

Neuroinflammation and Oxidative Stress in Individuals Affected by DiGeorge Syndrome

 and
and 
Abstract
1. Introduction
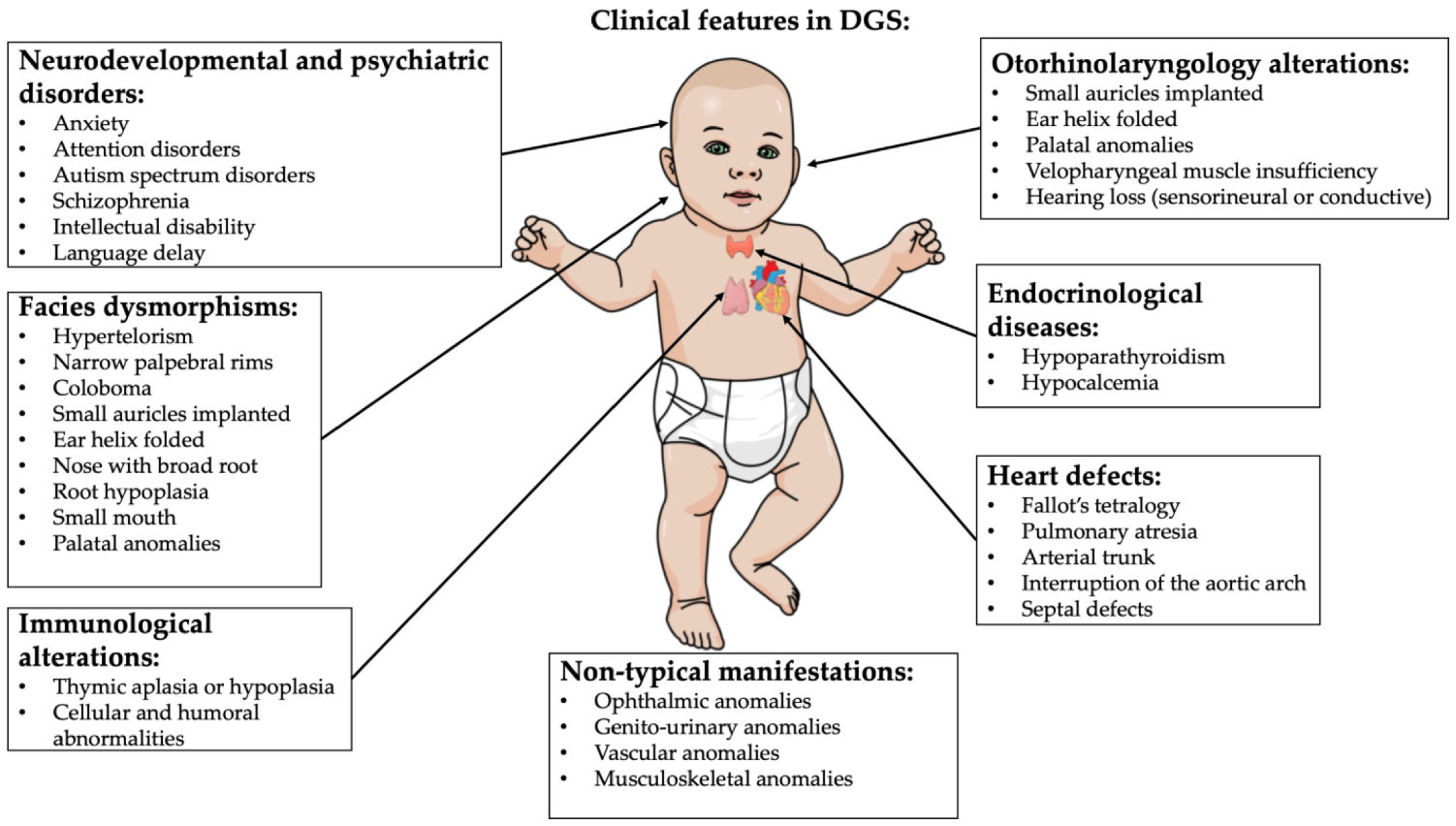
2. DiGeorge Syndrome
- Cardiac abnormality (commonly interrupted aortic arch, truncus arteriosus and tetralogy of Fallot)
- Abnormal facies
- Thymic aplasia or hypoplasia
- Cleft palate
- Hypocalcemia/hypoparathyroidism early in life
- -
- Palatal abnormalities (50%), particularly velopharyngeal incompetence, submucosal cleft palate and cleft palate; characteristic facial features (present in the majority of Caucasian individuals) including hypertelorism;
- -
- Cyanosis (bluish skin due to poor circulation of oxygen-rich blood);
- -
- Congenital heart disease (40% of individuals), particularly conotruncal malformations (interrupted aortic arch (50%), pulmonary atresia, persistent truncus arteriosus (34%), tetralogy of Fallot and ventricular septal defect);
- -
- Hearing loss (both conductive and sensorineural) (hearing loss with craniofacial syndromes);
- -
- Hypocalcemia (50%) (due to hypoparathyroidism);
- -
- Significant feeding problems (30%);
- -
- Learning difficulties (90%), including cognitive deficits and attention deficit disorders;
- -
- Laryngo-tracheo-esophageal anomalies;
- -
- Growth hormone deficiency;
- -
- Renal anomalies (37%);
- -
- Psychiatric disorders;
- -
- Autoimmune disorders;
- -
- Immunodeficiency present in about 75% of patients, related to thymic aplasia or hypoplasia determining alterations in both humoral and cell-mediated immune responses;
- -
- Immune disorders due to reduced T cell numbers;
- -
- Schizophrenia develops in 25–30% by adulthood;
- -
- Seizures (with or without hypocalcemia);
- -
- Skeletal abnormalities.
3. Oxidative Stress in DiGeorge Syndrome
4. Neuroinflammation

{kind=link}
{kind=link}
{kind=link}
| Neuroinflammation in DiGeorge Syndrome | |
|---|---|
| Clinical Manifestations: | Markers of Neuroinflammation: |
| Anxiety disorders | ↑ in CD3 and CD4 [52] |
| Autism-related disorders | ↑ in IL-12, IL- 6, IL-1β, and IFNγ [57] ↓ in IL-10 [57] |
| Cognitive problems (data also from animal models) | ↑ sarcoendoplasmic reticulum calcium-ATPase type 2 (SERCA2) [30,58] Alterations of synaptic plasticity [30,58] |
| Psychotic disorders | ↑ in IL-6 and IL1β [59] ↑ in neutrophil to leukocyte ratio (NLR) [60] ↓ in T-reg cells [59] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 22q11.2DS | 22q11.2 deletion syndrome |
| ASD | Autistic spectrum disorder |
| BDNF | Brain derived neurotropic factor |
| BBB | Blood–brain barrier |
| CCL2 | Chemokine ligand 2 |
| CCL5 | Chemokine ligand 5 |
| CXCL1 | Chemokine ligand 1 |
| CD3 | Cluster of differentiation 3 |
| CD4 | Cluster of differentiation 4 |
| COMT | catechol-O-methyltransferase |
| DGRC8 | DiGeorge syndrome critical region 8 |
| DGS | DiGeorge syndrome |
| GABA | Gamma-aminobutyric acid |
| IFNγ | Interferon gamma |
| IL-1 | Interleuchin-1 |
| IL-6 | Interleuchin-6 |
| IL-10 | Interleuchin-10 |
| IL-12 | Interleuchin-12 |
| miRNAs | microRNAs |
| NLR | Neutrophiles to leukocytes ratio |
| PGC-1α | PPARγ co-activator 1α |
