

The Link between Prostanoids and Cardiovascular Diseases

Abstract
1. Introduction
2. Arachidonic Acid Pathway
3. Bioavailability of Arachidonic Acid and PLA2 Enzymes
3.1. Cytosolic PLA2s
3.1.1. Cytosolic PLA2α
3.1.2. Cytosolic Ca2+-Dependent PLA2ζ and Ca2+-Independent iPLA2γ
3.2. Secretory PLA2s
3.2.1. Secretory PLA2-IIA
3.2.2. Secretory PLA2-V
4. Cyclooxygenases: The Key Point of Prostanoids Synthesis
5. Prostanoids
5.1. Prostaglandin D2 (PGD2)
Prostaglandin D Synthases and Receptors
5.2. Prostaglandin F2α (PGF2α)
Prostaglandin F Synthases and FP Receptor
5.3. Prostaglandin E2 (PGE2)
5.3.1. PGE2 and Inflammation
5.3.2. PGE2 and Atherosclerosis
5.3.3. PGE2 and Blood Pressure
5.3.4. PGE2 and Cardiac Health
5.3.5. PGE2 Synthases
5.3.6. Prostaglandin Dehydrogenase 1
5.3.7. Prostaglandin E2 Receptors
5.4. Thromboxane A2 (TxA2)
Thromboxane A Synthase 1 (TXAS) and TP Receptor
5.5. Prostacyclin PGI2
PGI2 Synthase and IP Receptor
6. Hemiketals
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| 15-PGDH | 15-hydroxyprostaglandin dehydrogenase/Prostaglandin Dehydrogenase 1 |
| 15d-PGJ2 | 15-Deoxy-∆-12,14-prostaglandin J2 |
| 5-LOX | 5-lipoxygenases |
| 5 S-HETE | 5S-hydroperoxyeicosatetraenoic acid |
| 5-OH-PGD2 | 5-hydroxy-prostaglandin D2 |
| 5-OH-PGE2 | 5-hydroxy-prostaglandin E2 |
| AC | Adenylyl cyclase |
| AMI | Acute myocardial infarction |
| ARIC | Atherosclerosis Risk in Community |
| CAD | Coronary artery disease |
| cAMP | Cyclic adenosine monophosphate |
| COX | Cyclooxygenase |
| cPGES | Cytosolic PGE2 synthase |
| CREB | cAMP response element-binding protein |
| CRP | C-reactive protein |
| CRTH2 | Chemoattractant receptor-homologous molecule expressed on Th2 lymphocytes |
| CVD | Cardiovascular disease |
| DP1 | PGD2 receptor subtype 1 |
| EETs | Epoxyeicosatrienoic acids |
| EP | E-type prostanoid receptor |
| ER | Endoplasmic reticulum |
| FP | Prostaglandin F receptor |
| H-PGDS | Hematopoietic prostaglandin D synthase |
| HETEs | Hydroxyeicosatetraenoic acids |
| HKD2 | Hemiketal D2 |
| HKE2 | Hemiketal E2 |
| I/R | Ischemia/reperfusion |
| IL | Interleukin |
| IP | Prostacyclin receptor |
| IP3 | Inositol trisphosphate |
| L-PGDS | Lipocalin-type prostaglandin D synthase |
| MACE | Major adverse cardiac events |
| mPGES | Microsomal PGE2 synthase |
| mPTP | Mitochondrial permeability transition pore |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSAID | Non-steroidal anti-inflammatory drug |
| PGD2 | Prostaglandin D2 |
| PGE2 | Prostaglandin E2 |
| PGF2α | Prostaglandin F2α |
| PGFS | Prostaglandin F synthase |
| PGG2 | Prostaglandin G2 |
| PGH2 | Prostaglandin H2 |
| PGI2 | Prostacyclin I2 |
| PGIS | Prostacyclin synthase |
| PKA | Protein kinase A/cAMP-dependent protein kinase |
| PLA2 | Phospholipase A2 |
| PPAR | Peroxisome proliferator-activated receptor |
| PUFA | Polyunsaturated fatty acid |
| ROS | Reactive oxygen species |
| SNPs | Single nucleotide polymorphisms |
| TF | Tissue factor |
| TGF-β | Transforming growth factor β |
| Th | T-helper cells |
| TNF-α | Tumor necrosis factor α |
| TP | Thromboxane prostanoid receptor |
| TxA2 | Thromboxane A2 |
| TXAS | Thromboxane A synthase 1 |
| VSMC | Vascular smooth muscle cell |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.; Coe, S.; Frayn, K. The Aetiology and Epidemiology of Cardiovascular Disease; Wiley: Hoboken, NJ, USA, 2018; pp. 1–28. [Google Scholar] [CrossRef]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.-D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Khan, H.; Xiao, J.; Cheang, W.S. Effects of Arachidonic Acid Metabolites on Cardiovascular Health and Disease. Int. J. Mol. Sci. 2021, 22, 12029. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.J.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and Its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Y.; Guo, Z.; Wang, M. Cardiovascular Biology of Prostanoids and Drug Discovery. Arter. Thromb. Vasc. Biol. 2020, 40, 1454–1463. [Google Scholar] [CrossRef]
- Piper, K.; Garelnabi, M. Eicosanoids: Atherosclerosis and Cardiometabolic Health. J. Clin. Transl. Endocrinol. 2020, 19, 100216. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G.; Rocca, B.; Patrono, C. The Key Contribution of Platelet and Vascular Arachidonic Acid Metabolism to the Pathophysiology of Atherothrombosis. Cardiovasc. Res. 2021, 117, 2001–2015. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Botta, E.; Holinstat, M. Eicosanoids in Inflammation in the Blood and the Vessel. Front. Pharmacol. 2022, 13, 997403. [Google Scholar] [CrossRef]
