
Specificity Proteins (SP) and Krüppel-like Factors (KLF) in Liver Physiology and Pathology


Abstract
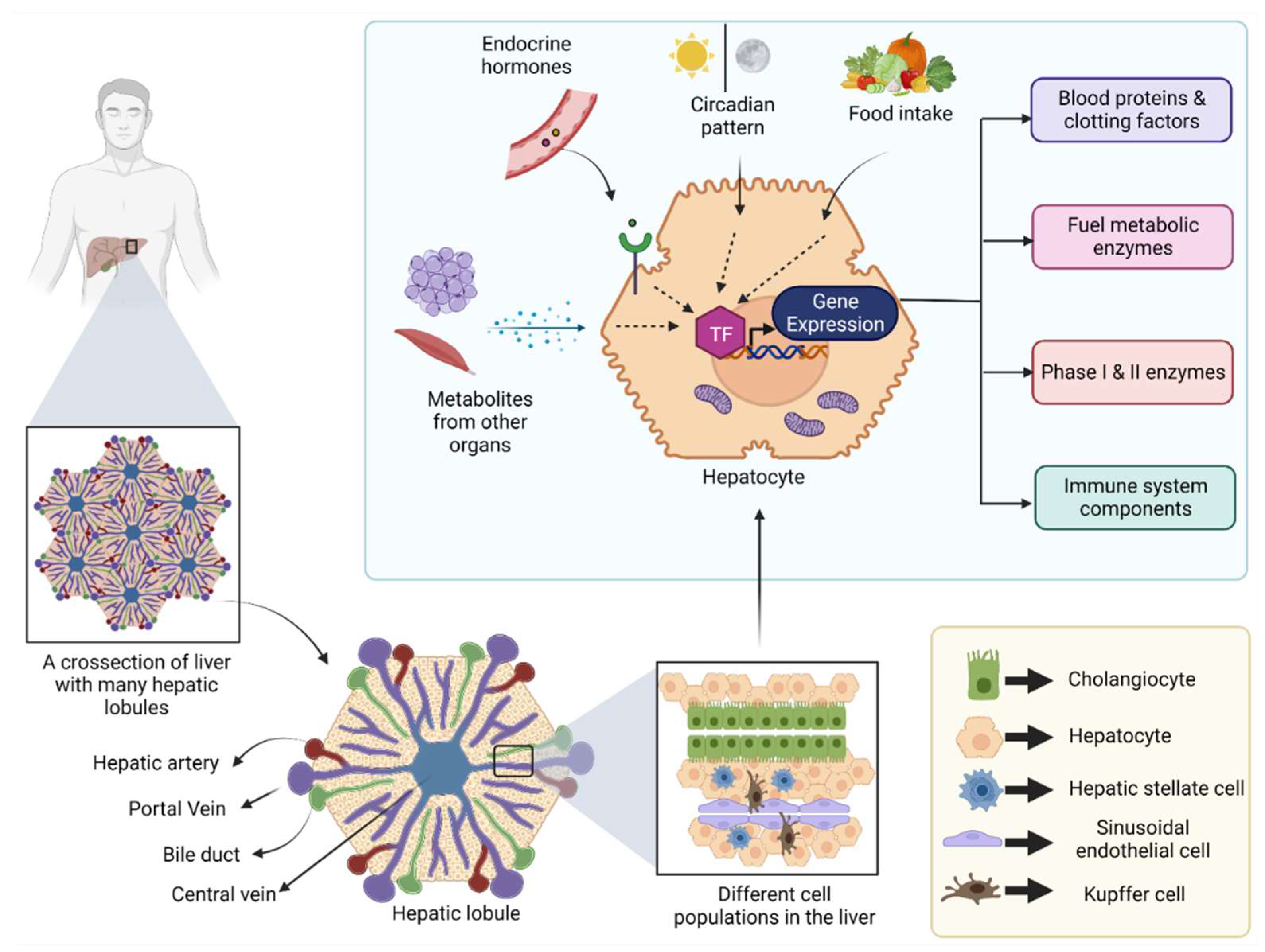
1. Introduction
2. SP and KLF Transcription Factors
- -
- KLF1, KLF2, KLF 4, KLF5, KLF6, and KLF7,
- -
- KLF3, KLF8, and KLF12 group,
- -
- KLF9, KLF10, KLF11, KLF13, KLF14, and KLF16 group [31].
- -
- SP1, SP2, SP3, and SP4,
- -
- SP5, SP6, SP7, SP8, and SP9 [32].
3. SP and KLF Transcription Factors in a Healthy Liver
3.1. Embryonic Liver Development
3.2. Glucose Metabolism
3.3. Fatty Acid and Cholesterol Metabolism
3.4. Amino Acid and Protein Metabolism
3.5. Bile Acid Synthesis and Metabolism
3.6. Blood Coagulation
3.7. Xenobiotic Metabolism
3.8. Liver Regeneration
4. SPs and KLFs in the Pathogenesis of Hepatic Diseases
4.1. Metabolic Diseases (Diabetes, Hyperlipidemia, and Hypercholesteremia)
4.2. Non-Alcoholic Fatty Liver Disease (NAFLD)
4.3. Hepatocellular Carcinoma (HCC)
4.4. Hepatic Fibrosis and Cirrhosis
4.5. Viral Hepatitis
4.6. Biliary Cholangitis
4.7. Hemochromatosis
4.8. Wilson’s Disease
4.9. Drug-Induced Hepatic Toxicity
4.10. Acute Hepatic Failure
5. Epilogue
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | Alpha smooth muscle actin |
| ABCA1 | ATP binding cassette subfamily A member 1 |
| ABCG1 | ATP binding cassette subfamily G member 1 |
| ACC | Acetyl-CoA carboxylase |
| AG | Alpha glucosidase |
| ALT1 | Alanine transaminase |
| AGPAT1 | 1-acylglycerol-3-phosphate O-acyltransferase 1 |
| Akt | AKT serine/threonine kinase 1 |
| Apo A-I | Apolipoprotein A-I |
| Apo A-IV | Apolipoprotein A-IV |
| AMPK | Adenosine monophosphate activated protein kinase |
| ApoE | Apolipoprotein E |
| AP-1 | Activator protein 1 |
| AhR | Aryl hydrocarbon receptor |
| Atg7 | Autophagy related 7 gene |
| ATP7B | ATPase copper transporting beta |
| Btd | Buttonhead box domain |
| Bmal1 | basic helix-loop-helix ARNT Like 1 |
| BA | Bile acids |
| Becn1 | Beclin1 |
| c/EBP | CCAAT-enhancer-binding proteins |
| ChREBP | Carbohydrate response element binding protein |
| CtBP | C-terminal-binding protein |
| CFU-E | Colony forming unit-Erythroid |
| CFU-MK | Colony forming unit-Megakaryocyte |
| CXCR4 | C-X-C chemokine receptor type 4 |
| CPS1 | Carbamoyl-phosphate synthase 1 |
| CYP450 | Cytochrome P450 |
| CYP1A1 | Cytochrome P450 family 1 subfamily A member 1 |
| CYP1A2 | Cytochrome P450 family 1 subfamily A member 2 |
| CYP2B10 | Cytochrome P450, family 2, subfamily b, polypeptide 10 |
| CYP2D6 | Cytochrome P450 family 2 subfamily D member 6 |
| CYP2E1 | Cytochrome P450 family 2 subfamily E member 1 |
| CYP3A4 | Cytochrome P450 family 3 subfamily A member 4 |
| CY4A10 | Cytochrome P450, family 4, subfamily a, polypeptide 10 |
| CYP4A14 | Cytochrome P450, family 4, subfamily a, polypeptide 14 |
| CYP7A1 | Cytochrome P450 family 7 subfamily A member 1 |
| CYP27A1 | Cytochrome P450 family 27 subfamily A member 1 |
| CYP13A8 | C.elegans cytochrome P450 family 13 subfamily A member 8 |
| CYP13A11 | C.elegans cytochrome P450 family 13 subfamily A member 11 |
| CYP33C9 | C.elegans cytochrome P450 family 33 subfamily C member 9 |
| CYP37A1 | C.elegans cytochrome P450 family 37 subfamily A member 1 |
| cAMP | 3',5'-cyclic adenosine monophosphate |
| CREB | cAMP response element-binding protein |
| CCL4 | Carbon tetrachloride |
| ChIP | Chromatin immunoprecipitation |
| CPT1A | Carnitine palmitoyltransferase 1A |
| CETP | Cholesteryl ester transfer protein |
| CA | Cholicacid |
| CDCA | Chenodeoxy cholicacid |
| CDX2 | Caudal-type homeobox 2 |
| c-MYC | MYC proto-oncogene |
| CSE | Cystathionine gamma-lyase |
| ccc DNA | Covalently closed circular DNA |
| DBP | D site of albumin promoter (albumin D-box) binding protein |
| DEX | Dexamethasone |
| DGAT1 | Diacylglycerol O-acyltransferase 1 |
| DDC | 3,5-diethoxycarbonyl-1,4-dihydrocollidine |
| ESLD | End stage liver disease |
| EKLF | Erythroid Krüppel-like factor |
| EMSA | Electrophoretic mobility shift assay |
| eNOS | Endothelial nitric oxide synthase |
| ERG | ETS related gene |
| ETF | ETF transcription factor |
| E2F | E2F family of transcription factors |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular signal-regulated kinases |
| FOXA2 | Forkhead Box A2 |
| FOXO1 | Forkhead box O1 |
| FOXO3a | Forkhead box O3 |
| Fam132a | Adipose-derived insulin-sensitizing factor; Adipolin |
| FGF15 | Fibroblast growth factor 15 |
| FGF21 | Fibroblast growth factor 21 |
| FASN | Fatty acid synthase |
| FBW7α | F-box and WD repeat domain containing 7 |
| FATP | Fatty acid transport protein |
| FXR | Farnesoid X receptor |
| GATA1 | GATA-binding factor 1 |
| GATA4 | GATA binding factor 4 |
| GATA6 | GATA-binding factor 6 |
| GLUT2 | Glucose transporter 2 |
| GCK | Glucokinase |
| G6PC | Glucose 6-phosphatase |
| GPAT1 | Glycerol-3-phosphate acyltransferase, mitochondrial |
| GSK-3β | Glycogen synthase kinase 3 beta |
| Gpbar1 | G protein-coupled bile acid receptor 1 |
| GST | Glutathione-s-transferase |
| HCC | Hepatocellular carcinoma |
| Hhex | Hematopoietically expressed homeobox |
| HNF4 | Hepatocyte nuclear factor 4 |
| HGF | Hepatocyte growth factor |
| HNF3β | Hepatocyte nuclear factor 3-beta |
| HFD | High fat diet |
| HDL | High density lipoprotein |
| HPD | 4-hydroxyphenylpyruvate dioxygenase |
| HUVEC | Human umbilical vein endothelial cells |
| HULC | Hepatocellular carcinoma up-regulated long non-coding RNA |
| HBV | Hepatitis B virus |
| HFE | Homeostatic iron regulator |
| InsR | Insulin receptor |
| IRS-1 | Insulin receptor substrate 1 |
| iPSCs | Induced pluripotent stem cells |
| KLFs | Krüppel-like family of transcription factors |
| LXR | The liver X receptor |
| LDL | Low density lipoprotein |
| LCAT | Lecithin-cholesterol acyltransferase |
| LncRNA | Long noncoding RNA |
| MCAD | Medium-chain acyl-CoA dehydrogenase |
| MAPK | Mitogen-activated protein kinases |
| MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| MMP-1 | Matrix metallopeptidase 1 |
| MMP-9 | Matrix metallopeptidase 9 |
| NASH | Nonalcoholic steatohepatitis |
| NAFLD | Nonalcoholic fatty liver disease |
| NEFA | Non-esterified fatty acids |
| NAG | N-Acetylglutamate |
| NAGS | N-Acetylglutamate Synthetase |
| NR0B2 | Nuclear receptor subfamily 0 group B member 2 |
| NF-κB | Nuclear factor kappa B |
| NCOA3 | Nuclear receptor coactivator 3 |
| OSM | Oncostatin M |
| OTC | Ornithine transcarbamylase |
| OCT-4 | Octamer-binding transcription factor 4 |
| ODN | Oligodeoxynucleotides |
| PARP1 | Poly ADP ribose polymerase 1 |
| PGC-1α | Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PCK1 | Phosphoenolpyruvate carboxykinase 1 |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PI3K | Phosphoinositide 3-kinase |
| PRODH | Proline dehydrogenase |
| PSC | Primary sclerosing cholangitis |
| PAI-I | Plasminogen activator inhibitor 1 |
| PON1 | Paraoxonase 1 |
| PCNA | Proliferating cell nuclear antigen |
| P53 | Tumor protein P53 |
| PDGF-β | Platelet derived growth factor-B |
| PXR | Pregnane X receptor |
| RORα | Retinoid-related orphan receptor alfa |
| RasGRP1 | RAS guanyl releasing protein 1 |
| RYBP | RING1 and YY1 binding protein |
| SREBF1 | Sterol regulatory element binding transcription factor 1 |
| SREBP-1C | Sterol regulatory element-binding protein 1 |
| SP | Specificity protein |
| SOX17 | SRY-Box transcription factor 17 |