| PRODH | Proline dehydrogenase |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| SERCA2 | Sarcoendoplasmic reticulum calcium-ATPase type 2 |
| SNPs | Single nucleotide polymorphisms |
| SOD | Superoxide dismutase |
| SSD | Schizophrenia spectrum disorder |
| TBX1 | T-box transcription factor 1 |
| TCA | tricarboxylic acid |
| TH 1 | T-helper 1 cells |
| TH 2 | T-helper 2 cells |
| TH 17 | T-helper 17 cells |
| TNF-α | Tumor necrosis factor alpha |
| TXNRD2 | Thioredoxin reductase 2 |
References
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed]
- Daw, S.C.M.; Taylor, C.; Kraman, M.; Call, K.; Mao, J.-I.; Schuffenhauer, S.; Meitinger, T.; Lipson, T.; Goodship, J.; Scambler, P. A common region of 10p deleted in DiGeorge and velocardiofacial syndromes. Nat. Genet. 1996, 13, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Schuffenhauer, S.; Lichtner, P.; Peykar-Derakhshandeh, P.; Murken, J.; Haas, O.A.; Back, E.; Wolff, G.; Zabel, B.; Barisic, I.; Rauch, A.; et al. Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus (DGS2). Eur. J. Hum. Genet. 1998, 6, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Meechan, D.W.; Karpinski, B.A.; Paronett, E.M.; Bryan, C.A.; Rutz, H.L.; Radin, E.A.; Lubin, N.; Bonner, E.R.; Popratiloff, A.; et al. Mitochondrial Dysfunction Leads to Cortical Under-Connectivity and Cognitive Impairment. Neuron 2019, 102, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.E. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol. Rev. 2019, 287, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Meechan, D.W.; Tucker, E.S.; Maynard, T.M.; LaMantia, A.S. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 16434–16445. [Google Scholar] [CrossRef]
- Conti, E.; Grifone, N.; Sarkozy, A.; Tandoi, C.; Marino, B.; Digilio, M.C.; Mingarelli, R.; Pizzuti, A.; Dallapiccola, B. DiGeorge subtypes of nonsyndromic conotruncal defects: Evidence against a major role of TBX1 gene. Eur. J. Hum. Genet. 2003, 11, 349–351. [Google Scholar] [CrossRef]
- Napoli, E.; Tassone, F.; Wong, S.; Angkustsiri, K.; Simon, T.J.; Song, G.; Giulivi, C. Mitochondrial citrate transporter-dependent metabolic signature in the 22q11.2 deletion syndrome. J. Biol. Chem. 2015, 290, 23240–23253. [Google Scholar] [CrossRef]
- Jonas, R.K.; Montojo, C.A.; Bearden, C.E. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol. Psychiatry 2014, 75, 351–360. [Google Scholar] [CrossRef]
- Toritsuka, M.; Kimoto, S.; Muraki, K.; Landek-Salgado, M.A.; Yoshida, A.; Yamamoto, N.; Horiuchi, Y.; Hiyama, H.; Tajinda, K.; Keni, N.; et al. Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc. Natl. Acad. Sci. USA 2013, 110, 17552–17557. [Google Scholar] [CrossRef]
- Sergi, C.; Serpi, M.; Müller-Navia, J.; Schnabel, P.A.; Hagl, S.; Otto, H.F.; Ulmer, H.E. CATCH 22 syndrome: Report of 7 infants with follow-up data and review of the recent advancements in the genetic knowledge of the locus 22q11. Pathologica 1999, 91, 166–172. [Google Scholar] [PubMed]
- Demczuk, S.; Aurias, A. DiGeorge syndrome and related syndromes associated with 22q11.2 deletions. A review. Ann. Genet. 1995, 38, 59–76. [Google Scholar] [PubMed]