- Powell, W.S.; Rokach, J. Biosynthesis, Biological Effects, and Receptors of Hydroxyeicosatetraenoic Acids (HETEs) and Oxoeicosatetraenoic Acids (Oxo-ETEs) Derived from Arachidonic Acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef]
- Shoieb, S.M.; El-Sherbeni, A.A.; El-Kadi, A.O.S. Subterminal Hydroxyeicosatetraenoic Acids: Crucial Lipid Mediators in Normal Physiology and Disease States. Chem. Biol. Interact. 2019, 299, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mäki-Petäjä, K.; Cheriyan, J.; McEniery, C.; Wilkinson, I.B. The Role of Epoxyeicosatrienoic Acids in the Cardiovascular System. Br. J. Clin. Pharmacol. 2015, 80, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Shindou, H.; Shimizu, T. Cytosolic Phospholipase A(2) and Lysophospholipid Acyltransferases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating Phospholipase A(2) Biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.L.; Lin, A.Y.; DeWitt, D.L. Interleukin-1 Alpha Induces the Accumulation of Cytosolic Phospholipase A2 and the Release of Prostaglandin E2 in Human Fibroblasts. J. Biol. Chem. 1992, 267, 23451–23454. [Google Scholar] [CrossRef]
- Hayakawa, M.; Ishida, N.; Takeuchi, K.; Shibamoto, S.; Hori, T.; Oku, N.; Ito, F.; Tsujimoto, M. Arachidonic Acid-Selective Cytosolic Phospholipase A2 Is Crucial in the Cytotoxic Action of Tumor Necrosis Factor. J. Biol. Chem. 1993, 268, 11290–11295. [Google Scholar] [CrossRef]
- Kerkelä, R.; Boucher, M.; Zaka, R.; Gao, E.; Harris, D.; Piuhola, J.; Song, J.; Serpi, R.; Woulfe, K.C.; Cheung, J.Y.; et al. Cytosolic Phospholipase A(2)α Protects against Ischemia/Reperfusion Injury in the Heart. Clin. Transl. Sci. 2011, 4, 236–242. [Google Scholar] [CrossRef]
- Saito, Y.; Watanabe, K.; Fujioka, D.; Nakamura, T.; Obata, J.; Kawabata, K.; Watanabe, Y.; Mishina, H.; Tamaru, S.; Kita, Y.; et al. Disruption of Group IVA Cytosolic Phospholipase A(2) Attenuates Myocardial Ischemia-Reperfusion Injury Partly through Inhibition of TNF-α-Mediated Pathway. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2018–H2030. [Google Scholar] [CrossRef]
- Elinder, L.S.; Dumitrescu, A.; Larsson, P.; Hedin, U.; Frostegård, J.; Claesson, H.E. Expression of Phospholipase A2 Isoforms in Human Normal and Atherosclerotic Arterial Wall. Arter. Thromb. Vasc. Biol. 1997, 17, 2257–2263. [Google Scholar] [CrossRef]
- Bonetti, A.; Allegri, L.; Baldan, F.; Contin, M.; Battistella, C.; Damante, G.; Marchini, M.; Ortolani, F. Critical Involvement of Calcium-Dependent Cytosolic Phospholipase A2α in Aortic Valve Interstitial Cell Calcification. Int. J. Mol. Sci. 2020, 21, 6398. [Google Scholar] [CrossRef]
- Hartiala, J.; Li, D.; Conti, D.V.; Vikman, S.; Patel, Y.; Tang, W.H.W.; Brennan, M.-L.; Newman, J.W.; Stephensen, C.B.; Armstrong, P.; et al. Genetic Contribution of the Leukotriene Pathway to Coronary Artery Disease. Hum. Genet. 2011, 129, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Hartiala, J.; Gilliam, E.; Vikman, S.; Campos, H.; Allayee, H. Association of PLA2G4A with Myocardial Infarction Is Modulated by Dietary PUFAs. Am. J. Clin. Nutr. 2012, 95, 959–965. [Google Scholar] [CrossRef] [PubMed]
- González, L.M.; Robles, N.R.; Mota-Zamorano, S.; Arévalo-Lorido, J.C.; Valdivielso, J.M.; López-Gómez, J.; Gervasini, G. Tag-SNPs in Phospholipase-Related Genes Modify the Susceptibility to Nephrosclerosis and Its Associated Cardiovascular Risk. Front. Pharmacol. 2022, 13, 817020. [Google Scholar] [CrossRef] [PubMed]
- Adler, D.H.; Cogan, J.D.; Phillips, J.A.; Schnetz-Boutaud, N.; Milne, G.L.; Iverson, T.; Stein, J.A.; Brenner, D.A.; Morrow, J.D.; Boutaud, O.; et al. Inherited Human CPLA(2alpha) Deficiency Is Associated with Impaired Eicosanoid Biosynthesis, Small Intestinal Ulceration, and Platelet Dysfunction. J. Clin. Investig. 2008, 118, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Duvernay, M.T.; Matafonov, A.; Lindsley, C.W.; Hamm, H.E. Platelet Lipidomic Profiling: Novel Insight into Cytosolic Phospholipase A2α Activity and Its Role in Human Platelet Activation. Biochemistry 2015, 54, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Liu, X.; Cedars, A.M.; Yang, K.; Kiebish, M.A.; Joseph, S.M.; Kelley, J.; Jenkins, C.M.; Gross, R.W. Heart Failure-Induced Activation of Phospholipase IPLA(2)γ Generates Hydroxyeicosatetraenoic Acids Opening the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 2018, 293, 115–129. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The Role of the Mitochondrial Permeability Transition Pore in Heart Disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef]