| SR-B1 | The scavenger receptor, class B type 1 |
| SCD1 | Stearoyl-Coenzyme A desaturase 1 |
| SSW | Sweetened sucrose water |
| SULT1A1 | Sulfotransferase family 1A member 1 |
| SULT2B1B | Sulfotransferase family 2B member 1 |
| SOX-2 | (Sex determining region Y)-box 2 |
| SMAD3 | SMAD family member 3 |
| TAT | Tyrosine aminotransferase |
| TGs | Triglycerides |
| TDO2 | Tryptophan 2,3-Dioxygenase |
| TGR-5 | Takeda G protein-coupled receptor 5 |
| TSS | Transcriptional start site |
| TM | Thrombomodulin |
| TF | Tissue factor |
| TGF-β | Transforming growth factor beta |
| TERT | Telomerase reverse transcriptase |
| TIMP-1 | Tissue inhibitor matrix metalloproteinase 1 |
| TNF-α | Tumor necrosis factor α |
| UPR | Unfolded protein response |
| UGT1A8 | UDP glucuronosyltransferase family 1 member A8 |
| UGT1A9 | UDP Glucuronosyltransferase Family 1 Member A9 |
| UGT1A10 | UDP Glucuronosyltransferase Family 1 Member A10 |
| VCAM-1VLDL | Vascular cell adhesion molecule-1Very low-density lipoprotein |
| WD | Wilsons disease |
| WNT | Proto-oncogene Wnt |
| WAT | White adipose tissue |
| XRE | Xenobiotic response element |
| YY1 | Yin Yang 1 |
| ZDHHC7 | Zinc finger DHHC domain-containing protein 7 |
| ZBP-89 | Zinc-finger binding protein-89 |
References
- Kalra, A.; Yetiskul, E.; Wehrle, C.J.; Tuma, F. Physiology, Liver. 2018. Available online: https://europepmc.org/article/nbk/nbk535438 (accessed on 7 September 2022).
- Barrett, K.E. Chapter 10. Functional Anatomy of the Liver and Biliary System. In Gastrointestinal Physiology, 2e; The McGraw-Hill Companies: New York, NY, USA, 2014; Available online: https://accessmedicine.mhmedical.com/content.aspx? (accessed on 27 February 2023).
- Ding, C.; Li, Y.; Guo, F.; Jiang, Y.; Ying, W.; Li, D.; Yang, D.; Xia, X.; Liu, W.; Zhao, Y. A cell-type-resolved liver proteome. Mol. Cell. Proteom. 2016, 15, 3190–3202. [Google Scholar] [CrossRef]
- Lautt, W.W. Hepatic circulation: Physiology and pathophysiology. In Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Publishers: San Rafael, CA, USA, 2009; pp. 1–174. [Google Scholar]
- Atashgahi, S.; Shetty, S.A.; Smidt, H.; de Vos, W.M. Flux, Impact, and Fate of Halogenated Xenobiotic Compounds in the Gut. Front. Physiol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of hepatotoxicity. Toxicol. Sci. 2002, 65, 166–176. [Google Scholar] [CrossRef]
- Grant, L.M.; Rockey, D.C. Drug-induced liver injury. Curr. Opin. Gastroenterol. 2012, 28, 198–202. [Google Scholar] [CrossRef]
- Sharma, A.; Nagalli, S. Chronic liver disease. In StatPearls [Internet]; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Haep, N.; Florentino, R.M.; Squires, J.E.; Bell, A.; Soto-Gutierrez, A. The Inside-Out of End-Stage Liver Disease: Hepatocytes are the Keystone. In Seminars in Liver Disease; Thieme Medical Publishers, Inc.: New York, NY, USA, 2021; pp. 213–224. [Google Scholar]
- Cheemerla, S.; Balakrishnan, M. Global epidemiology of chronic liver disease. Clin. Liver Dis. 2021, 17, 365. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Dufour, J.-F.; Anstee, Q.M.; Bugianesi, E.; Harrison, S.; Loomba, R.; Paradis, V.; Tilg, H.; Wong, V.W.-S.; Zelber-Sagi, S. Current therapies and new developments in NASH. Gut 2022, 71, 2123–2134. [Google Scholar] [CrossRef]
- Tapper, E.B.; Ufere, N.N.; Huang, D.Q.; Loomba, R. Current and emerging therapies for the management of cirrhosis and its complications. Aliment. Pharmacol. Ther. 2022, 55, 1099–1115. [Google Scholar] [CrossRef]
- Schmid, P.; Schulz, W.A. Coexpression of the c-myc protooncogene with alpha-fetoprotein and albumin in fetal mouse liver. Differentiation 1990, 45, 96–102. [Google Scholar] [CrossRef]
- Cascio, S.; Zaret, K.S. Hepatocyte differentiation initiates during endodermal-mesenchymal interactions prior to liver formation. Development 1991, 113, 217–225. [Google Scholar] [CrossRef]
- Lin, H.V.; Accili, D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011, 14, 9–19. [Google Scholar] [CrossRef]
- Shimizu, N.; Maruyama, T.; Yoshikawa, N.; Matsumiya, R.; Ma, Y.; Ito, N.; Tasaka, Y.; Kuribara-Souta, A.; Miyata, K.; Oike, Y.; et al. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat. Commun. 2015, 6, 6693. [Google Scholar] [CrossRef]
- Chun, S.K.; Masri, S. Circadian Control of Transcriptional and Metabolic Rhythms in Primary Hepatocytes. Methods Mol. Biol. 2022, 2482, 169–179. [Google Scholar] [CrossRef]
- Bideyan, L.; Nagari, R.; Tontonoz, P. Hepatic transcriptional responses to fasting and feeding. Genes Dev. 2021, 35, 635–657. [Google Scholar] [CrossRef] [PubMed]
- Schrem, H.; Klempnauer, J.; Borlak, J. Liver-enriched transcription factors in liver function and development. Part I: The hepatocyte nuclear factor network and liver-specific gene expression. Pharm. Rev. 2002, 54, 129–158. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef]
- Yang, H.; Arif, M.; Yuan, M.; Li, X.; Shong, K.; Türkez, H.; Nielsen, J.; Uhlén, M.; Borén, J.; Zhang, C.; et al. A network-based approach reveals the dysregulated transcriptional regulation in non-alcoholic fatty liver disease. iScience 2021, 24, 103222. [Google Scholar] [CrossRef]
- Xu, X.; Poulsen, K.L.; Wu, L.; Liu, S.; Miyata, T.; Song, Q.; Wei, Q.; Zhao, C.; Lin, C.; Yang, J. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target 2022, 7, 287. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct. Target 2022, 7, 206. [Google Scholar] [CrossRef]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Caramori, G. Chapter 31 - Transcription Factors. In Asthma and COPD, 2nd ed.; Barnes, P.J., Drazen, J.M., Rennard, S.I., Thomson, N.C., Eds.; Academic Press: Oxford, UK, 2009; pp. 373–380. [Google Scholar] [CrossRef]
- Swamynathan, S.K. Krüppel-like factors: Three fingers in control. Hum. Genom. 2010, 4, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Hoffman, M.; Goldberg, I.J.; Drosatos, K. Krüppel-like factors: Crippling and un-crippling metabolic pathways. JACC Basic Transl. Sci. 2018, 3, 132–156. [Google Scholar] [CrossRef] [PubMed]
- Presnell, J.S.; Schnitzler, C.E.; Browne, W.E. KLF/SP Transcription Factor Family Evolution: Expansion, Diversification, and Innovation in Eukaryotes. Genome Biol. Evol. 2015, 7, 2289–2309. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Bouwman, P.; Philipsen, S. Regulation of the activity of Sp1-related transcription factors. Mol. Cell. Endocrinol. 2002, 195, 27–38. [Google Scholar] [CrossRef]
- Blom-Dahl, D.; Córdoba, S.; Gabilondo, H.; Carr-Baena, P.; Díaz-Benjumea, F.J.; Estella, C. In vivo analysis of the evolutionary conserved BTD-box domain of Sp1 and Btd during Drosophila development. Dev. Biol. 2020, 466, 77–89. [Google Scholar] [CrossRef]
- Schöck, F.; Sauer, F.; Jäckle, H.; Purnell, B.A. Drosophila head segmentation factor buttonhead interacts with the same TATA box-binding protein-associated factors and in vivo DNA targets as human Sp1 but executes a different biological program. Proc. Natl. Acad. Sci. USA 1999, 96, 5061–5065. [Google Scholar] [CrossRef]
- Treichel, D.; Schöck, F.; Jäckle, H.; Gruss, P.; Mansouri, A. mBtd is required to maintain signaling during murine limb development. Genes Dev. 2003, 17, 2630–2635. [Google Scholar] [CrossRef]