- Motahari, Z.; Moody, S.A.; Maynard, T.M.; LaMantia, A.-S. In the line-up: Deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects? J. Neurodev. Disord. 2019, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Fiksinski, A.M.; Hoftman, G.D.; Vorstman, J.A.S.; Bearden, C.E. A genetics-first approach to understanding autism and schizophrenia spectrum disorders: The 22q11.2 deletion syndrome. Mol. Psychiatry 2023, 28, 341–353. [Google Scholar] [CrossRef]
- Sullivan, K.E. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial Syndrome. Immunol. Allergy Clin. N. Am. 2008, 28, 353–366. [Google Scholar] [CrossRef]
- McLean-Tooke, A.; Spickett, G.P.; Gennery, A.R. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand. J. Immunol. 2007, 66, 1–7. [Google Scholar] [CrossRef]
- Cirillo, A.; Lioncino, M.; Maratea, A.; Passariello, A.; Fusco, A.; Fratta, F.; Monda, E.; Caiazza, M.; Signore, G.; Esposito, A.; et al. Clinical Manifestations of 22q11.2 Deletion Syndrome. Heart Fail Clin. 2022, 18, 155–164. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Signer, R.; Saitta, S.C. Immune and Genetic Features of the Chromosome 22q11.2 Deletion (DiGeorge Syndrome). Curr. Allergy Asthma Rep. 2018, 18, 75. [Google Scholar] [CrossRef]
- Bayat, M.; Bayat, A. Neurological manifestation of 22q11.2 deletion syndrome. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2022, 43, 1695–1700. [Google Scholar] [CrossRef]
- Goldmuntz, E. 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 64–72. [Google Scholar] [CrossRef]
- Burn, J. Closing time for CATCH22. J. Med. Genet. 1999, 36, 737–738. [Google Scholar] [CrossRef] [PubMed]
- McDonald-Mcginn, D.M.; Sullivan, K.E. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore) 2011, 90, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Moon, E.; Park, J.M.; Lee, B.D.; Lee, Y.M.; Jeong, H.J.; Kim, S.Y.; Lee, K.; Suh, H. Various psychiatric manifestation in digeorge syndrome (22q11.2 deletion syndrome): A case report. Clin Psychopharmacol. Neurosci. 2020, 18, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Fomin, A.B.F.; Pastorino, A.C.; Kim, C.A.; Pereira, A.C.; Carneiro-Sampaio, M.; Abe Jacob, C.M. DiGeorge Syndrome: A not so rare disease. Clinics 2010, 65, 865–869. [Google Scholar] [CrossRef]
- Muchova, J.; Zitnanova, I.; Durackova, Z. Oxidative stress and Down syndrome. Do antioxidants play a role in therapy? Physiol. Res. 2014, 63, 535–542. [Google Scholar] [CrossRef]
- Jung, Y.-D.; Park, S.-K.; Kang, D.; Hwang, S.; Kang, M.-H.; Hong, S.-W.; Moon, J.-H.; Shin, J.-S.; Jin, D.-H.; You, D.; et al. Epigenetic regulation of miR-29a/miR-30c/DNMT3A axis controls SOD2 and mitochondrial oxidative stress in human mesenchymal stem cells. Redox Biol. 2020, 37, 101716. [Google Scholar] [CrossRef]
- Crowe, D.A.; Goodwin, S.J.; Blackman, R.K.; Sakellaridi, S.; Sponheim, S.R.; MacDonald, A.W.; Chafee, M.V. Prefrontal neurons transmit signals to parietal neurons that reflect executive control of cognition. Nat. Neurosci. 2013, 16, 1484–1491. [Google Scholar] [CrossRef]
- Just, M.A.; Cherkassky, V.L.; Keller, T.A.; Minshew, N.J. Cortical activation and synchronization during sentence comprehension in high-functioning autism: Evidence of underconnectivity. Brain 2004, 127, 1811–1821. [Google Scholar] [CrossRef]