- Moon, S.H.; Jenkins, C.M.; Kiebish, M.A.; Sims, H.F.; Mancuso, D.J.; Gross, R.W. Genetic Ablation of Calcium-Independent Phospholipase A(2)γ (IPLA(2)γ) Attenuates Calcium-Induced Opening of the Mitochondrial Permeability Transition Pore and Resultant Cytochrome c Release. J. Biol. Chem. 2012, 287, 29837–29850. [Google Scholar] [CrossRef]
- Moon, S.H.; Mancuso, D.J.; Sims, H.F.; Liu, X.; Nguyen, A.L.; Yang, K.; Guan, S.; Dilthey, B.G.; Jenkins, C.M.; Weinheimer, C.J.; et al. Cardiac Myocyte-Specific Knock-out of Calcium-Independent Phospholipase A2γ (IPLA2γ) Decreases Oxidized Fatty Acids during Ischemia/Reperfusion and Reduces Infarct Size. J. Biol. Chem. 2016, 291, 19687–19700. [Google Scholar] [CrossRef]
- Hara, S.; Yoda, E.; Sasaki, Y.; Nakatani, Y.; Kuwata, H. Calcium-Independent Phospholipase A2γ (IPLA2γ) and Its Roles in Cellular Functions and Diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 861–868. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Kuwata, H.; Yoshihara, K.; Masuda, S.; Shimbara, S.; Oh-ishi, S.; Murakami, M.; Kudo, I. Induction of Distinct Sets of Secretory Phospholipase A(2) in Rodents during Inflammation. Biochim. Biophys. Acta 2003, 1635, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, L.H.; Duchez, A.-C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets Release Mitochondria Serving as Substrate for Bactericidal Group IIA-Secreted Phospholipase A2 to Promote Inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- Duchez, A.-C.; Boudreau, L.H.; Naika, G.S.; Bollinger, J.; Belleannée, C.; Cloutier, N.; Laffont, B.; Mendoza-Villarroel, R.E.; Lévesque, T.; Rollet-Labelle, E.; et al. Platelet Microparticles Are Internalized in Neutrophils via the Concerted Activity of 12-Lipoxygenase and Secreted Phospholipase A2-IIA. Proc. Natl. Acad. Sci. USA 2015, 112, E3564–E3573. [Google Scholar] [CrossRef] [PubMed]
- Menschikowski, M.; Kasper, M.; Lattke, P.; Schiering, A.; Schiefer, S.; Stockinger, H.; Jaross, W. Secretory Group II Phospholipase A2 in Human Atherosclerotic Plaques. Atherosclerosis 1995, 118, 173–181. [Google Scholar] [CrossRef]
- Sartipy, P.; Johansen, B.; Gâsvik, K.; Hurt-Camejo, E. Molecular Basis for the Association of Group IIA Phospholipase A(2) and Decorin in Human Atherosclerotic Lesions. Circ. Res. 2000, 86, 707–714. [Google Scholar] [CrossRef]
- Ghesquiere, S.A.I.; Gijbels, M.J.J.; Anthonsen, M.; van Gorp, P.J.J.; van der Made, I.; Johansen, B.; Hofker, M.H.; de Winther, M.P.J. Macrophage-Specific Overexpression of Group IIa SPLA2 Increases Atherosclerosis and Enhances Collagen Deposition. J. Lipid Res. 2005, 46, 201–210. [Google Scholar] [CrossRef]
- Kimura-Matsumoto, M.; Ishikawa, Y.; Komiyama, K.; Tsuruta, T.; Murakami, M.; Masuda, S.; Akasaka, Y.; Ito, K.; Ishiguro, S.; Morita, H.; et al. Expression of Secretory Phospholipase A2s in Human Atherosclerosis Development. Atherosclerosis 2008, 196, 81–91. [Google Scholar] [CrossRef]
- Dutour, A.; Achard, V.; Sell, H.; Naour, N.; Collart, F.; Gaborit, B.; Silaghi, A.; Eckel, J.; Alessi, M.-C.; Henegar, C.; et al. Secretory Type II Phospholipase A2 Is Produced and Secreted by Epicardial Adipose Tissue and Overexpressed in Patients with Coronary Artery Disease. J. Clin. Endocrinol. Metab. 2010, 95, 963–967. [Google Scholar] [CrossRef]
- Muller, O.; Ntalianis, A.; Wijns, W.; Delrue, L.; Dierickx, K.; Auer, R.; Rodondi, N.; Mangiacapra, F.; Trana, C.; Hamilos, M.; et al. Association of Biomarkers of Lipid Modification with Functional and Morphological Indices of Coronary Stenosis Severity in Stable Coronary Artery Disease. J. Cardiovasc. Transl. Res. 2013, 6, 536–544. [Google Scholar] [CrossRef]
