.R.; Wunderlich, F.T. Macrophage Function in Obesity-Induced Inflammation and Insulin Resistance. PflügersArch. Eur. J. Physiol. 2017, 469, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Ji, H.-H.; Li, Y.-J.; Guo, S.-D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 679797. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.T.; Kothari, V.; Galdo, J. Hypoglycemic Agents and Potential Anti-Inflammatory Activity. J. Inflamm. Res. 2016, 9, 27. [Google Scholar] [CrossRef]
- Gage, M.C.; Bécares, N.; Louie, R.; Waddington, K.E.; Zhang, Y.; Tittanegro, T.H.; Rodríguez-Lorenzo, S.; Jathanna, A.; Pourcet, B.; Pello, O.M.; et al. Disrupting LXRα Phosphorylation Promotes FoxM1 Expression and Modulates Atherosclerosis by Inducing Macrophage Proliferation. Proc. Natl. Acad. Sci. USA 2018, 115, E6556–E6565. [Google Scholar] [CrossRef]
- Neuhofer, A.; Wernly, B.; Leitner, L.; Sarabi, A.; Sommer, N.G.; Staffler, G.; Zeyda, M.; Stulnig, T.M. An Accelerated Mouse Model for Atherosclerosis and Adipose Tissue Inflammation. Cardiovasc. Diabetol. 2014, 13, 23. [Google Scholar] [CrossRef]
- Pendse, A.A.; Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Maeda, N. Apolipoprotein E Knock-out and Knock-in Mice: Atherosclerosis, Metabolic Syndrome, and Beyond. J. Lipid Res. 2009, 50, S178–S182. [Google Scholar] [CrossRef]
- Dupasquier, C.M.C.; Dibrov, E.; Kneesh, A.L.; Cheung, P.K.M.; Lee, K.G.Y.; Alexander, H.K.; Yeganeh, B.K.; Moghadasian, M.H.; Pierce, G.N. Dietary Flaxseed Inhibits Atherosclerosis in the LDL Receptor-Deficient Mouse in Part through Antiproliferative and Anti-Inflammatory Actions. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2394–H2402. [Google Scholar] [CrossRef]
- Grassin-Delyle, S.; Abrial, C.; Salvator, H.; Brollo, M.; Naline, E.; Devillier, P. The Role of Toll-like Receptors in the Production of Cytokines by Human Lung Macrophages. J. Innate Immun. 2020, 12, 63–73. [Google Scholar] [CrossRef]
- Papadopoulos, G.; Weinberg, E.O.; Massari, P.; Gibson, F.C.; Wetzler, L.M.; Morgan, E.F.; Genco, C.A. Macrophage-Specific TLR2 Signaling Mediates Pathogen-Induced TNF-Dependent Inflammatory Oral Bone Loss. J. Immunol. 2013, 190, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin Inhibits Monocyte-To-Macrophage Differentiation via AMPK-Mediated Inhibition of STAT3 Activation: Potential Role in Atherosclerosis. Diabetes 2014, 64, 2028–2041. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. 2011, 122, 253–270. [Google Scholar] [CrossRef]
- Higgins, J.W.; Bedwell, D.W.; Zamek-Gliszczynski, M.J. Ablation of Both Organic Cation Transporter (Oct)1 and Oct2 Alters Metformin Pharmacokinetics but Has No Effect on Tissue Drug Exposure and Pharmacodynamics. Drug Metab. Dispos. 2012, 40, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Qiu, S.; Zhu, X.; Lin, H.; Li, L. OCT1-Mediated Metformin Uptake Regulates Pancreatic Stellate Cell Activity. Cell. Physiol. Biochem. 2018, 47, 1711–1720. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The Mechanisms of Action of Metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Houzet, L.; Defrance, P.; Droogmans, L.; Huez, G.; Kruys, V. Tumor necrosis factor-α mRNA remains unstable and hypoadenylated upon stimulation of macrophages by lipopolysaccharides. Eur. J. Biochem. 2000, 267, 6004–6012. [Google Scholar] [CrossRef]