- Cheng, T.-L.; Wang, Z.; Liao, Q.; Zhu, Y.; Zhou, W.-H.; Xu, W.; Qiu, Z. MeCP2 Suppresses Nuclear MicroRNA Processing and Dendritic Growth by Regulating the DGCR8/Drosha Complex. Dev. Cell. 2014, 28, 547–560. [Google Scholar] [CrossRef]
- Earls, L.R.; Gaines Fricke, R.; Yu, J.; Berry, R.B.; Baldwin, L.T.; Zakharenko, S.S. Age-dependent microRNA control of synaptic plasticity in 22q11 deletion syndrome and schizophrenia. J. Neurosc. 2012, 32, 14132–14144. [Google Scholar] [CrossRef]
- Maynard, T.; Meechan, D.; Dudevoir, M.; Gopalakrishna, D.; Peters, A.; Heindel, C.; Sugimoto, T.; Wu, Y.; Lieberman, J.; LaMantia, A.-S. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol. Cell. Neurosci. 2008, 39, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Padula, M.C.; Schaer, M.; Scariati, E.; Schneider, M.; Van De Ville, D.; Debbané, M.; Eliez, S. Structural and functional connectivity in the default mode network in 22q11.2 deletion syndrome. J. Neurodev. Disord. 2015, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Mukai, J.; Liu, H.; Burt, R.; Swor, D.E.; Lai, W.-S.; Karayiorgou, M.; Gogos, J.A. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat. Genet. 2004, 36, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.-J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Lönn, M.E.; Schütte, L.D.; Funke, M.; Godoy, J.R.; Eitner, S.; Hudemann, C.; Lillig, C.H. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J. Biol. Chem. 2010, 285, 40699–40705. [Google Scholar] [CrossRef]
- Liu, H.; Abecasis, G.R.; Heath, S.C.; Knowles, A.; Demars, S.; Chen, Y.-J.; Roos, J.L.; Rapoport, J.L.; Gogos, J.A.; Karayiorgou, M. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc. Natl. Acad. Sci. USA 2002, 99, 16859–16864. [Google Scholar] [CrossRef]
- Liu, H.; Heath, S.C.; Sobin, C.; Roos, J.L.; Galke, B.L.; Blundell, M.L.; Lenane, M.; Robertson, B.; Wijsman, E.M.; Rapoport, J.L.; et al. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc. Natl. Acad. Sci. USA 2002, 99, 3717–3722. [Google Scholar] [CrossRef]
- Micangeli, G.; Menghi, M.; Profeta, G.; Tarani, F.; Mariani, A.; Petrella, C.; Barbato, C.; Ferraguti, G.; Ceccanti, M.; Tarani, L.; et al. The Impact of Oxidative Stress on Pediatrics Syndromes. Antioxidants 2022, 11, 1983. [Google Scholar] [CrossRef]
- Profeta, G.; Micangeli, G.; Tarani, F.; Paparella, R.; Ferraguti, G.; Spaziani, M.; Isidori, A.M.; Menghi, M.; Ceccanti, M.; Fiore, M.; et al. Sexual Developmental Disorders in Pediatrics. Clin. Ter. 2022, 173, 475–488. [Google Scholar] [CrossRef]
- Fiore, M.; Arani, L.T.; Ceci, F.M.; Carito, V.; Ferraguti, G.; Petrella, C.; Greco, A.; Ralli, M.; Minni, A.; Spaziani, M.; et al. Neuroimmune Dysregulation in Prepubertal and Adolescent Individuals Affected by Klinefelter Syndrome. Endocr. Metab. Immune Disord. Drug Targets 2022, 23, 105–114. [Google Scholar] [CrossRef]