- Akinkuolie, A.O.; Lawler, P.R.; Chu, A.Y.; Caulfield, M.; Mu, J.; Ding, B.; Nyberg, F.; Glynn, R.J.; Ridker, P.M.; Hurt-Camejo, E.; et al. Group IIA Secretory Phospholipase A2, Vascular Inflammation, and Incident Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 1182–1190. [Google Scholar] [CrossRef]
- Exeter, H.J.; Folkersen, L.; Palmen, J.; Franco-Cereceda, A.; Cooper, J.A.; Kalea, A.Z.; Hooft, F.V.; Eriksson, P.; Humphries, S.E.; Talmud, P.J. Functional Analysis of Two PLA2G2A Variants Associated with Secretory Phospholipase A2-IIA Levels. PLoS ONE 2012, 7, e41139. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.V.; Simon, T.; Exeter, H.J.; Folkersen, L.; Asselbergs, F.W.; Guardiola, M.; Cooper, J.A.; Palmen, J.; Hubacek, J.A.; Carruthers, K.F.; et al. Secretory Phospholipase A(2)-IIA and Cardiovascular Disease: A Mendelian Randomization Study. J. Am. Coll. Cardiol. 2013, 62, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Komiyama, K.; Masuda, S.; Murakami, M.; Akasaka, Y.; Ito, K.; Akishima-Fukasawa, Y.; Kimura, M.; Fujimoto, A.; Kudo, I.; et al. Expression of Type V Secretory Phospholipase A in Myocardial Remodelling after Infarction. Histopathology 2005, 47, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Fujioka, D.; Saito, Y.; Kobayashi, T.; Nakamura, T.; Obata, J.; Kawabata, K.; Watanabe, K.; Watanabe, Y.; Mishina, H.; et al. Group V Secretory Phospholipase A2 Plays a Pathogenic Role in Myocardial Ischaemia-Reperfusion Injury. Cardiovasc. Res. 2011, 90, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.V.; Exeter, H.J.; Folkersen, L.; Nelson, C.P.; Guardiola, M.; Cooper, J.A.; Sofat, R.; Boekholdt, S.M.; Khaw, K.-T.; Li, K.-W.; et al. Novel Genetic Approach to Investigate the Role of Plasma Secretory Phospholipase A2 (SPLA2)-V Isoenzyme in Coronary Heart Disease: Modified Mendelian Randomization Analysis Using PLA2G5 Expression Levels. Circ. Cardiovasc. Genet. 2014, 7, 144–150. [Google Scholar] [CrossRef]
- Vargas-Alarcón, G.; Posadas-Romero, C.; Villarreal-Molina, T.; Alvarez-León, E.; Angeles-Martinez, J.; Soto, M.E.; Monroy-Muñoz, I.; Juárez, J.G.; Sánchez-Ramírez, C.J.; Ramirez-Bello, J.; et al. The (G>A) Rs11573191 Polymorphism of PLA2G5 Gene Is Associated with Premature Coronary Artery Diseas.se in the Mexican Mestizo Population: The Genetics of Atherosclerotic Disease Mexican Study. Biomed Res. Int. 2014, 2014, 931361. [Google Scholar] [CrossRef]
- Biringer, R.G. The Enzymology of the Human Prostanoid Pathway. Mol. Biol. Rep. 2020, 47, 4569–4586. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.L.; Ricciotti, E. Druggable Prostanoid Pathway. Adv. Exp. Med. Biol. 2020, 1274, 29–54. [Google Scholar] [CrossRef]
- Tanabe, T.; Tohnai, N. Cyclooxygenase Isozymes and Their Gene Structures and Expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar] [CrossRef]
- Belton, O.; Byrne, D.; Kearney, D.; Leahy, A.; Fitzgerald, D.J. Cyclooxygenase-1 and -2-Dependent Prostacyclin Formation in Patients with Atherosclerosis. Circulation 2000, 102, 840–845. [Google Scholar] [CrossRef]
- Brock, T.G.; McNish, R.W.; Peters-Golden, M. Arachidonic Acid Is Preferentially Metabolized by Cyclooxygenase-2 to Prostacyclin and Prostaglandin E2. J. Biol. Chem. 1999, 274, 11660–11666. [Google Scholar] [CrossRef]
- Schrör, K. Aspirin and Platelets: The Antiplatelet Action of Aspirin and Its Role in Thrombosis Treatment and Prophylaxis. Semin. Thromb. Hemost. 1997, 23, 349–356. [Google Scholar] [CrossRef]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Do Selective Cyclo-Oxygenase-2 Inhibitors and Traditional Non-Steroidal Anti-Inflammatory Drugs Increase the Risk of Atherothrombosis? Meta-Analysis of Randomised Trials. BMJ 2006, 332, 1302–1308. [Google Scholar] [CrossRef]
- Barbieri, S.S.; Amadio, P.; Gianellini, S.; Tarantino, E.; Zacchi, E.; Veglia, F.; Howe, L.R.; Weksler, B.B.; Mussoni, L.; Tremoli, E. Cyclooxygenase-2-Derived Prostacyclin Regulates Arterial Thrombus Formation by Suppressing Tissue Factor in a Sirtuin-1-Dependent-Manner. Circulation 2012, 126, 1373–1384. [Google Scholar] [CrossRef]
- Yu, Y.; Ricciotti, E.; Scalia, R.; Tang, S.Y.; Grant, G.; Yu, Z.; Landesberg, G.; Crichton, I.; Wu, W.; Puré, E.; et al. Vascular COX-2 Modulates Blood Pressure and Thrombosis in Mice. Sci. Transl. Med. 2012, 4, 132ra54. [Google Scholar] [CrossRef]