- Athanikar, J.N.; Sanchez, H.B.; Osborne, T.F. Promoter selective transcriptional synergy mediated by sterol regulatory element binding protein and Sp1: A critical role for the Btd domain of Sp1. Mol. Cell Biol. 1997, 17, 5193–5200. [Google Scholar] [CrossRef]
- Black, A.R.; Black, J.D.; Azizkhan-Clifford, J. Sp1 and krüppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 2001, 188, 143–160. [Google Scholar] [CrossRef]
- Parkins, A.C.; Sharpe, A.H.; Orkin, S.H. Lethal β-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature 1995, 375, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.T.; Veselits, M.L.; Leiden, J.M. LKLF: A transcriptional regulator of single-positive T cell quiescence and survival. Science 1997, 277, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Zorn, A.M. Liver development. In StemBook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2008. [Google Scholar] [CrossRef]
- Arora-Gupta, N.; Hagedorn, C.H. Liver, Development. In Encyclopedia of Gastroenterology; Johnson, L.R., Ed.; Elsevier: New York, NY, USA, 2004; pp. 530–533. [Google Scholar] [CrossRef]
- Gordillo, M.; Evans, T.; Gouon-Evans, V. Orchestrating liver development. Development 2015, 142, 2094–2108. [Google Scholar] [CrossRef] [PubMed]
- Krüger, I.; Vollmer, M.; Simmons, D.; Elsässer, H.P.; Philipsen, S.; Suske, G. Sp1/Sp3 compound heterozygous mice are not viable: Impaired erythropoiesis and severe placental defects. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 2235–2244. [Google Scholar] [CrossRef]
- Nuez, B.; Michalovich, D.; Bygrave, A.; Ploemacher, R.; Grosveld, F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nat. Int. Wkly. J. Sci. 1995, 375, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Kubo, A.; Liu, H.; Akita, K.; Laub, F.; Ramirez, F.; Keller, G.; Friedman, S.L. Developmental regulation of yolk sac hematopoiesis by Kruppel-like factor 6. Blood 2006, 107, 1357–1365. [Google Scholar] [CrossRef]
- Zhao, X.; Monson, C.; Gao, C.; Gouon-Evans, V.; Matsumoto, N.; Sadler, K.C.; Friedman, S.L. Klf6/copeb is required for hepatic outgrowth in zebrafish and for hepatocyte specification in mouse ES cells. Dev. Biol. 2010, 344, 79–93. [Google Scholar] [CrossRef]
- Lavallée, G.; Andelfinger, G.; Nadeau, M.; Lefebvre, C.; Nemer, G.; Horb, M.E.; Nemer, M. The Kruppel-like transcription factor KLF13 is a novel regulator of heart development. EMBO J. 2006, 25, 5201–5213. [Google Scholar] [CrossRef]
- Anzai, K.; Tsuruya, K.; Ida, K.; Kagawa, T.; Inagaki, Y.; Kamiya, A. Kruppel-like factor 15 induces the development of mature hepatocyte-like cells from hepatoblasts. Sci. Rep. 2021, 11, 18551. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [PubMed]
- Lemaigre, F.P.; Rousseau, G.G. Transcriptional control of genes that regulate glycolysis and gluconeogenesis in adult liver. Biochem. J. 1994, 303 Pt 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Bell-Anderson, K.S.; Funnell, A.P.; Williams, H.; Mat Jusoh, H.; Scully, T.; Lim, W.F.; Burdach, J.G.; Mak, K.S.; Knights, A.J.; Hoy, A.J.; et al. Loss of Krüppel-like factor 3 (KLF3/BKLF) leads to upregulation of the insulin-sensitizing factor adipolin (FAM132A/CTRP12/C1qdc2). Diabetes 2013, 62, 2728–2737. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Gastaldelli, A.; Vetter, D.; Patman, G.L.; Pascoe, L.; Hannivoort, R.A.; Lee, U.E.; Fiel, I.; Munoz, U.; Ciociaro, D.; et al. Glucokinase links Kruppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease. Hepatology 2012, 55, 1083–1093. [Google Scholar] [CrossRef]
- Cui, A.; Fan, H.; Zhang, Y.; Zhang, Y.; Niu, D.; Liu, S.; Liu, Q.; Ma, W.; Shen, Z.; Shen, L.; et al. Dexamethasone-induced Krüppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J. Clin. Investig. 2019, 129, 2266–2278. [Google Scholar] [CrossRef]
- Gans, I.M.; Grendler, J.; Babich, R.; Jayasundara, N.; Coffman, J.A. Glucocorticoid-Responsive Transcription Factor Krüppel-Like Factor 9 Regulates fkbp5 and Metabolism. Front. Cell Dev. Biol. 2021, 9, 727037. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Q.; Sun, L.; Zhang, H.; Yao, L.; Cui, X.; Gao, Y.; Fang, F.; Chang, Y. KLF10 transcription factor regulates hepatic glucose metabolism in mice. Diabetologia 2017, 60, 2443–2452. [Google Scholar] [CrossRef]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Krüppel-like factor-10 is directly regulated by carbohydrate response element-binding protein in rat primary hepatocytes. Biochem. Biophys. Res. Commun. 2011, 412, 638–643. [Google Scholar] [CrossRef]
- Wang, L.; Tong, X.; Gu, F.; Zhang, L.; Chen, W.; Cheng, X.; Xie, L.; Chang, Y.; Zhang, H. The KLF14 transcription factor regulates hepatic gluconeogenesis in mice. J. Biol. Chem. 2017, 292, 21631–21642. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.R.; Fan, L.; Jain, M.K. Taking KLF9 to "Cort" for crimes against metabolism. J. Clin. Investig. 2019, 129, 2178–2180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Q.; Jiao, T.; Cui, A.; Sun, X.; Fang, W.; Xie, L.; Liu, Y.; Fang, F.; Chang, Y. Involvement of KLF11 in hepatic glucose metabolism in mice via suppressing of PEPCK-C expression. PLoS ONE 2014, 9, e89552. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.; Wang, B.; Orihuela, Y.; Hong, E.G.; Fisch, S.; Haldar, S.; Cline, G.W.; Kim, J.K.; Peroni, O.D.; Kahn, B.B.; et al. Regulation of gluconeogenesis by Krüppel-like factor 15. Cell Metab. 2007, 5, 305–312. [Google Scholar] [CrossRef]
- Teshigawara, K.; Ogawa, W.; Mori, T.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; Inoue, H.; Miyake, K.; Sakaue, H.; Kasuga, M. Role of Krüppel-like factor 15 in PEPCK gene expression in the liver. Biochem. Biophys. Res. Commun. 2005, 327, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Takashima, M.; Ogawa, W.; Hayashi, K.; Inoue, H.; Kinoshita, S.; Okamoto, Y.; Sakaue, H.; Wataoka, Y.; Emi, A.; Senga, Y.; et al. Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes 2010, 59, 1608–1615. [Google Scholar] [CrossRef]
- Chiefari, E.; Foti, D.P.; Sgarra, R.; Pegoraro, S.; Arcidiacono, B.; Brunetti, F.S.; Greco, M.; Manfioletti, G.; Brunetti, A. Transcriptional Regulation of Glucose Metabolism: The Emerging Role of the HMGA1 Chromatin Factor. Front. Endocrinol. 2018, 9, 357. [Google Scholar] [CrossRef]
- Chen, S.; Li, H.; Zhang, J.; Jiang, S.; Zhang, M.; Xu, Y.; Dong, K.; Yang, Y.; Fang, Q.; Jia, W. Identification of Sp1 as a Transcription Activator to Regulate Fibroblast Growth Factor 21 Gene Expression. Biomed. Res. Int. 2017, 2017, 8402035. [Google Scholar] [CrossRef]
- Moreno-Aliaga, M.J.; Swarbrick, M.M.; Lorente-Cebrián, S.; Stanhope, K.L.; Havel, P.J.; Martínez, J.A. Sp1-mediated transcription is involved in the induction of leptin by insulin-stimulated glucose metabolism. J. Mol. Endocrinol. 2007, 38, 537–546. [Google Scholar] [CrossRef]
- Egea, M.; Metón, I.; Baanante, I.V. Sp1 and Sp3 regulate glucokinase gene transcription in the liver of gilthead sea bream (Sparus aurata). J. Mol. Endocrinol. 2007, 38, 481–492. [Google Scholar] [CrossRef]
- Mounier, C.; Posner, B.I. Transcriptional regulation by insulin: From the receptor to the gene. Can. J. Physiol. Pharm. 2006, 84, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Dupriez, V.J.; Darville, M.I.; Antoine, I.V.; Gegonne, A.; Ghysdael, J.; Rousseau, G.G. Characterization of a hepatoma mRNA transcribed from a third promoter of a 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-encoding gene and controlled by ets oncogene-related products. Proc. Natl. Acad. Sci. USA 1993, 90, 8224–8228. [Google Scholar] [CrossRef]