- Fiore, M.; Petrella, C.; Coriale, G.; Rosso, P.; Fico, E.; Ralli, M.; Greco, A.; De Vincentiis, M.; Minni, A.; Polimeni, A.; et al. Markers of Neuroinflammation in the Serum of Prepubertal Children with Fetal Alcohol Spectrum Disorders. CNS Neurol. Disord. Drug Targets 2022, 21, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Tarani, L.; Carito, V.; Ferraguti, G.; Petrella, C.; Greco, A.; Ralli, M.; Messina, M.P.; Rasio, D.; De Luca, E.; Putotto, C.; et al. Neuroinflammatory Markers in the Serum of Prepubertal Children with down Syndrome. J. Immunol. Res. 2020, 2020, 6937154. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef]
- Dingezweni, S. The blood–brain barrier. South African J Anaesth Analg 2020, 26, S32–S34. [Google Scholar] [CrossRef]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef]
- Bors, L.A.; Erdö, F. Overcoming the blood-brain barrier. Challenges and tricks for CNS drug delivery. Sci. Pharm. 2019, 87, 6. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and psychiatric illness. J. Neuroinflammation 2013, 10, 43. [Google Scholar] [CrossRef]
- Ousley, O.; Evans, A.N.; Fernandez-Carriba, S.; Smearman, E.L.; Rockers, K.; Morrier, M.J.; Evans, D.W.; Coleman, K.; Cubells, J. Examining the overlap between autism spectrum disorder and 22q11.2 deletion syndrome. Int. J. Mol. Sci. 2017, 18, 1071. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Crowley, T.B.; Gallagher, S.; Bailey, A.; McGinn, D.; Zackai, E.; Gur, R.E.; McGinn, D.M.; Sullivan, K.E. Increased T-cell counts in patients with 22q11.2 deletion syndrome who have anxiety. Am. J. Med. Genet. Part A 2020, 182, 1815–1818. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Engchuan, W.; Hestand, M.S.; Heung, T.; Holleman, A.M.; Johnston, H.R.; Monfeuga, T.; McDonald-McGinn, D.M.; Gur, R.E.; Morrow, B.E.; et al. Genetic contributors to risk of schizophrenia in the presence of a 22q11.2 deletion. Mol. Psychiatry 2021, 26, 4496–4510. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, D.; Law, A.J.; Frisch, A.; Chen, J.; Zarchi, O.; Michaelovsky, E.; Ren-Patterson, R.; Lipska, B.K.; Carmel, M.; Kolachana, B.; et al. Biological effects of COMT haplotypes and psychosis risk in 22q11.2 deletion syndrome. Biol. Psychiatry 2014, 75, 406–413. [Google Scholar] [CrossRef]
- Duarte, M.; Afonso, J.; Moreira, A.; Antunes, D.; Ferreira, C.; Correia, H.; Marques, M.; Sequeira, S. Hyperprolinemia as a clue in the diagnosis of a patient with psychiatric manifestations. Brain Dev. 2017, 39, 539–541. [Google Scholar] [CrossRef]
- Bender, H.-U.; Almashanu, S.; Steel, G.; Hu, C.-A.; Lin, W.-W.; Willis, A.; Pulver, A.; Valle, D. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 2005, 76, 409–420. [Google Scholar] [CrossRef]
- Ross, H.E.; Guo, Y.; Coleman, K.; Ousley, O.; Miller, A.H. Association of IL-12p70 and IL-6: IL-10 ratio with autism-related behaviors in 22q11.2 deletion syndrome: A preliminary report. Brain Behav. Immun. 2013, 31, 76–81. [Google Scholar] [CrossRef]
- Earls, L.R.; Bayazitov, I.T.; Fricke, R.G.; Berry, R.B.; Illingworth, E.; Mittleman, G.; Zakharenko, S.S. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J. Neurosci. 2010, 30, 15843–15855. [Google Scholar] [CrossRef]