- Li, W.; Xu, J.; Wang, X.; Chen, J.; Zhang, C.; Sun, K.; Hui, R. Cyclooxygenase-2 (COX-2) G-765C Is a Protective Factor for Coronary Artery Disease but Not for Ischemic Stroke: A Meta-Analysis. Atherosclerosis 2009, 207, 492–495. [Google Scholar] [CrossRef]
- Wang, H.; Fu, Y.; Liu, D.; Zhang, M.; Zhang, G.; Wu, W.; Yang, S.; Li, C.; Zhang, H. The COX-2 Rs20417 Polymorphism and Risk of Coronary Artery Disease: Evidence from 17,621 Subjects. Heart Lung Circ. 2014, 23, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Papafili, A.; Hill, M.R.; Brull, D.J.; McAnulty, R.J.; Marshall, R.P.; Humphries, S.E.; Laurent, G.J. Common Promoter Variant in Cyclooxygenase-2 Represses Gene Expression: Evidence of Role in Acute-Phase Inflammatory Response. Arter. Thromb. Vasc. Biol. 2002, 22, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; González, J.I.; Bulló, M.; Carrasco, P.; Portolés, O.; Díez-Espino, J.; Covas, M.I.; Ruíz-Gutierrez, V.; Gómez-Gracia, E.; Arós, F.; et al. Polymorphisms Cyclooxygenase-2 -765G>C and Interleukin-6 -174G>C Are Associated with Serum Inflammation Markers in a High Cardiovascular Risk Population and Do Not Modify the Response to a Mediterranean Diet Supplemented with Virgin Olive Oil or Nuts. J. Nutr. 2009, 139, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Orbe, J.; Beloqui, O.; Rodriguez, J.A.; Belzunce, M.S.; Roncal, C.; Páramo, J.A. Protective Effect of the G-765C COX-2 Polymorphism on Subclinical Atherosclerosis and Inflammatory Markers in Asymptomatic Subjects with Cardiovascular Risk Factors. Clin. Chim. Acta 2006, 368, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Toniato, E.; Martinotti, S.; Fazia, M.; Iezzi, A.; Cuccurullo, C.; Pini, B.; Ursi, S.; Vitullo, G.; Averna, M.; et al. A Polymorphism in the Cyclooxygenase 2 Gene as an Inherited Protective Factor against Myocardial Infarction and Stroke. JAMA 2004, 291, 2221–2228. [Google Scholar] [CrossRef]
- Colaizzo, D.; Fofi, L.; Tiscia, G.; Guglielmi, R.; Cocomazzi, N.; Prencipe, M.; Margaglione, M.; Toni, D. The COX-2 G/C -765 Polymorphism May Modulate the Occurrence of Cerebrovascular Ischemia. Blood Coagul. Fibrinolysis 2006, 17, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Eikelboom, J.; Anand, S.S.; Eriksson, N.; Gerstein, H.C.; Mehta, S.; Connolly, S.J.; Rose, L.; Ridker, P.M.; Wallentin, L.; et al. Association of Cyclooxygenase-2 Genetic Variant with Cardiovascular Disease. Eur. Heart J. 2014, 35, 2242–2248a. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Kim, S.H.; Cha, D.H.; Lim, S.W.; Moon, J.Y.; Kim, J.O.; Ryu, C.S.; Park, H.S.; Sung, J.H.; Kim, N.K. Association of COX2 -765G>C Promoter Polymorphism and Coronary Artery Disease in Korean Population. Genes Genom. 2019, 41, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Ma, Y.-T.; Yang, Y.-N.; Fu, Z.-Y.; Li, X.-M.; Huang, D.; Ma, X.; Chen, B.; Liu, F. Interaction between COX-2 G-765C and Smoking in Relation to Coronary Artery Disease in a Chinese Uighur Population. Clin. Chem. Lab. Med. 2011, 49, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; North, K.E.; Bray, M.S.; Couper, D.J.; Heiss, G.; Zeldin, D.C. Cyclooxygenase Polymorphisms and Risk of Cardiovascular Events: The Atherosclerosis Risk in Communities (ARIC) Study. Clin. Pharmacol. Ther. 2008, 83, 52–60. [Google Scholar] [CrossRef]
- Kohsaka, S.; Volcik, K.A.; Folsom, A.R.; Wu, K.K.; Ballantyne, C.M.; Willerson, J.T.; Boerwinkle, E. Increased Risk of Incident Stroke Associated with the Cyclooxygenase 2 (COX-2) G-765C Polymorphism in African-Americans: The Atherosclerosis Risk in Communities Study. Atherosclerosis 2008, 196, 926–930. [Google Scholar] [CrossRef]
- Huuskonen, K.H.; Kunnas, T.A.; Tanner, M.M.; Mikkelsson, J.; Ilveskoski, E.; Karhunen, P.J.; Nikkari, S.T. COX-2 Gene Promoter Polymorphism and Coronary Artery Disease in Middle-Aged Men: The Helsinki Sudden Death Study. Mediat. Inflamm. 2008, 2008, 289453. [Google Scholar] [CrossRef]
- Hegener, H.H.; Diehl, K.A.; Kurth, T.; Gaziano, J.M.; Ridker, P.M.; Zee, R.Y.L. Polymorphisms of Prostaglandin-Endoperoxide Synthase 2 Gene, and Prostaglandin-E Receptor 2 Gene, C-Reactive Protein Concentrations and Risk of Atherothrombosis: A Nested Case-Control Approach. J. Thromb. Haemost. 2006, 4, 1718–1722. [Google Scholar] [CrossRef]