- Huang, J.; Borensztajn, J.; Reddy, J.K. Hepatic lipid metabolism. In Molecular Pathology of Liver Diseases; Springer: Berlin/Heidelberg, Germany, 2011; pp. 133–146. [Google Scholar]
- Chen, J.L.; Lu, X.J.; Zou, K.L.; Ye, K. Krüppel-like factor 2 promotes liver steatosis through upregulation of CD36. J. Lipid Res. 2014, 55, 32–40. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, H.J.; Na, H.; Nam, M.W.; Kim, J.Y.; Kim, J.S.; Koo, S.H.; Lee, M.O. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep. 2017, 20, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chen, C.; Zhang, B.; Huang, H.; Wu, G.; Wen, J.; Liu, J. Induction of Krüppel-like factor 4 by high-density lipoproteins promotes the expression of scavenger receptor class B type I. FEBS J. 2010, 277, 3780–3788. [Google Scholar] [CrossRef]
- Kumadaki, S.; Karasawa, T.; Matsuzaka, T.; Ema, M.; Nakagawa, Y.; Nakakuki, M.; Saito, R.; Yahagi, N.; Iwasaki, H.; Sone, H.; et al. Inhibition of ubiquitin ligase F-box and WD repeat domain-containing 7α (Fbw7α) causes hepatosteatosis through Krüppel-like factor 5 (KLF5)/peroxisome proliferator-activated receptor γ2 (PPARγ2) pathway but not SREBP-1c protein in mice. J. Biol. Chem. 2011, 286, 40835–40846. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Vetter, D.; Ishida, J.; Hannivoort, R.A.; Lang, U.E.; Kocabayoglu, P.; Fiel, M.I.; Muñoz, U.; Patman, G.L.; Ge, F.; et al. Post-transcriptional activation of PPAR alpha by KLF6 in hepatic steatosis. J. Hepatol. 2013, 58, 1000–1006. [Google Scholar] [CrossRef]
- Zhou, S.S.; Zhang, Y.L.; Chang, Y.S. [KLF9 regulates hepatic lipid metabolism via inducing CD36 expression]. Sheng Li Xue Bao 2021, 73, 772–780. [Google Scholar] [PubMed]
- Ruberto, A.A.; Gréchez-Cassiau, A.; Guérin, S.; Martin, L.; Revel, J.S.; Mehiri, M.; Subramaniam, M.; Delaunay, F.; Teboul, M. KLF10 integrates circadian timing and sugar signaling to coordinate hepatic metabolism. Elife 2021, 10, e65574. [Google Scholar] [CrossRef]
- Lee, J.; Oh, A.R.; Lee, H.Y.; Moon, Y.A.; Lee, H.J.; Cha, J.Y. Deletion of KLF10 Leads to Stress-Induced Liver Fibrosis upon High Sucrose Feeding. Int. J. Mol. Sci. 2020, 22, 331. [Google Scholar] [CrossRef]
- Yang, S.; Jia, L.; Xiang, J.; Yang, G.; Qiu, S.; Kang, L.; Zheng, P.; Liang, Z.; Lu, Y. KLF10 promotes nonalcoholic steatohepatitis progression through transcriptional activation of zDHHC7. EMBO Rep. 2022, 23, e54229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Q.; Yang, M.; Zhu, B.; Cui, Y.; Xue, Y.; Gong, N.; Cui, A.; Wang, M.; Shen, L.; et al. Mouse KLF11 regulates hepatic lipid metabolism. J. Hepatol. 2013, 58, 763–770. [Google Scholar] [CrossRef]
- Yang, M.; Ren, Y.; Lin, Z.; Tang, C.; Jia, Y.; Lai, Y.; Zhou, T.; Wu, S.; Liu, H.; Yang, G.; et al. Krüppel-like factor 14 increases insulin sensitivity through activation of PI3K/Akt signal pathway. Cell Signal 2015, 27, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Fan, Y.; Zhang, J.; Lomberk, G.A.; Zhou, Z.; Sun, L.; Mathison, A.J.; Garcia-Barrio, M.T.; Zhang, J.; Zeng, L.; et al. Perhexiline activates KLF14 and reduces atherosclerosis by modulating ApoA-I production. J. Clin. Investig. 2015, 125, 3819–3830. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Krüppel-like factor 16 (KLF16) targets PPARα to improve steatohepatitis and insulin resistance. Gut 2021, 70, 2183–2195. [Google Scholar] [CrossRef]
- Jung, D.Y.; Chalasani, U.; Pan, N.; Friedline, R.H.; Prosdocimo, D.A.; Nam, M.; Azuma, Y.; Maganti, R.; Yu, K.; Velagapudi, A.; et al. KLF15 is a molecular link between endoplasmic reticulum stress and insulin resistance. PLoS ONE 2013, 8, e77851. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Yahagi, N.; Aita, Y.; Murayama, Y.; Sawada, Y.; Piao, X.; Toya, N.; Oya, Y.; Shikama, A.; Takarada, A.; et al. KLF15 Enables Rapid Switching between Lipogenesis and Gluconeogenesis during Fasting. Cell Rep. 2016, 16, 2373–2386. [Google Scholar] [CrossRef]
- Fan, L.; Sweet, D.R.; Fan, E.K.; Prosdocimo, D.A.; Madera, A.; Jiang, Z.; Padmanabhan, R.; Haldar, S.M.; Vinayachandran, V.; Jain, M.K. Transcription factors KLF15 and PPARδ cooperatively orchestrate genome-wide regulation of lipid metabolism in skeletal muscle. J. Biol. Chem. 2022, 298, 101926. [Google Scholar] [CrossRef]
- Fan, L.; Sweet, D.R.; Prosdocimo, D.A.; Vinayachandran, V.; Chan, E.R.; Zhang, R.; Ilkayeva, O.; Lu, Y.; Keerthy, K.S.; Booth, C.E.; et al. Muscle Krüppel-like factor 15 regulates lipid flux and systemic metabolic homeostasis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.K.; Ngo, T.T.; Athanikar, J.N.; Rosenfeld, J.M.; Osborne, T.F. Co-stimulation of promoter for low density lipoprotein receptor gene by sterol regulatory element-binding protein and Sp1 is specifically disrupted by the yin yang 1 protein. J. Biol. Chem. 1999, 274, 13025–13032. [Google Scholar] [CrossRef]
- Hoppe, K.L.; Francone, O.L. Binding and functional effects of transcription factors Sp1 and Sp3 on the proximal human lecithin:cholesterol acyltransferase promoter. J. Lipid Res. 1998, 39, 969–977. [Google Scholar] [CrossRef]
- Mizutani, T.; Yamada, K.; Minegishi, T.; Miyamoto, K. Transcriptional regulation of rat scavenger receptor class B type I gene. J. Biol. Chem. 2000, 275, 22512–22519. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, S.; Kan, H.Y.; Reardon-Alulis, C.; Zannis, V. The SP1 sites of the human apoCIII enhancer are essential for the expression of the apoCIII gene and contribute to the hepatic and intestinal expression of the apoA-I gene in transgenic mice. Nucleic Acids Res. 2000, 28, 4919–4929. [Google Scholar] [CrossRef]
- Zheng, X.L.; Matsubara, S.; Diao, C.; Hollenberg, M.D.; Wong, N.C. Activation of apolipoprotein AI gene expression by protein kinase A and kinase C through transcription factor, Sp1. J. Biol. Chem. 2000, 275, 31747–31754. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, Y.; Guo, Z.; Zhou, L.; Okoro, E.U.; Yang, H. Transcriptional regulation of ATP-binding cassette transporter A1 expression by a novel signaling pathway. J. Biol. Chem. 2011, 286, 8917–8923. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T.; Porsch-Ozcürümez, M.; Heimerl, S.; Probst, M.; Moehle, C.; Taher, M.; Borsukova, H.; Kielar, D.; Kaminski, W.E.; Dittrich-Wengenroth, E.; et al. Identification of sterol-independent regulatory elements in the human ATP-binding cassette transporter A1 promoter: Role of Sp1/3, E-box binding factors, and an oncostatin M-responsive element. J. Biol. Chem. 2002, 277, 14443–14450. [Google Scholar] [CrossRef]
- Langmann, T.; Porsch-Ozcürümez, M.; Unkelbach, U.; Klucken, J.; Schmitz, G. Genomic organization and characterization of the promoter of the human ATP-binding cassette transporter-G1 (ABCG1) gene. Biochim. Biophys. Acta 2000, 1494, 175–180. [Google Scholar] [CrossRef]
- Chang, D.J.; Paik, Y.K.; Leren, T.P.; Walker, D.W.; Howlett, G.J.; Taylor, J.M. Characterization of a human apolipoprotein E gene enhancer element and its associated protein factors. J. Biol. Chem. 1990, 265, 9496–9504. [Google Scholar] [CrossRef]
- Le Goff, W.; Guerin, M.; Petit, L.; Chapman, M.J.; Thillet, J. Regulation of human CETP gene expression: Role of SP1 and SP3 transcription factors at promoter sites -690, -629, and -37. J. Lipid Res. 2003, 44, 1322–1331. [Google Scholar] [CrossRef]