- Vergaelen, E.; Schiweck, C.; Van Steeland, K.; Counotte, J.; Veling, W.; Swillen, A.; Drexhage, H.; Claes, S. A pilot study on immuno-psychiatry in the 22q11.2 deletion syndrome: A role for Th17 cells in psychosis? Brain Behav. Immun. 2018, 70, 88–95. [Google Scholar] [CrossRef]
- Mekori-Domachevsky, E.; Taler, M.; Weinberger, R.; Guri, Y.; Dar, S.; Shani, S.; Dekel, I.; Weizman, A.; Gothelf, D. Neutrophils to lymphocytes ratio and psychosis in 22q11.2 deletion syndrome—Clinical and scientific implications. Schizophr. Res. 2021, 231, 164–169. [Google Scholar] [CrossRef]
- Bearden, C.E.; Jawad, A.F.; Lynch, D.R.; Monterossso, J.R.; Sokol, S.; McDonald-McGinn, D.M.; Saitta, S.C.; Harris, S.E.; Moss, E.; Wang, P.; et al. Effects of COMT genotype on behavioral symptomatology in the 22q11.2 Deletion Syndrome. Child Neuropsychol. 2005, 11, 109–117. [Google Scholar] [CrossRef]
- Mekori-Domachevsky, E.; Taler, M.; Shoenfeld, Y.; Gurevich, M.; Sonis, P.; Weisman, O.; Weizman, A.; Gothelf, D. Elevated Proinflammatory Markers in 22q11.2 Deletion Syndrome Are Associated With Psychosis and Cognitive Deficits. J. Clin. Psychiatry 2017, 78, 9052. [Google Scholar] [CrossRef] [PubMed]
- Hidding, E.; Swaab, H.; de Sonneville, L.M.J.; van Engeland, H.; Vorstman, J.A.S. The role of COMT and plasma proline in the variable penetrance of autistic spectrum symptoms in 22q11.2 deletion syndrome. Clin. Genet. 2016, 90, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; Saravanababu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of Oxidative Stress and Antioxidants in Autism. Adv. Neurobiol. 2020, 24, 193–206. [Google Scholar] [CrossRef]
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative Stress in Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2314–2332. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Liao, X.; Li, Y. Genetic associations between voltage-gated calcium channels and autism spectrum disorder: A systematic review. Mol. Brain 2020, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Earls, L.R.; Zakharenko, S.S. A synaptic function approach to investigating complex psychiatric diseases. Neuroscientist 2014, 20, 257–271. [Google Scholar] [CrossRef]
- Zehnder, T.; Petrelli, F.; Romanos, J.; Figueiredo, E.C.D.O.; Lewis, T.L.; Déglon, N.; Polleux, F.; Santello, M.; Bezzi, P. Mitochondrial biogenesis in developing astrocytes regulates astrocyte maturation and synapse formation. Cell Rep. 2021, 35, 108952. [Google Scholar] [CrossRef]
- Campbell, I.L.; Hofer, M.J.; Pagenstecher, A. Transgenic models for cytokine-induced neurological disease. Biochim. Biophys. Acta Mol. Basis. Dis. 2010, 1802, 903–917. [Google Scholar] [CrossRef]
- Aloe, L.; Fiore, M. TNF-α expressed in the brain of transgenic mice lowers central tyroxine hydroxylase immunoreactivity and alters grooming behavior. Neurosci. Lett. 1997, 238, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Carito, V.; Ceccanti, M.; Cestari, V.; Natella, F.; Bello, C.; Coccurello, R.; Mancinelli, R.; Fiore, M. Olive polyphenol effects in a mouse model of chronic ethanol addiction. Nutrition 2017, 33, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Petrella, C.; Carito, V.; Carere, C.; Ferraguti, G.; Ciafrè, S.; Natella, F.; Bello, C.; Greco, A.; Ralli, M.; Mancinelli, R.; et al. Oxidative stress inhibition by resveratrol in alcohol-dependent mice. Nutrition 2020, 79–80, 110783. [Google Scholar] [CrossRef] [PubMed]