- Montali, A.; Barillà, F.; Tanzilli, G.; Vestri, A.; Fraioli, A.; Gaudio, C.; Martino, F.; Mezzetti, A.; Cipollone, F.; Arca, M. Functional Rs20417 SNP (-765G>C) of Cyclooxygenase-2 Gene Does Not Predict the Risk of Recurrence of Ischemic Events in Coronary Patients: Results of a 7-Year Prospective Study. Cardiology 2010, 115, 236–242. [Google Scholar] [CrossRef]
- Lemaitre, R.N.; Rice, K.; Marciante, K.; Bis, J.C.; Lumley, T.S.; Wiggins, K.L.; Smith, N.L.; Heckbert, S.R.; Psaty, B.M. Variation in Eicosanoid Genes, Non-Fatal Myocardial Infarction and Ischemic Stroke. Atherosclerosis 2009, 204, e58–e63. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kaul, S.; Al-Hazzani, A.; Alshatwi, A.A.; Jyothy, A.; Munshi, A. Association of COX-2 Rs20417 with Aspirin Resistance. J. Thromb. Thrombolysis 2013, 35, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Bottone, F.G.; Krahn, J.M.; Li, L.; Mohrenweiser, H.W.; Cook, M.E.; Petrovich, R.M.; Bell, D.A.; Eling, T.E.; Zeldin, D.C. Identification and Functional Characterization of Polymorphisms in Human Cyclooxygenase-1 (PTGS1). Pharm. Genom. 2007, 17, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Licis, N.; Krivmane, B.; Latkovskis, G.; Erglis, A. A Common Promoter Variant of the Gene Encoding Cyclooxygenase-1 (PTGS1) Is Related to Decreased Incidence of Myocardial Infarction in Patients with Coronary Artery Disease. Thromb. Res. 2011, 127, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Motovska, Z.; Kvasnicka, J.; Hajkova, J.; Kala, P.; Simek, S.; Bobcikova, P.; Petr, R.; Bilkova, D.; Poloczek, M.; Miklik, R.; et al. Platelet Gene Polymorphisms and Risk of Bleeding in Patients Undergoing Elective Coronary Angiography: A Genetic Substudy of the PRAGUE-8 Trial. Atherosclerosis 2010, 212, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Momary, K.M.; Shapiro, N.L.; Brace, L.D.; Shord, S.S.; Grossi, E.; Viana, M.A.; Helgason, C.M.; Nutescu, E.A.; Cavallari, L.H. Influence of Cyclooxygenase-1 Genotype on Ex Vivo Aspirin Response in Patients at Risk for Stroke. Cerebrovasc. Dis. 2009, 27, 585–593. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Z.; Sun, W.; Bai, W.; Sun, W.; Zhang, Y.; Wang, X.; Cai, B.; Xie, X.; Duan, Z.; et al. Impacts of COX-1 Gene Polymorphisms on Vascular Outcomes in Patients with Ischemic Stroke and Treated with Aspirin. Gene 2014, 546, 172–176. [Google Scholar] [CrossRef]
- Zheng, H.; Fu, L.; Xu, Y.; Zhang, T.F.; Che, D.; Li, J.Q.; Zhou, H.; Jiang, Z.; Lin, K.; Zhang, L.; et al. The PTGS1 (Rs1330344) CC Genotype Contributes to Susceptibility to Kawasaki Disease in Southern Chinese Children. Angiology 2022, 33197221118343. [Google Scholar] [CrossRef]
- Zhao, L.; Fang, J.; Zhou, M.; Zhou, J.; Yu, L.; Chen, N.; He, L. Interaction between COX-1 and COX-2 Increases Susceptibility to Ischemic Stroke in a Chinese Population. BMC Neurol. 2019, 19, 291. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and Inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Yao, C.; Narumiya, S. Prostaglandin-Cytokine Crosstalk in Chronic Inflammation. Br. J. Pharmacol. 2019, 176, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Soter, N.A.; Diamond, P.T.; Austen, K.F.; Oates, J.A.; Roberts, L.J. Prostaglandin D2 Generation after Activation of Rat and Human Mast Cells with Anti-IgE. J. Immunol. 1982, 129, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Ujihara, M.; Horiguchi, Y.; Ikai, K.; Hayaishi, O. The Major Source of Endogenous Prostaglandin D2 Production Is Likely Antigen-Presenting Cells. Localization of Glutathione-Requiring Prostaglandin D Synthetase in Histiocytes, Dendritic, and Kupffer Cells in Various Rat Tissues. J. Immunol. 1989, 143, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ogawa, K.; Sugamura, K.; Nakamura, M.; Takano, S.; Nagata, K. Cutting Edge: Differential Production of Prostaglandin D2 by Human Helper T Cell Subsets. J. Immunol. 2000, 164, 2277–2280. [Google Scholar] [CrossRef]
- Kong, D.; Yu, Y. Prostaglandin D2 Signaling and Cardiovascular Homeostasis. J. Mol. Cell. Cardiol. 2022, 167, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, F.A.; Wynalda, M.A. Albumin-Catalyzed Metabolism of Prostaglandin D2. Identification of Products Formed in Vitro. J. Biol. Chem. 1983, 258, 11713–11718. [Google Scholar] [CrossRef]
