
The PPAR-γ Agonist Pioglitazone Modulates Proliferation and Migration in HUVEC, HAOSMC and Human Arteriovenous Fistula-Derived Cells
 ,
,  , ,
, ,  ,
,  , ,
, ,  ,
, 
Abstract
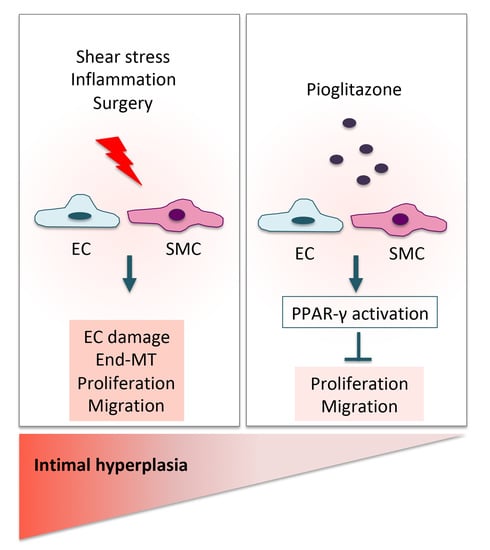
1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Altered Distribution and Expression of PPAR-γ in Failed AVF Veins and Cells
2.3. The PPAR-γ Agonist Pioglitazone Affects Proliferation and Migration Property in Endothelial and Smooth Muscle Cells
2.4. Characterization of a Primary Cell Model Isolated from Failed AVFs
2.5. Pioglitazone Mitigated IH Pathogenic Mechanisms in AVF T0 and T1 Cell Model
2.6. PPAR-γ Inhibition Reversed the Regulatory Effects of Pioglitazone in AVFCs T1 Proliferation and Migration
3. Discussion
4. Materials and Methods
4.1. Study Design and Sample Collection
4.2. Immunohistochemistry
4.3. Cell Cultures and Treatments
4.4. Immunofluorescence
4.5. Cell Growth and Proliferation Assay
4.6. In Vitro Migration Assay
4.7. Gene Expression Analysis
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, H.; Patel, S.; Hanisch, J.J.; Santana, J.M.; Hashimoto, T.; Bai, H.; Kudze, T.; Foster, T.R.; Guo, J.; Yatsula, B.; et al. Future Research Directions to Improve Fistula Maturation and Reduce Access Failure. Semin. Vasc. Surg. 2016, 29, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Roy-Chaudhury, P.; Spergel, L.M.; Besarab, A.; Asif, A.; Ravani, P. Biology of Arteriovenous Fistula Failure. J. Nephrol. 2007, 20, 150–163. [Google Scholar] [PubMed]
- Roy-Chaudhury, P.; Arend, L.; Zhang, J.; Krishnamoorthy, M.; Wang, Y.; Banerjee, R.; Samaha, A.; Munda, R. Neointimal Hyperplasia in Early Arteriovenous Fistula Failure. Am. J. Kidney Dis. 2007, 50, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.-S.; Chi, W.-C.; Lin, C.-H.; Liu, Y.-C.; Wu, Y.-W. Association of Early Failure of Arteriovenous Fistula with Mortality in Hemodialysis Patients. Sci. Rep. 2021, 11, 5699. [Google Scholar] [CrossRef]
- Nantakool, S.; Srisuwan, T.; Reanpang, T.; Rerkasem, K.; Prasannarong, M. A Randomized Controlled Trial of the Effect of Postoperative Hand Exercise Training on Arteriovenous Fistula Maturation in Patients with Chronic Kidney Disease. J. Vasc. Surg. 2022, 75, 230–237. [Google Scholar] [CrossRef]
- Ma, S.; Duan, S.; Liu, Y.; Wang, H. Intimal Hyperplasia of Arteriovenous Fistula. Ann. Vasc. Surg. 2022, 85, 444–453. [Google Scholar] [CrossRef]
- Ando, J.; Yamamoto, K. Effects of Shear Stress and Stretch on Endothelial Function. Antioxid. Redox Signal. 2011, 15, 1389–1403. [Google Scholar] [CrossRef]
- Carracedo, M.; Artiach, G.; Arnardottir, H.; Bäck, M. The Resolution of Inflammation through Omega-3 Fatty Acids in Atherosclerosis, Intimal Hyperplasia, and Vascular Calcification. Semin. Immunopathol. 2019, 41, 757–766. [Google Scholar] [CrossRef]
- Tian, D.; Zeng, X.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, Y. Protective Effect of Rapamycin on Endothelial-to-Mesenchymal Transition in HUVECs through the Notch Signaling Pathway. Vasc. Pharmacol. 2019, 113, 20–26. [Google Scholar] [CrossRef]