- Shurety, W.; Merino-Trigo, A.; Brown, D.; Hume, D.A.; Stow, J.L. Localization and Post-Golgi Traffiing of Tumor Necrosis Factor-alpha in Macrophages. J. Interferon Cytokine Res. 2000, 20, 427–438. [Google Scholar] [CrossRef]
- Bauer, M.; Weis, S.; Netea, M.G.; Wetzker, R. Remembering Pathogen Dose: Long-Term Adaptation in Innate Immunity. Trends Immunol. 2018, 39, 438–445. [Google Scholar] [CrossRef]
- Makimura, Y.; Asai, Y.; Taiji, Y.; Sugiyama, A.; Tamai, R.; Ogawa, T. Correlation between Chemical Structure and Biological Activities of Porphyromonas Gingivalis Synthetic Lipopeptide Derivatives. Clin. Exp. Immunol. 2006, 146, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Latz, E. The Inflammasomes: Mechanisms of Activation and Function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Reyes, J.L.; Iannuzzi, J.; Leung, G.; McKay, D.M. The Pro-Inflammatory Cytokine, Interleukin-6, Enhances the Polarization of Alternatively Activated Macrophages. PLoS ONE 2014, 9, e94188. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Ohishi, M.; Mori, H.; Murakami, M.; Chinen, T.; Aki, D.; Hanada, T.; Takeda, K.; Akira, S.; Hoshijima, M.; et al. IL-6 Induces an Anti-Inflammatory Response in the Absence of SOCS3 in Macrophages. Nat. Immunol. 2003, 4, 551–556. [Google Scholar] [CrossRef]
- Nackiewicz, D.; Dan, M.; He, W.; Kim, R.; Salmi, A.; Rütti, S.; Westwell-Roper, C.; Cunningham, A.; Speck, M.; Schuster-Klein, C.; et al. TLR2/6 and TLR4-Activated Macrophages Contribute to Islet Inflammation and Impair Beta Cell Insulin Gene Expression via IL-1 and IL-6. Diabetologia 2014, 57, 1645–1654. [Google Scholar] [CrossRef]
- Hunt, D.; Drake, L.A.; Drake, J.R. Murine Macrophage TLR2-FcγR Synergy via FcγR Licensing of IL-6 Cytokine MRNA Ribosome Binding and Translation. PLoS ONE 2018, 13, e0200764. [Google Scholar] [CrossRef]
- Su, X.; Yu, Y.; Zhong, Y.; Giannopoulou, E.G.; Hu, X.; Liu, H.; Cross, J.R.; Rätsch, G.; Rice, C.M.; Ivashkiv, L.B. Interferon-γ Regulates Cellular Metabolism and MRNA Translation to Potentiate Macrophage Activation. Nat. Immunol. 2015, 16, 838–849. [Google Scholar] [CrossRef]
- Wu, C.; Xue, Y.; Wang, P.; Lin, L.; Liu, Q.; Li, N.; Xu, J.; Cao, X. IFN-γ Primes Macrophage Activation by Increasing Phosphatase and Tensin Homolog via Downregulation of MiR-3473b. J. Immunol. 2014, 193, 3036–3044. [Google Scholar] [CrossRef]
- Hu, X.; Chakravarty, S.D.; Ivashkiv, L.B. Regulation of Interferon and Toll-like Receptor Signaling during Macrophage Activation by Opposing Feedforward and Feedback Inhibition Mechanisms. Immunol. Rev. 2008, 226, 41–56. [Google Scholar] [CrossRef]
- Schleicher, U. Minute Numbers of Contaminant CD8+ T Cells or CD11b+CD11c+ NK Cells Are the Source of IFN-γ in IL-12/IL-18-Stimulated Mouse Macrophage Populations. Blood 2004, 105, 1319–1328. [Google Scholar] [CrossRef]
- Fultz, M.J.; Barber, S.A.; Dieffenbach, C.W.; Vogel, S.N. Induction of IFN-γ in Macrophages by Lipopolysaccharide. Int. Immunol. 1993, 5, 1383–1392. [Google Scholar] [CrossRef]
- Benson, S.A.; Ernst, J.D. TLR2-Dependent Inhibition of Macrophage Responses to IFN-γ Is Mediated by Distinct, Gene-Specific Mechanisms. PLoS ONE 2009, 4, e6329. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Heo, Y.J.; Choi, S.-E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Anti-Inflammatory Effects of Empagliflozin and Gemigliptin on LPS-Stimulated Macrophage via the IKK/NF-ΚB, MKK7/JNK, and JAK2/STAT1 Signalling Pathways. J. Immunol. Res. 2021, 2021, 9944880. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Jelaska, A.; Zeller, C.; Kim, G.; Broedl, U.C.; Woerle, H.J.; EMPA-REG BASALTM trial investigators. Impact of empagliflozin added on to basal insulin in type 2 diabetes inadequately controlled on basal insulin: A 78-week randomized, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2015, 17, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.; Woerle, H.-J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef]