- Holgate, S.T.; Burns, G.B.; Robinson, C.; Church, M.K. Anaphylactic- and Calcium-Dependent Generation of Prostaglandin D2 (PGD2), Thromboxane B2, and Other Cyclooxygenase Products of Arachidonic Acid by Dispersed Human Lung Cells and Relationship to Histamine Release. J. Immunol. 1984, 133, 2138–2144. [Google Scholar] [CrossRef]
- Murray, J.J.; Tonnel, A.B.; Brash, A.R.; Roberts, L.J.; Gosset, P.; Workman, R.; Capron, A.; Oates, J.A. Release of Prostaglandin D2 into Human Airways during Acute Antigen Challenge. N. Engl. J. Med. 1986, 315, 800–804. [Google Scholar] [CrossRef]
- Matsuoka, T.; Hirata, M.; Tanaka, H.; Takahashi, Y.; Murata, T.; Kabashima, K.; Sugimoto, Y.; Kobayashi, T.; Ushikubi, F.; Aze, Y.; et al. Prostaglandin D2 as a Mediator of Allergic Asthma. Science 2000, 287, 2013–2017. [Google Scholar] [CrossRef]
- Fujitani, Y.; Kanaoka, Y.; Aritake, K.; Uodome, N.; Okazaki-Hatake, K.; Urade, Y. Pronounced Eosinophilic Lung Inflammation and Th2 Cytokine Release in Human Lipocalin-Type Prostaglandin D Synthase Transgenic Mice. J. Immunol. 2002, 168, 443–449. [Google Scholar] [CrossRef]
- Fajt, M.L.; Gelhaus, S.L.; Freeman, B.; Uvalle, C.E.; Trudeau, J.B.; Holguin, F.; Wenzel, S.E. Prostaglandin D₂ Pathway Upregulation: Relation to Asthma Severity, Control, and TH2 Inflammation. J. Allergy Clin. Immunol. 2013, 131, 1504–1512. [Google Scholar] [CrossRef]
- Zuo, S.; Kong, D.; Wang, C.; Liu, J.; Wang, Y.; Wan, Q.; Yan, S.; Zhang, J.; Tang, J.; Zhang, Q.; et al. CRTH2 Promotes Endoplasmic Reticulum Stress-Induced Cardiomyocyte Apoptosis through m-Calpain. EMBO Mol. Med. 2018, 10, e8237. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Wang, B.; Liu, J.; Kong, D.; Cui, H.; Jia, Y.; Wang, C.; Xu, X.; Chen, G.; Wang, Y.; et al. ER-Anchored CRTH2 Antagonizes Collagen Biosynthesis and Organ Fibrosis via Binding LARP6. EMBO J. 2021, 40, e107403. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Pillinger, M.H. 15d-PGJ2: The Anti-Inflammatory Prostaglandin? Clin. Immunol. 2005, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Rajakariar, R.; Hilliard, M.; Lawrence, T.; Trivedi, S.; Colville-Nash, P.; Bellingan, G.; Fitzgerald, D.; Yaqoob, M.M.; Gilroy, D.W. Hematopoietic Prostaglandin D2 Synthase Controls the Onset and Resolution of Acute Inflammation through PGD2 and 15-DeoxyDelta12 14 PGJ2. Proc. Natl. Acad. Sci. USA 2007, 104, 20979–20984. [Google Scholar] [CrossRef]
- Li, J.; Guo, C.; Wu, J. 15-Deoxy-∆-12,14-Prostaglandin J2 (15d-PGJ2), an Endogenous Ligand of PPAR-γ: Function and Mechanism. PPAR Res. 2019, 2019, 7242030. [Google Scholar] [CrossRef]
- Fu, M.; Zhang, J.; Zhu, X.; Myles, D.E.; Willson, T.M.; Liu, X.; Chen, Y.E. Peroxisome Proliferator-Activated Receptor Gamma Inhibits Transforming Growth Factor Beta-Induced Connective Tissue Growth Factor Expression in Human Aortic Smooth Muscle Cells by Interfering with Smad3. J. Biol. Chem. 2001, 276, 45888–45894. [Google Scholar] [CrossRef]
- Eguez, C.; Clark, M.A.; O’Connor, A.T. 15-Deoxy-Δ-12,14-Prostaglandin J2 Effects in Vascular Smooth Muscle Cells: Implications in Vascular Smooth Muscle Cell Proliferation and Contractility. Prostaglandins Other Lipid Mediat. 2021, 156, 106583. [Google Scholar] [CrossRef]
- Koyani, C.N.; Windischhofer, W.; Rossmann, C.; Jin, G.; Kickmaier, S.; Heinzel, F.R.; Groschner, K.; Alavian-Ghavanini, A.; Sattler, W.; Malle, E. 15-Deoxy-Δ12,14-PGJ₂ Promotes Inflammation and Apoptosis in Cardiomyocytes via the DP2/MAPK/TNFα Axis. Int. J. Cardiol. 2014, 173, 472–480. [Google Scholar] [CrossRef][Green Version]
- Kong, D.; Shen, Y.; Liu, G.; Zuo, S.; Ji, Y.; Lu, A.; Nakamura, M.; Lazarus, M.; Stratakis, C.A.; Breyer, R.M.; et al. PKA Regulatory IIα Subunit Is Essential for PGD2-Mediated Resolution of Inflammation. J. Exp. Med. 2016, 213, 2209–2226. [Google Scholar] [CrossRef]