- Liang, A.; Wang, Y.; Han, G.; Truong, L.; Cheng, J. Chronic Kidney Disease Accelerates Endothelial Barrier Dysfunction in a Mouse Model of an Arteriovenous Fistula. Am. J. Physiol. Renal. Physiol. 2013, 304, F1413–F1420. [Google Scholar] [CrossRef]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.-R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to Mesenchymal Transition Is Common in Atherosclerotic Lesions and Is Associated with Plaque Instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Ciavarella, C.; Motta, I.; Vasuri, F.; Fittipaldi, S.; Valente, S.; Pollutri, D.; Ricci, F.; Gargiulo, M.; Pasquinelli, G. Involvement of MiR-30a-5p and MiR-30d in Endothelial to Mesenchymal Transition and Early Osteogenic Commitment under Inflammatory Stress in HUVEC. Biomolecules 2021, 11, 226. [Google Scholar] [CrossRef]
- Hao, Y.-M.; Yuan, H.-Q.; Ren, Z.; Qu, S.-L.; Liu, L.-S.; Wei, D.-H.; Yin, K.; Fu, M.; Jiang, Z.-S. Endothelial to Mesenchymal Transition in Atherosclerotic Vascular Remodeling. Clin. Chim. Acta 2019, 490, 34–38. [Google Scholar] [CrossRef]
- Moonen, J.-R.A.J.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.A.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-Mesenchymal Transition Contributes to Fibro-Proliferative Vascular Disease and Is Modulated by Fluid Shear Stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. PPARgamma: A Nuclear Regulator of Metabolism, Differentiation, and Cell Growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef]
- Rosen, E.D.; Hsu, C.-H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPα Induces Adipogenesis through PPARγ: A Unified Pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef]
- Wu, Z.; Xie, Y.; Morrison, R.F.; Bucher, N.L.; Farmer, S.R. PPARgamma Induces the Insulin-Dependent Glucose Transporter GLUT4 in the Absence of C/EBPalpha during the Conversion of 3T3 Fibroblasts into Adipocytes. J. Clin. Investig. 1998, 101, 22–32. [Google Scholar] [CrossRef]
- Tan, M.H. Current Treatment of Insulin Resistance in Type 2 Diabetes Mellitus. Int. J. Clin. Pract. Suppl. 2000, 113, 54–62. [Google Scholar]
- Ciavarella, C.; Motta, I.; Valente, S.; Pasquinelli, G. Pharmacological (or Synthetic) and Nutritional Agonists of PPAR-γ as Candidates for Cytokine Storm Modulation in COVID-19 Disease. Molecules 2020, 25, 2076. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Gao, C.-Y.; Fang, C.-Q.; Wang, Y.-J.; Gao, D.; Yao, G.-E.; Xiang, J.; Wang, J.-Z.; Li, J.-C. PPARγ Attenuates Intimal Hyperplasia by Inhibiting TLR4-Mediated Inflammation in Vascular Smooth Muscle Cells. Cardiovasc. Res. 2011, 92, 484–493. [Google Scholar] [CrossRef]
- Yang, H.-M.; Kim, B.-K.; Kim, J.-Y.; Kwon, Y.-W.; Jin, S.; Lee, J.-E.; Cho, H.-J.; Lee, H.-Y.; Kang, H.-J.; Oh, B.-H.; et al. PPARγ Modulates Vascular Smooth Muscle Cell Phenotype via a Protein Kinase G-Dependent Pathway and Reduces Neointimal Hyperplasia after Vascular Injury. Exp. Mol. Med. 2013, 45, e65. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.; Tricò, D.; Mengozzi, A.; Natali, A. Rethinking Pioglitazone as a Cardioprotective Agent: A New Perspective on an Overlooked Drug. Cardiovasc. Diabetol. 2021, 20, 109. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Inzucchi, S.; Abdul-Ghani, M.; Nissen, S.E. Pioglitazone: The Forgotten, Cost-Effective Cardioprotective Drug for Type 2 Diabetes. Diabetes Vasc. Dis. Res. 2019, 16, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Pioglitazone Enhances Cytokine-Induced Apoptosis in Vascular Smooth Muscle Cells and Reduces Intimal Hyperplasia. Available online: https://www.ahajournals.org/doi/epub/10.1161/hc3001.092040 (accessed on 6 February 2023).