- Walker, V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes. Metab. 2009, 11, 823–835. [Google Scholar] [CrossRef]
- Heibel, S.K.; Lopez, G.Y.; Panglao, M.; Sodha, S.; Mariño-Ramírez, L.; Tuchman, M.; Caldovic, L. Transcriptional regulation of N-acetylglutamate synthase. PLoS ONE 2012, 7, e29527. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Yahagi, N.; Aita, Y.; Mehrazad-Saber, Z.; Ho, M.H.; Huyan, Y.; Murayama, Y.; Shikama, A.; Masuda, Y.; Izumida, Y.; et al. FoxO-KLF15 pathway switches the flow of macronutrients under the control of insulin. iScience 2021, 24, 103446. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, D.; Scheer, F.A.; Ripperger, J.A.; Haldar, S.M.; Lu, Y.; Prosdocimo, D.A.; Eapen, S.J.; Eapen, B.L.; Cui, Y.; Mahabeleshwar, G.H.; et al. Klf15 orchestrates circadian nitrogen homeostasis. Cell Metab. 2012, 15, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Houten, S.M. Peroxisomes and bile acid biosynthesis. Biochim. Biophys. Acta 2006, 1763, 1427–1440. [Google Scholar] [CrossRef]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef]
- Han, S.; Zhang, R.; Jain, R.; Shi, H.; Zhang, L.; Zhou, G.; Sangwung, P.; Tugal, D.; Atkins, G.B.; Prosdocimo, D.A.; et al. Circadian control of bile acid synthesis by a KLF15-Fgf15 axis. Nat. Commun. 2015, 6, 7231. [Google Scholar] [CrossRef]
- Wang, G.; Wu, B.; Cui, Y.; Zhang, B.; Jiang, C.; Wang, H. Teneligliptin Promotes Bile Acid Synthesis and Attenuates Lipid Accumulation in Obese Mice by Targeting the KLF15-Fgf15 Pathway. Chem. Res. Toxicol. 2020, 33, 2164–2171. [Google Scholar] [CrossRef]
- Sydor, S.; Manka, P.; van Buren, L.; Theurer, S.; Schwertheim, S.; Best, J.; Heegsma, J.; Saeed, A.; Vetter, D.; Schlattjan, M.; et al. Hepatocyte KLF6 expression affects FXR signalling and the clinical course of primary sclerosing cholangitis. Liver Int. 2020, 40, 2172–2181. [Google Scholar] [CrossRef]
- Chintalapati, C.; Wöhler, C.; Ehlting, C.; Bode, J.; Häussinger, D.; Keitel, V. Differential regulation of G-protein coupled bile acid receptor (Gpbar-1) by Sp1/KLF5 family transcription factors. Z Gastroenterol. 2015, 53, A2_18. [Google Scholar] [CrossRef]
- Ni, Z.; Min, Y.; Han, C.; Yuan, T.; Lu, W.; Ashktorab, H.; Smoot, D.T.; Wu, Q.; Wu, J.; Zeng, W.; et al. TGR5-HNF4α axis contributes to bile acid-induced gastric intestinal metaplasia markers expression. Cell Death Discov. 2020, 6, 56. [Google Scholar] [CrossRef]
- Kujovich, J.L. Coagulopathy in liver disease: A balancing act. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Ceelie, H.; Spaargaren-Van Riel, C.C.; De Jong, M.; Bertina, R.M.; Vos, H.L. Functional characterization of transcription factor binding sites for HNF1-alpha, HNF3-beta (FOXA2), HNF4-alpha, Sp1 and Sp3 in the human prothrombin gene enhancer. J. Thromb. Haemost. 2003, 1, 1688–1698. [Google Scholar] [CrossRef]
- Hung, H.-L.; Pollak, E.S.; Kudaravalli, R.D.; Arruda, V.; Chu, K.; High, K.A. Regulation of human coagulation factor X gene expression by GATA-4 and the Sp family of transcription factors. Blood 2001, 97, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, D.R.; Chukwumezie, B.N.; Wilberding, J.A.; Rosen, E.D.; Castellino, F.J. Characterization of Transcriptional Regulatory Elements in the Promoter Region of the Murine Blood Coagulation Factor VII Gene*. J. Biol. Chem. 1998, 273, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.A.; Pollak, E.S.; High, K.A.; Bauer, K.A. Severe factor VII deficiency due to a mutation disrupting an Sp1 binding site in the factor VII promoter. Blood 1998, 92, 1639–1645. [Google Scholar] [CrossRef]
- Oeth, P.; Parry, G.C.; Mackman, N. Regulation of the tissue factor gene in human monocytic cells. Role of AP-1, NF-kappa B/Rel, and Sp1 proteins in uninduced and lipopolysaccharide-induced expression. Arter. Thromb. Vasc. Biol. 1997, 17, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Moll, T.; Czyz, M.; Holzmüller, H.; Hofer-Warbinek, R.; Wagner, E.; Winkler, H.; Bach, F.H.; Hofer, E. Regulation of the Tissue Factor Promoter in Endothelial Cells: BINDING OF NFκB-, AP-1-, AND Sp1-LIKE TRANSCRIPTION FACTORS (∗). J. Biol. Chem. 1995, 270, 3849–3857. [Google Scholar] [CrossRef]
- de Wolf, C.J.; Cupers, R.M.; Bertina, R.M.; Vos, H.L. The constitutive expression of anticoagulant protein S is regulated through multiple binding sites for Sp1 and Sp3 transcription factors in the protein S gene promoter. J. Biol. Chem. 2006, 281, 17635–17643. [Google Scholar] [CrossRef] [PubMed]
- Tatewaki, H.; Tsuda, H.; Kanaji, T.; Yokoyama, K.; Hamasaki, N. Characterization of the human protein S gene promoter: A possible role of transcription factors Sp1 and HNF3 in liver. Thromb. Haemost. 2003, 90, 1029–1039. [Google Scholar] [CrossRef]
- Lin, Z.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A., Jr.; Balasubramanian, V.; García-Cardeña, G.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005, 96, e48–e57. [Google Scholar] [CrossRef] [PubMed]
- Novodvorsky, P.; Chico, T.J. The role of the transcription factor KLF2 in vascular development and disease. Prog. Mol. Biol. Transl. Sci. 2014, 124, 155–188. [Google Scholar] [CrossRef] [PubMed]
- Peghaire, C.; Dufton, N.P.; Lang, M.; Salles, C., II; Ahnström, J.; Kalna, V.; Raimondi, C.; Pericleous, C.; Inuabasi, L.; Kiseleva, R.; et al. The transcription factor ERG regulates a low shear stress-induced anti-thrombotic pathway in the microvasculature. Nat. Commun. 2019, 10, 5014. [Google Scholar] [CrossRef] [PubMed]
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerszten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like Factor 4 Regulates Endothelial Inflammation*. J. Biol. Chem. 2007, 282, 13769–13779. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, X.; Xie, S.; Zhou, R. Transcriptome analysis of Klf15-mediated inhibitory functions in a mouse deep venous thrombosis model. Int. J. Mol. Med. 2020, 45, 1735–1752. [Google Scholar] [CrossRef]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic metabolism, disposition, and regulation by receptors: From biochemical phenomenon to predictors of major toxicities. Toxicol Sci 2011, 120 (Suppl. 1), S49–S75. [Google Scholar] [CrossRef]
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: Implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar] [CrossRef]
- Danielson, P.B. The cytochrome P450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef]
- Jover, R.; Bort, R.; Gómez-Lechón, M.J.; Castell, J.V. Cytochrome P450 regulation by hepatocyte nuclear factor 4 in human hepatocytes: A study using adenovirus-mediated antisense targeting. Hepatology 2001, 33, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Jiang, J.; Ye, W.; Chen, R.; Deng, Y.; Wen, J. Sp1, Instead of AhR, Regulates the Basal Transcription of Porcine CYP1A1 at the Proximal Promoter. Front. Pharm. 2018, 9, 927. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Yao, P.L.; Goudarzi, M.; Murray, I.A.; Balandaram, G.; Gonzalez, F.J.; Perdew, G.H.; Fornace, A.J., Jr.; Peters, J.M. Regulation of Cytochrome P450 2B10 (CYP2B10) Expression in Liver by Peroxisome Proliferator-activated Receptor-β/δ Modulation of SP1 Promoter Occupancy. J. Biol. Chem. 2016, 291, 25255–25263. [Google Scholar] [CrossRef]