- Al Jobori, H.; Daniele, G.; Adams, J.; Cersosimo, E.; Solis-Herrera, C.; Triplitt, C.; DeFronzo, R.; Abdul-Ghani, M. Empagliflozin Treatment Is Associated With Improved β-Cell Function in Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2018, 103, 1402–1407. [Google Scholar] [CrossRef]
- Dror, E.; Dalmas, E.; Meier, D.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283. [Google Scholar] [CrossRef]
- Gage, M.C. Measuring Apoptotic Cell Engulfment (Efferocytosis) Efficiency. Methods Mol. Biol. 2019, 1951, 143–152. [Google Scholar] [CrossRef]
- Pourcet, B.; Gage, M.C.; León, T.E.; Waddington, K.E.; Pello, O.M.; Steffensen, K.R.; Castrillo, A.; Valledor, A.F.; Pineda-Torra, I. The Nuclear Receptor LXR Modulates Interleukin-18 Levels in Macrophages through Multiple Mechanisms. Sci. Rep. 2016, 6, 25481. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Arefin, A.; Ismail Ema, T.; Islam, T.; Saddam Hossen, M.; Islam, T.; Al Azad, S.; Nasir Uddin Badal, M.; Aminul Islam, M.; Biswas, P.; Alam, N.U.; et al. Target Specificity of Selective Bioactive Compounds in Blocking α-Dystroglycan Receptor to Suppress Lassa Virus Infection: An In Silico Approach. J. Biomed. Res. 2021, 35, 459. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An Integrated Modeling System. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. A New Model for Calculating Atomic Charges in Molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx; Methods in Molecular Biology, 1263; Humana Press: New York, NY, USA, 2014; pp. 243–250. ISBN 978-1-4939-2269-7. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Protein | Ligand | Potential H-Bond Formation | Predicted Amino Acid Residue Interaction (Number) | Predicted Binding Energy (kcal/mol) |
|---|---|---|---|---|
| TLR2 | Zymosan | 5 | R423 (1) V425 (1) D444 (1) S445 (1) S447 (1) | −4.2 |
| TLR2 | Empagliflozin | 6 | V425 (2) S445 (4) | −6.0 |
| Target Protein | Ligand | Potential H-Bond Formation | Predicted Amino Acid Residue Interaction (Number) | Predicted Binding Energy (kcal/mol) |
|---|---|---|---|---|
| DECTIN1 | Zymosan | 8 | H126 (1) K128 (1) S129 (1) Y131 (1) N159 (3) E241 (1) | −5.0 |
| DECTIN1 | Empagliflozin | 3 | Y131 (2) N159 (1) | −6.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arefin, A.; Gage, M.C. Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression. Int. J. Mol. Sci. 2023, 24, 4785. https://doi.org/10.3390/ijms24054785
Arefin A, Gage MC. Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression. International Journal of Molecular Sciences. 2023; 24(5):4785. https://doi.org/10.3390/ijms24054785
Chicago/Turabian StyleArefin, Adittya, and Matthew C. Gage. 2023. "Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression" International Journal of Molecular Sciences 24, no. 5: 4785. https://doi.org/10.3390/ijms24054785
APA StyleArefin, A., & Gage, M. C. (2023). Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression. International Journal of Molecular Sciences, 24(5), 4785. https://doi.org/10.3390/ijms24054785