- Osman, I.; Fairaq, A.; Segar, L. Pioglitazone Attenuates Injury-Induced Neointima Formation in Mouse Femoral Artery Partially through the Activation of AMP-Activated Protein Kinase. Pharmacology 2017, 100, 64–73. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, M.-J.; Li, B.-H.; Chen, L.; Pi, Y.; Yin, Y.-W.; Long, C.-Y.; Wang, X.; Sun, M.-J.; Chen, X.; et al. PPARγ Inhibits VSMC Proliferation and Migration via Attenuating Oxidative Stress through Upregulating UCP2. PLoS ONE 2016, 11, e0154720. [Google Scholar] [CrossRef]
- Spigoni, V.; Picconi, A.; Cito, M.; Ridolfi, V.; Bonomini, S.; Casali, C.; Zavaroni, I.; Gnudi, L.; Metra, M.; Cas, A.D. Pioglitazone Improves In Vitro Viability and Function of Endothelial Progenitor Cells from Individuals with Impaired Glucose Tolerance. PLoS ONE 2012, 7, e48283. [Google Scholar] [CrossRef]
- Mauro, R.; Rocchi, C.; Vasuri, F.; Pini, A.; Croci Chiocchini, A.L.; Ciavarella, C.; La Manna, G.; Pasquinelli, G.; Faggioli, G.; Gargiulo, M. Tissue Ki67 Proliferative Index Expression and Pathological Changes in Hemodialysis Arteriovenous Fistulae: Preliminary Single-Center Results. J. Vasc. Access 2021, 24, 11297298211015496. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, A.; Luo, J.; Liang, M.; Han, G.; Mitch, W.E.; Cheng, J. Blocking Notch in Endothelial Cells Prevents Arteriovenous Fistula Failure Despite CKD. J. Am. Soc. Nephrol. 2014, 25, 773–783. [Google Scholar] [CrossRef]
- Shih, Y.-C.; Chen, P.-Y.; Ko, T.-M.; Huang, P.-H.; Ma, H.; Tarng, D.-C. MMP-9 Deletion Attenuates Arteriovenous Fistula Neointima through Reduced Perioperative Vascular Inflammation. Int. J. Mol. Sci. 2021, 22, 5448. [Google Scholar] [CrossRef]
- Ji, Z.; Li, J.; Wang, J. Jujuboside B Inhibits Neointimal Hyperplasia and Prevents Vascular Smooth Muscle Cell Dedifferentiation, Proliferation, and Migration via Activation of AMPK/PPAR-γ Signaling. Front. Pharmacol. 2021, 12, 1360. [Google Scholar] [CrossRef]
- Pei, F.; Pei, H.; Su, C.; Du, L.; Wang, J.; Xie, F.; Yin, Q.; Gao, Z. Fisetin Alleviates Neointimal Hyperplasia via PPARγ/PON2 Antioxidative Pathway in SHR Rat Artery Injury Model. Oxidative Med. Cell. Longev. 2021, 2021, 6625517. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | |
|---|---|---|
| GAPDH | FWD 5′AATGGGCAGCCGTTAGGAAA 3′ REV 5′ AGGAGAAATCGGGCCAGCTA 3′ | |
| MMP-9 | FWD 5′ GAACCAATCTCACCGACAG 3′ REV 5′ GCCACCCGAGTGTAACCAT 3′ | |
| NF-kB | FWD 5′ AGGCTATCAGTCAGCGCATC 3′ REV 5′ TCCCCACGCTGCTCTTCTAT 3′ | |
| PPAR-γ | FWD 5′ GTGGTAGGTAAGGAAGGGGC 3′ REV 5′ GGCTGACTCTCGTTTGAGAA 3′ | |
| SLUG | FWD 5′ TTCAACGCCTCCAAAAAGCC 3′ REV 5′ GATGGGGCTGTATGCTCCTG 3′ | |
| TNF-α | FWD 5′ CAGGGACCTCTCTCTAATCA 3′ REV 5′ TTGAGGGTTTGCTACAACAT 3′ | |
| VIMENTIN | FWD 5′ATCGATGTGGATGTTTCCAA 3′ REV 5′ TTGTACCATTCTTCTGCCTC 3′ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciavarella, C.; Motta, I.; Vasuri, F.; Palumbo, T.; Lisi, A.P.; Costa, A.; Astolfi, A.; Valente, S.; Versura, P.; Fornasiero, E.F.; et al. The PPAR-γ Agonist Pioglitazone Modulates Proliferation and Migration in HUVEC, HAOSMC and Human Arteriovenous Fistula-Derived Cells. Int. J. Mol. Sci. 2023, 24, 4424. https://doi.org/10.3390/ijms24054424
Ciavarella C, Motta I, Vasuri F, Palumbo T, Lisi AP, Costa A, Astolfi A, Valente S, Versura P, Fornasiero EF, et al. The PPAR-γ Agonist Pioglitazone Modulates Proliferation and Migration in HUVEC, HAOSMC and Human Arteriovenous Fistula-Derived Cells. International Journal of Molecular Sciences. 2023; 24(5):4424. https://doi.org/10.3390/ijms24054424
Chicago/Turabian StyleCiavarella, Carmen, Ilenia Motta, Francesco Vasuri, Teresa Palumbo, Anthony Paul Lisi, Alice Costa, Annalisa Astolfi, Sabrina Valente, Piera Versura, Eugenio F. Fornasiero, and et al. 2023. "The PPAR-γ Agonist Pioglitazone Modulates Proliferation and Migration in HUVEC, HAOSMC and Human Arteriovenous Fistula-Derived Cells" International Journal of Molecular Sciences 24, no. 5: 4424. https://doi.org/10.3390/ijms24054424
APA StyleCiavarella, C., Motta, I., Vasuri, F., Palumbo, T., Lisi, A. P., Costa, A., Astolfi, A., Valente, S., Versura, P., Fornasiero, E. F., Mauro, R., Gargiulo, M., & Pasquinelli, G. (2023). The PPAR-γ Agonist Pioglitazone Modulates Proliferation and Migration in HUVEC, HAOSMC and Human Arteriovenous Fistula-Derived Cells. International Journal of Molecular Sciences, 24(5), 4424. https://doi.org/10.3390/ijms24054424