- Bombail, V.; Taylor, K.; Gibson, G.G.; Plant, N. Role of Sp1, C/EBP alpha, HNF3, and PXR in the basal- and xenobiotic-mediated regulation of the CYP3A4 gene. Drug Metab. Dispos. 2004, 32, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Chen, Q.; Liu, X.; Wen, J.; Jiang, J.; Deng, Y. Role of Specificity Protein 1, Hepatocyte Nuclear Factor 1α, and Pregnane X Receptor in the Basal and Rifampicin-Induced Transcriptional Regulation of Porcine Cytochrome P450 3A46. Drug Metab. Dispos. 2015, 43, 1458–1467. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, J.; Hu, Z.; Ye, W.; Yuan, Q.; Li, M.; Wen, J.; Deng, Y. Coordinated Transcriptional Regulation of Cytochrome P450 3As by Nuclear Transcription Factor Y and Specificity Protein 1. Mol. Pharm. 2019, 95, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.H.; Pan, X.; Zhang, W.; McLachlan, A.; Urrutia, R.; Jeong, H. Krüppel-like factor 9 promotes hepatic cytochrome P450 2D6 expression during pregnancy in CYP2D6-humanized mice. Mol. Pharm. 2014, 86, 727–735. [Google Scholar] [CrossRef]
- Herholz, M.; Cepeda, E.; Baumann, L.; Kukat, A.; Hermeling, J.; Maciej, S.; Szczepanowska, K.; Pavlenko, V.; Frommolt, P.; Trifunovic, A. KLF-1 orchestrates a xenobiotic detoxification program essential for longevity of mitochondrial mutants. Nat. Commun. 2019, 10, 3323. [Google Scholar] [CrossRef]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharm. Exp. 2013, 345, 419–429. [Google Scholar] [CrossRef]
- Gregory, P.A.; Gardner-Stephen, D.A.; Lewinsky, R.H.; Duncliffe, K.N.; Mackenzie, P.I. Cloning and Characterization of the Human UDP-glucuronosyltransferase 1A8, 1A9, and 1A10 Gene Promoters: DIFFERENTIAL REGULATION THROUGH AN INITIATOR-LIKE REGION*. J. Biol. Chem. 2003, 278, 36107–36114. [Google Scholar] [CrossRef]
- Hempel, N.; Wang, H.; LeCluyse, E.L.; McManus, M.E.; Negishi, M. The human sulfotransferase SULT1A1 gene is regulated in a synergistic manner by Sp1 and GA binding protein. Mol. Pharm. 2004, 66, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Higashi, Y.; Luu, C.; Shimizu, C.; Strott, C.A. Sp1 elements in SULT2B1b promoter and 5'-untranslated region of mRNA: Sp1/Sp2 induction and augmentation by histone deacetylase inhibition. FEBS Lett. 2005, 579, 3639–3645. [Google Scholar] [CrossRef] [PubMed]
- Moffat, G.J.; McLaren, A.W.; Wolf, C.R. Sp1-mediated transcriptional activation of the human Pi class glutathione S-transferase promoter. J. Biol. Chem. 1996, 271, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver regeneration. J. Cell Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Zellmer, S.; Schmidt-Heck, W.; Godoy, P.; Weng, H.; Meyer, C.; Lehmann, T.; Sparna, T.; Schormann, W.; Hammad, S.; Kreutz, C.; et al. Transcription factors ETF, E2F, and SP-1 are involved in cytokine-independent proliferation of murine hepatocytes. Hepatology 2010, 52, 2127–2136. [Google Scholar] [CrossRef]
- Manavski, Y.; Abel, T.; Hu, J.; Kleinlützum, D.; Buchholz, C.J.; Belz, C.; Augustin, H.G.; Boon, R.A.; Dimmeler, S. Endothelial transcription factor KLF2 negatively regulates liver regeneration via induction of activin A. Proc. Natl. Acad. Sci. USA 2017, 114, 3993–3998. [Google Scholar] [CrossRef]
- Nandan, M.O.; Yang, V.W. The role of Krüppel-like factors in the reprogramming of somatic cells to induced pluripotent stem cells. Histol. Histopathol. 2009, 24, 1343–1355. [Google Scholar] [CrossRef]
- Ji, W.; Shi, H.; Shen, H.; Kong, J.; Song, J.; Bian, H.; Lv, X. Mechanism of KLF4 Protection against Acute Liver Injury via Inhibition of Apelin Signaling. Oxid. Med. Cell Longev. 2019, 2019, 6140360. [Google Scholar] [CrossRef]
- Heo, S.H.; Jeong, E.S.; Lee, K.S.; Seo, J.H.; Lee, W.K.; Choi, Y.K. Knockout of krüppel-like factor 10 suppresses hepatic cell proliferation in a partially hepatectomized mouse model. Oncol. Lett. 2017, 13, 4843–4848. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, W.; InSug, O.; Williams, J.B.; Dong, Q.; Park, E.A.; Raghow, R.; Unterman, T.G.; Elam, M.B. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 2012, 287, 20132–20143. [Google Scholar] [CrossRef] [PubMed]
- Diels, S.; Cuypers, B.; Tvarijonaviciute, A.; Derudas, B.; Van Dijck, E.; Verrijken, A.; Van Gaal, L.F.; Laukens, K.; Lefebvre, P.; Ceron, J.J.; et al. A targeted multi-omics approach reveals paraoxonase-1 as a determinant of obesity-associated fatty liver disease. Clin. Epigenetics 2021, 13, 158. [Google Scholar] [CrossRef]
- Iloani, N.; Hafeez, A.; Bao, S.; Dulemba, V.; Lambring, C.; Sankpal, U.T.; Basha, R. Chapter 12 - Association of specificity protein 1 with hepatocellular carcinoma. In Theranostics and Precision Medicine for the Management of Hepatocellular Carcinoma; Nagaraju, G.P., Vadde, R., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 2, pp. 185–193. [Google Scholar] [CrossRef]
- Li, W.; Yan, Y.; Zheng, Z.; Zhu, Q.; Long, Q.; Sui, S.; Luo, M.; Chen, M.; Li, Y.; Hua, Y.; et al. Targeting the NCOA3-SP1-TERT axis for tumor growth in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1011. [Google Scholar] [CrossRef] [PubMed]
- Gandhy, S.U.; Imanirad, P.; Jin, U.H.; Nair, V.; Hedrick, E.; Cheng, Y.; Corton, J.C.; Kim, K.; Safe, S. Specificity protein (Sp) transcription factors and metformin regulate expression of the long non-coding RNA HULC. Oncotarget 2015, 6, 26359–26372. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, L.; Shen, S.; Li, J.; Lu, H.; Mo, W.; Dang, Y.; Luo, D.; Chen, G.; Feng, Z. Sp1 cooperates with Sp3 to upregulate MALAT1 expression in human hepatocellular carcinoma. Oncol. Rep. 2015, 34, 2403–2412. [Google Scholar] [CrossRef]
- Kong, X.; Xu, P.; Cai, W.J.; Wang, H.G.; Li, B.B.; Huang, G.L.; He, Z.W.; Chen, G.; Ye, C.G. ZBP-89 and Sp1 contribute to Bak expression in hepatocellular carcinoma cells. BMC Cancer 2018, 18, 419. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, Y.; Chen, K.Q.; An, G.; Ji, S.Y.; Chen, Q.K. Anti-fibrotic effects via regulation of transcription factor Sp1 on hepatic stellate cells. Cell Physiol. Biochem. 2012, 29, 51–60. [Google Scholar] [CrossRef]
- Xia, Y.; Guo, H. Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antivir. Res. 2020, 180, 104824. [Google Scholar] [CrossRef]
- Park, J.H.; Jo, J.H.; Kim, K.H.; Kim, S.J.; Lee, W.R.; Park, K.K.; Park, J.B. Antifibrotic effect through the regulation of transcription factor using ring type-Sp1 decoy oligodeoxynucleotide in carbon tetrachloride-induced liver fibrosis. J. Gene Med. 2009, 11, 824–833. [Google Scholar] [CrossRef]
- Steensels, S.; Qiao, J.; Ersoy, B.A. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites 2020, 10, 283. [Google Scholar] [CrossRef]
- Lin, J.; Tan, H.; Nie, Y.; Wu, D.; Zheng, W.; Lin, W.; Zhu, Z.; Yang, B.; Chen, X.; Chen, T. Krüppel-like factor 2 inhibits hepatocarcinogenesis through negative regulation of the Hedgehog pathway. Cancer Sci. 2019, 110, 1220–1231. [Google Scholar] [CrossRef]
- Zou, K.; Lu, X.; Ye, K.; Wang, C.; You, T.; Chen, J. Krüppel-like factor 2 promotes cell proliferation in hepatocellular carcinoma through up-regulation of c-myc. Cancer Biol. 2016, 17, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Sahoo, S.; McMullen, I.; Saxena, A.N.; Venugopal, S.K.; Somarelli, J.A.; Jolly, M.K. KLF4 Induces Mesenchymal-Epithelial Transition (MET) by Suppressing Multiple EMT-Inducing Transcription Factors. Cancers 2021, 13, 5135. [Google Scholar] [CrossRef]