- Alba, G.; Dakhaoui, H.; Santa-Maria, C.; Palomares, F.; Cejudo-Guillen, M.; Geniz, I.; Sobrino, F.; la Paz, S.M.-D.; Lopez-Enriquez, S. Nutraceuticals as Potential Therapeutic Modulators in Immunometabolism. Nutrients 2023, 15, 411. [Google Scholar] [CrossRef]
- Vivarelli, S.; Costa, C.; Teodoro, M.; Giambò, F.; Tsatsakis, A.M.; Fenga, C. Polyphenols: A route from bioavailability to bioactivity addressing potential health benefits to tackle human chronic diseases. Arch. Toxicol. 2022, 97, 3–38. [Google Scholar] [CrossRef]
- Gasmi, A.; Mujawdiya, P.K.; Noor, S.; Lysiuk, R.; Darmohray, R.; Piscopo, S.; Lenchyk, L.; Antonyak, H.; Dehtiarova, K.; Shanaida, M.; et al. Polyphenols in Metabolic Diseases. Molecules 2022, 27, 6280. [Google Scholar] [CrossRef]



| Oxidative Stress in DiGeorge Syndrome | |
|---|---|
| Genetic or Enzymatic Alterations: | Markers of Oxidative Stress: |
| Increase in mitochondria-associated oxidative stress in layer 2/3 projection neurons in mouse models | Disruption of layer 2/3 projection neurons related to association cortico-cortical connections [4] |
| Overproduction of modified proteins such as Met sulfoxide from Met, indole-3-lactate from Trp and aminomalonate Overproduction of hexoses such as 5-hydroxymethyl-2-furanoic acid and hexuronic acid in children | Inhibition of Complex IV, Complex V and creatine kinase [8] |
| Loss of DGCR8 gene in animal models | Inhibition of superoxide dismutase 2 (SOD2) [29,30] Increase in ROS [29,30] Accelerated senescence [29,30] |
| Loss of TXNRD2 gene in children | Reduction in antioxidant defense [8,31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menghi, M.; Micangeli, G.; Tarani, F.; Putotto, C.; Pirro, F.; Mariani, A.; Petrella, C.; Pulvirenti, F.; Cinicola, B.; Colloridi, F.; et al. Neuroinflammation and Oxidative Stress in Individuals Affected by DiGeorge Syndrome. Int. J. Mol. Sci. 2023, 24, 4242. https://doi.org/10.3390/ijms24044242
Menghi M, Micangeli G, Tarani F, Putotto C, Pirro F, Mariani A, Petrella C, Pulvirenti F, Cinicola B, Colloridi F, et al. Neuroinflammation and Oxidative Stress in Individuals Affected by DiGeorge Syndrome. International Journal of Molecular Sciences. 2023; 24(4):4242. https://doi.org/10.3390/ijms24044242
Chicago/Turabian StyleMenghi, Michela, Ginevra Micangeli, Francesca Tarani, Carolina Putotto, Federica Pirro, Alessandro Mariani, Carla Petrella, Federica Pulvirenti, Bianca Cinicola, Fiorenza Colloridi, and et al. 2023. "Neuroinflammation and Oxidative Stress in Individuals Affected by DiGeorge Syndrome" International Journal of Molecular Sciences 24, no. 4: 4242. https://doi.org/10.3390/ijms24044242
APA StyleMenghi, M., Micangeli, G., Tarani, F., Putotto, C., Pirro, F., Mariani, A., Petrella, C., Pulvirenti, F., Cinicola, B., Colloridi, F., Tarani, L., & Fiore, M. (2023). Neuroinflammation and Oxidative Stress in Individuals Affected by DiGeorge Syndrome. International Journal of Molecular Sciences, 24(4), 4242. https://doi.org/10.3390/ijms24044242