- Li, T.; Niu, L.; Li, M.; Liu, Y.; Xu, Z.; Gao, X.; Liu, D. Effects of small interfering RNA-mediated downregulation of the Krüppel-like factor 4 gene on collagen metabolism in human hepatic stellate cells. Mol. Med. Rep. 2015, 12, 3972–3978. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Chien, Y.; Chen, Y.J.; Chen, S.F.; Chang, Y.L.; Chiang, C.H.; Jeng, S.Y.; Chang, C.M.; Wang, M.L.; Chen, L.K.; et al. Reprogramming induced pluripotent stem cells in the absence of c-Myc for differentiation into hepatocyte-like cells. Biomaterials 2011, 32, 5994–6005. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.M.; Liao, Y.W.; Chiang, C.H.; Chen, Y.J.; Lai, Y.H.; Chang, Y.L.; Chen, H.L.; Jeng, S.Y.; Hsieh, J.H.; Peng, C.H.; et al. Improvement of carbon tetrachloride-induced acute hepatic failure by transplantation of induced pluripotent stem cells without reprogramming factor c-Myc. Int. J. Mol. Sci. 2012, 13, 3598–3617. [Google Scholar] [CrossRef]
- An, T.; Dong, T.; Zhou, H.; Chen, Y.; Zhang, J.; Zhang, Y.; Li, Z.; Yang, X. The transcription factor Krüppel-like factor 5 promotes cell growth and metastasis via activating PI3K/AKT/Snail signaling in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2019, 508, 159–168. [Google Scholar] [CrossRef]
- Okada, H.; Yamada, M.; Kamimoto, K.; Kok, C.Y.; Kaneko, K.; Ema, M.; Miyajima, A.; Itoh, T. The transcription factor Klf5 is essential for intrahepatic biliary epithelial tissue remodeling after cholestatic liver injury. J. Biol. Chem. 2018, 293, 6214–6229. [Google Scholar] [CrossRef]
- Wen, P.H.; Wang, D.Y.; Zhang, J.K.; Wang, Z.H.; Pan, J.; Shi, X.Y.; Yang, H.; Zhang, S.J.; Guo, W.Z. Kruppel-like factor 6 suppresses growth and invasion of hepatocellular carcinoma cells in vitro and in vivo. Int. J. Immunopathol. Pharm. 2016, 29, 666–675. [Google Scholar] [CrossRef]
- Sirach, E.; Bureau, C.; Péron, J.M.; Pradayrol, L.; Vinel, J.P.; Buscail, L.; Cordelier, P. KLF6 transcription factor protects hepatocellular carcinoma-derived cells from apoptosis. Cell Death Differ. 2007, 14, 1202–1210. [Google Scholar] [CrossRef]
- Ghiassi-Nejad, Z.; Hernandez-Gea, V.; Woodrell, C.; Lang, U.E.; Dumic, K.; Kwong, A.; Friedman, S.L. Reduced hepatic stellate cell expression of Kruppel-like factor 6 tumor suppressor isoforms amplifies fibrosis during acute and chronic rodent liver injury. Hepatology 2013, 57, 786–796. [Google Scholar] [CrossRef]
- Selvaraj, S.; Oh, J.H.; Spanel, R.; Länger, F.; Han, H.Y.; Lee, E.H.; Yoon, S.; Borlak, J. The pathogenesis of diclofenac induced immunoallergic hepatitis in a canine model of liver injury. Oncotarget 2017, 8, 107763–107824. [Google Scholar] [CrossRef]
- Sydor, S.; Manka, P.; Best, J.; Jafoui, S.; Sowa, J.P.; Zoubek, M.E.; Hernandez-Gea, V.; Cubero, F.J.; Kälsch, J.; Vetter, D.; et al. Krüppel-like factor 6 is a transcriptional activator of autophagy in acute liver injury. Sci. Rep. 2017, 7, 8119. [Google Scholar] [CrossRef]
- Cheng, S.; Zhang, X.; Xu, Y.; Dai, X.; Li, J.; Zhang, T.; Chen, X. Krüppel-like factor 8 regulates VEGFA expression and angiogenesis in hepatocellular carcinoma. Sci. Rep. 2018, 8, 17415. [Google Scholar] [CrossRef] [PubMed]
- Leclère, P.S.; Rousseau, D.; Patouraux, S.; Guérin, S.; Bonnafous, S.; Gréchez-Cassiau, A.; Ruberto, A.A.; Luci, C.; Subramaniam, M.; Tran, A.; et al. MCD diet-induced steatohepatitis generates a diurnal rhythm of associated biomarkers and worsens liver injury in Klf10 deficient mice. Sci. Rep. 2020, 10, 12139. [Google Scholar] [CrossRef]
- Chen, C.C.; Xie, X.M.; Zhao, X.K.; Zuo, S.; Li, H.Y. Krüppel-like Factor 13 Promotes HCC Progression by Transcriptional Regulation of HMGCS1-mediated Cholesterol Synthesis. J Clin Transl Hepatol 2022, 10, 1125–1137. [Google Scholar] [CrossRef]
- Witka, B.Z.; Oktaviani, D.J.; Marcellino, M.; Barliana, M.I.; Abdulah, R. Type 2 Diabetes-Associated Genetic Polymorphisms as Potential Disease Predictors. Diabetes Metab Syndr Obes 2019, 12, 2689–2706. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Q.; Chen, A.; Liu, N.; Chen, N.; Chen, X.; Zhu, L.; Xia, B.; Gong, Y.; Chen, X. KLF15-activating Twist2 ameliorated hepatic steatosis by inhibiting inflammation and improving mitochondrial dysfunction via NF-κB-FGF21 or SREBP1c-FGF21 pathway. FASEB J. 2019, 33, 14254–14269. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, T.; Tian, Y.; Zheng, B.; Ou, J.H.; Huang, E.J.; Yen, T.S. Krüppel-like factor 15 activates hepatitis B virus gene expression and replication. Hepatology 2011, 54, 109–121. [Google Scholar] [CrossRef]
- Tian, L.L.; Zhang, J.; Wang, Z.Z.; Chen, S.C.; Zou, X.B.; Yu, Z.K.; Kang, C.C. KLF15 reduces the level of apoptosis in mouse liver induced by sepsis by inhibiting p38MAPK/ERK1/2 signaling pathway. Eur. Rev. Med. Pharm. Sci. 2020, 24, 10819–10828. [Google Scholar] [CrossRef]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef]
- Majumdar, G.; Harrington, A.; Hungerford, J.; Martinez-Hernandez, A.; Gerling, I.C.; Raghow, R.; Solomon, S. Insulin dynamically regulates calmodulin gene expression by sequential o-glycosylation and phosphorylation of sp1 and its subcellular compartmentalization in liver cells. J. Biol. Chem. 2006, 281, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Q.; Kitamoto, T.; Hou, J.; Qin, J.; Accili, D. Identification of Insulin-Responsive Transcription Factors That Regulate Glucose Production by Hepatocytes. Diabetes 2019, 68, 1156–1167. [Google Scholar] [CrossRef]
- Liang, H.; Balas, B.; Tantiwong, P.; Dube, J.; Goodpaster, B.H.; O'Doherty, R.M.; DeFronzo, R.A.; Richardson, A.; Musi, N.; Ward, W.F. Whole body overexpression of PGC-1alpha has opposite effects on hepatic and muscle insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E945–E954. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Vallejo, L.; Guatibonza-García, V.; Mantzoros, C.S. Recent guidelines for Non-Alcoholic Fatty Liver disease (NAFLD)/ Fatty Liver Disease (FLD): Are they already outdated and in need of supplementation? Metabolism 2022, 136, 155248. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Zhou, H.J.; Chen, X.P.; Ren, G.Y.; Ruan, X.X.; Zhang, Y.; Zhang, R.L.; Chen, J. Loss of expression of Kruppel-like factor 6 in primary hepatocellular carcinoma and hepatoma cell lines. J. Exp. Clin. Cancer Res. 2007, 26, 117–124. [Google Scholar]
- Pan, X.C.; Chen, Z.; Chen, F.; Chen, X.H.; Jin, H.Y.; Xu, X.Y. Inactivation of the tumor suppressor Krüppel-like factor 6 (KLF6) by mutation or decreased expression in hepatocellular carcinomas. J. Zhejiang Univ. Sci. B 2006, 7, 830–836. [Google Scholar] [CrossRef]
- Xue, M.; Zhou, C.; Zheng, Y.; Zhang, Z.; Wang, S.; Fu, Y.; Atyah, M.; Xue, X.; Zhu, L.; Dong, Q.; et al. The association between KLF4 as a tumor suppressor and the prognosis of hepatocellular carcinoma after curative resection. Aging 2020, 12, 15566–15580. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr. 2014, 3, 344–363. [Google Scholar] [CrossRef]
- Carter, J.K.; Friedman, S.L. Hepatic Stellate Cell-Immune Interactions in NASH. Front. Endocrinol. 2022, 13, 867940. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, Q.N.; Wu, J.F.; Ai, W.B.; Ma, L. Analysis of key genes and related transcription factors in liver fibrosis based on bioinformatic technology. Int. J. Clin. Exp. Pathol. 2021, 14, 444–454. [Google Scholar] [PubMed]
- Castaneda, D.; Gonzalez, A.J.; Alomari, M.; Tandon, K.; Zervos, X.B. From hepatitis A to E: A critical review of viral hepatitis. World J. Gastroenterol. 2021, 27, 1691–1715. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef]
- Li, J.; Ou, J.H. Differential regulation of hepatitis B virus gene expression by the Sp1 transcription factor. J. Virol. 2001, 75, 8400–8406. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Umeyama, H.; Iwadate, M.; Taguchi, Y.H.; Yano, Y.; Honda, T.; Itami-Matsumoto, S.; Kozuka, R.; Enomoto, M.; Tamori, A.; et al. Development of a novel anti-hepatitis B virus agent via Sp1. Sci. Rep. 2020, 10, 47. [Google Scholar] [CrossRef]
- Lin, Y.C.; Hsu, E.C.; Ting, L.P. Repression of hepatitis B viral gene expression by transcription factor nuclear factor-kappaB. Cell Microbiol. 2009, 11, 645–660. [Google Scholar] [CrossRef]
- Sarcognato, S.; Sacchi, D.; Grillo, F.; Cazzagon, N.; Fabris, L.; Cadamuro, M.; Cataldo, I.; Covelli, C.; Mangia, A.; Guido, M. Autoimmune biliary diseases: Primary biliary cholangitis and primary sclerosing cholangitis. Pathologica 2021, 113, 170–184. [Google Scholar] [CrossRef]
- Pietrangelo, A. Hemochromatosis: An endocrine liver disease. Hepatology 2007, 46, 1291–1301. [Google Scholar] [CrossRef]
- Mura, C.; Le Gac, G.; Jacolot, S.; Férec, C. Transcriptional regulation of the human HFE gene indicates high liver expression and erythropoiesis coregulation. FASEB J. 2004, 18, 1922–1924. [Google Scholar] [CrossRef] [PubMed]
- Pelham, C.; Jimenez, T.; Rodova, M.; Rudolph, A.; Chipps, E.; Islam, M.R. Regulation of HFE expression by poly(ADP-ribose) polymerase-1 (PARP1) through an inverted repeat DNA sequence in the distal promoter. Biochim. Biophys. Acta 2013, 1829, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P. Wilson's Disease. Clin. Gastroenterol. Hepatol. 2005, 3, 726–733. [Google Scholar] [CrossRef]
- Wooton-Kee, C.R.; Robertson, M.; Zhou, Y.; Dong, B.; Sun, Z.; Kim, K.H.; Liu, H.; Xu, Y.; Putluri, N.; Saha, P.; et al. Metabolic dysregulation in the Atp7b(-/-) Wilson's disease mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Chen, H.H.; Aiba, I.; Hossain, A.; Liang, Z.D.; Klomp, L.W.; Kuo, M.T. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol. Pharm. 2008, 74, 705–713. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Hamilton, J.P. Drug-induced Liver Injury. US Gastroenterol. Hepatol. Rev. 2010, 6, 73–80. [Google Scholar] [PubMed]
- Shah, F.; Medvedev, A.; Wassermann, A.M.; Brodney, M.; Zhang, L.; Makarov, S.; Stanton, R.V. The Identification of Pivotal Transcriptional Factors Mediating Cell Responses to Drugs With Drug-Induced Liver Injury Liabilities. Toxicol. Sci. 2018, 162, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Toma, D.; Lazar, O.; Bontas, E. Acute Liver Failure. Liver Dis. 2019, 10, 369–380. [Google Scholar] [CrossRef]
- De Simone, V.; Cortese, R. Transcriptional regulation of liver-specific gene expression. Curr. Opin. Cell Biol. 1991, 3, 960–965. [Google Scholar] [CrossRef]
- Costa, R.H.; Kalinichenko, V.V.; Holterman, A.X.; Wang, X. Transcription factors in liver development, differentiation, and regeneration. Hepatology 2003, 38, 1331–1347. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. Sussing out statins in cirrhosis—KLF2 is the key. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 64. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcription Factor | Insulin Resistance/NAFLD | HCC | Hepatic Fibrosis | Biliary Cholangitis | Hepatitis | Drug Induced Hepatotoxicity | Acute Liver Toxicity/Failure |
|---|---|---|---|---|---|---|---|
| SP1 | ↑ SREBP-1c → ↑ Hepatic lipogenesis [150] ↓ Methylation of PON1 promoter → ↑ NAFLD [151] | ↑ RasGRP1, RYBP, CSE, TERT → ↑ HCC [152,153] ↑ HULC and MALAT1 lncRNAs → ↑ HCC [154,155] ↑ Bak → ↓ HCC [156] | ↑ TGF-β → ↑ Fibrosis [157] | - | ↑ HBV ccc DNA replication → ↑ Hepatitis B [158] | - | ↑ TGF-β → ↑ Liver failure [159] |
| SP3 | - | ↑ HULC and MALAT1 lncRNAs and ↓ SP1-Bak induction →↑ HCC [154,155,156] | - | - | - | - | - |
| KLF2 | ↑ CD36 → ↑ steatohepatitis [72] Predicted link to fibrotic NASH (by IPA analysis of GSE130970) [160] | ↓ Hedgehog and TGF-β → ↓ HCC [161] ↑ c-Myc → ↑ HCC [162] | - | - | - | - | ↑ Activin A secretion → ↑ Hepatic damage [146] |
| KLF3 | ↓ Fam132a → ↑ Insulin resistance [53] | - | - | - | - | - | - |
| KLF4 | ↓ Slug → ↓ EMT → ↓ HCC [163] | ↑ TGF-β and ↑MMP/TIMP → ↑ Fibrosis [164] | - | - | - | ↑ Liver regeneration → ↓ Liver damage [165,166] | |
| KLF5 | - | ↑ PI3K/Akt/Snail → ↑ EMT → ↑ HCC [167] | - | ↑ Ductal regeneration → ↓ Cholangitis [168]. | - | - | - |
| KLF6 | ↓miR10b → ↑ PPARα → ↑ Trb3→ ↑ Insulin resistance [76] KLF6-IVS1-27Gwt (no splicing) → ↑ KLF6-IVS1-27G>A (splice variant) →↓ NAFLD [54] | ↓ PCNA, MMP-9 → ↓ HCC [169] ↓ p53 and↑ BCL-XL → ↑HCC [170] | ↑ α1 collagen → ↑HSC activation → ↑ Fibrosis [171] | ↓ NR0B2 → ↑ Bile duct ligation induced hepatic injury [109] | - | ↑ Diclofenac induced hepatotoxicity [172] | ↑ Autophagy → ↑ Hepatic damage [173] |
| KLF8 | - | ↑ Wnt/β-catenin → ↑ HCC [174] | - | - | - | - | - |
| KLF9 | ↑ CD36 → ↑ Steatohepatitis [77] ↑ PGC-1α → ↑ Hepatic gluconeogenesis → ↑ Insulin resistance [55] ↑ PGC-1α → ↑ Glucocorticoid induced diabetes [55] | - | - | - | - | - | - |
| KLF10 | ↑ Pepck → ↑ Hepatic gluconeogenesis → ↑ Insulin resistance [57] ↑ zDHHC7 → ↑ CD36 mediated FA uptake → ↑ Steatohepatitis [80] ↓ Hepatocyte apoptosis → ↓ Steatohepatitis by methionine and choline deficient diet [175] | - | ↓ TGF-β → ↓Fibrosis [79] | - | - | - | - |
| KLF11 | ↑ FA oxidation → ↓ Steatohepatitis [81] ↓ PEPCK, PGC1A → ↓ Hepatic gluconeogenesis [61] | - | - | - | - | - | - |
| KLF13 | ↑ Expression → ↑ NAFLD in humans [23] | ↑ Cholesterol synthesis → ↑ HCC [176] | - | - | - | - | - |
| KLF14 | ↑ PGC-1α → ↑ Hepatic gluconeogenesis → ↑ Insulin resistance [59] ↑ PI3K/Akt → ↑ Hepatic insulin sensitivity [82] Gene polymorphism in KLF14 → ↑ type II diabetes [177] | - | - | - | - | - | - |
| KLF15 | ↑Twist2 → ↓ Steatohepatitis [178] ↑ Recruitment of RIP40 corepressor to the SREBF-1 promoter → ↓ Hepatic lipogenesis [86] ↑ ER stress → ↑ Insulin resistance [85] | - | - | - | ↑ Core and surface genes expression → ↑Hepatitis B [179] | - | ↓ p38 MAPK/ERK1/2 → ↓ Hepatic injury [180] |
| KLF16 | ↑ PPARα → ↑ FA oxidation → ↓ Steatohepatitis and NAFLD [84] | - | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yerra, V.G.; Drosatos, K. Specificity Proteins (SP) and Krüppel-like Factors (KLF) in Liver Physiology and Pathology. Int. J. Mol. Sci. 2023, 24, 4682. https://doi.org/10.3390/ijms24054682
Yerra VG, Drosatos K. Specificity Proteins (SP) and Krüppel-like Factors (KLF) in Liver Physiology and Pathology. International Journal of Molecular Sciences. 2023; 24(5):4682. https://doi.org/10.3390/ijms24054682
Chicago/Turabian StyleYerra, Veera Ganesh, and Konstantinos Drosatos. 2023. "Specificity Proteins (SP) and Krüppel-like Factors (KLF) in Liver Physiology and Pathology" International Journal of Molecular Sciences 24, no. 5: 4682. https://doi.org/10.3390/ijms24054682
APA StyleYerra, V. G., & Drosatos, K. (2023). Specificity Proteins (SP) and Krüppel-like Factors (KLF) in Liver Physiology and Pathology. International Journal of Molecular Sciences, 24(5), 4682. https://doi.org/10.3390/ijms24054682