- Liu, J.; Lin, K.; Guo, C.; Gao, H.; Yao, Y.; Lin, D. Expression and Purification of Cysteine Mutation Isoforms of Rat Lipocalin-Type Prostaglandin D Synthase for Nuclear Magnetic Resonance Study. Acta Biochim. Biophys. Sin. 2008, 40, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Eguchi, N.; Oda, H.; Seiki, K.; Kijima, Y.; Matsu-ura, Y.; Urade, Y.; Hayaishi, O. Expression of Lipocalin-Type Prostaglandin D Synthase (Beta-Trace) in Human Heart and Its Accumulation in the Coronary Circulation of Angina Patients. Proc. Natl. Acad. Sci. USA 1997, 94, 14689–14694. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Ujihara, M.; Horiguchi, Y.; Igarashi, M.; Nagata, A.; Ikai, K.; Hayaishi, O. Mast Cells Contain Spleen-Type Prostaglandin D Synthetase. J. Biol. Chem. 1990, 265, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-L.; Ricciotti, E.; Liang, X.; Grosser, T.; Grant, G.R.; FitzGerald, G.A. Lipocalin-Like Prostaglandin D Synthase but Not Hemopoietic Prostaglandin D Synthase Deletion Causes Hypertension and Accelerates Thrombogenesis in Mice. J. Pharmacol. Exp. Ther. 2018, 367, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Ragolia, L.; Palaia, T.; Hall, C.E.; Maesaka, J.K.; Eguchi, N.; Urade, Y. Accelerated Glucose Intolerance, Nephropathy, and Atherosclerosis in Prostaglandin D2 Synthase Knock-out Mice. J. Biol. Chem. 2005, 280, 29946–29955. [Google Scholar] [CrossRef]
- Tokudome, S.; Sano, M.; Shinmura, K.; Matsuhashi, T.; Morizane, S.; Moriyama, H.; Tamaki, K.; Hayashida, K.; Nakanishi, H.; Yoshikawa, N.; et al. Glucocorticoid Protects Rodent Hearts from Ischemia/Reperfusion Injury by Activating Lipocalin-Type Prostaglandin D Synthase-Derived PGD2 Biosynthesis. J. Clin. Investig. 2009, 119, 1477–1488. [Google Scholar] [CrossRef]
- Katsumata, Y.; Shinmura, K.; Sugiura, Y.; Tohyama, S.; Matsuhashi, T.; Ito, H.; Yan, X.; Ito, K.; Yuasa, S.; Ieda, M.; et al. Endogenous Prostaglandin D2 and Its Metabolites Protect the Heart against Ischemia-Reperfusion Injury by Activating Nrf2. Hypertension 2014, 63, 80–87. [Google Scholar] [CrossRef]
- Zhou, Y.; Shaw, N.; Li, Y.; Zhao, Y.; Zhang, R.; Liu, Z.-J. Structure-Function Analysis of Human l-Prostaglandin D Synthase Bound with Fatty Acid Molecules. FASEB J. 2010, 24, 4668–4677. [Google Scholar] [CrossRef][Green Version]
- Walch, L.; Labat, C.; Gascard, J.P.; de Montpreville, V.; Brink, C.; Norel, X. Prostanoid Receptors Involved in the Relaxation of Human Pulmonary Vessels. Br. J. Pharmacol. 1999, 126, 859–866. [Google Scholar] [CrossRef]
- He, Y.; Zuo, C.; Jia, D.; Bai, P.; Kong, D.; Chen, D.; Liu, G.; Li, J.; Wang, Y.; Chen, G.; et al. Loss of DP1 Aggravates Vascular Remodeling in Pulmonary Arterial Hypertension via MTORC1 Signaling. Am. J. Respir. Crit. Care Med. 2020, 201, 1263–1276. [Google Scholar] [CrossRef]
- Zou, F.; Li, Y.; Zhang, S.; Zhang, J. DP1 (Prostaglandin D2 Receptor 1) Activation Protects Against Vascular Remodeling and Vascular Smooth Muscle Cell Transition to Myofibroblasts in Angiotensin II-Induced Hypertension in Mice. Hypertension 2022, 79, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, J.; Shen, Y.; Liu, G.; Zuo, S.; Tao, B.; Ji, Y.; Lu, A.; Lazarus, M.; Breyer, R.M.; et al. Niacin Promotes Cardiac Healing after Myocardial Infarction through Activation of the Myeloid Prostaglandin D2 Receptor Subtype 1. J. Pharmacol. Exp. Ther. 2017, 360, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K. Prostaglandin F Synthase. Prostaglandins Other Lipid Mediat. 2002, 68–69, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Poyser, N.L. The Control of Prostaglandin Production by the Endometrium in Relation to Luteolysis and Menstruation. Prostaglandins Leukot. Essent. Fat. Acids 1995, 53, 147–195. [Google Scholar] [CrossRef]
- Li, W.-J.; Lu, J.-W.; Zhang, C.-Y.; Wang, W.-S.; Ying, H.; Myatt, L.; Sun, K. PGE2 vs PGF2α in Human Parturition. Placenta 2021, 104, 208–219. [Google Scholar] [CrossRef]
- Basu, S. Novel Cyclooxygenase-Catalyzed Bioactive Prostaglandin F2alpha from Physiology to New Principles in Inflammation. Med. Res. Rev. 2007, 27, 435–468. [Google Scholar] [CrossRef]
- Basu, S. Bioactive Eicosanoids: Role of Prostaglandin F(2α) and F₂-Isoprostanes in Inflammation and Oxidative Stress Related Pathology. Mol. Cells 2010, 30, 383–391. [Google Scholar] [CrossRef]
- Takayama, K.; Yuhki, K.; Ono, K.; Fujino, T.; Hara, A.; Yamada, T.; Kuriyama, S.; Karibe, H.; Okada, Y.; Takahata, O.; et al. Thromboxane A2 and Prostaglandin F2alpha Mediate Inflammatory Tachycardia. Nat. Med. 2005, 11, 562–566. [Google Scholar] [CrossRef]
- Jovanović, N.; Pavlović, M.; Mircevski, V.; Du, Q.; Jovanović, A. An Unexpected Negative Inotropic Effect of Prostaglandin F2alpha in the Rat Heart. Prostaglandins Other Lipid Mediat. 2006, 80, 110–119. [Google Scholar] [CrossRef]
- Dorn, G.W.; Becker, M.W.; Davis, M.G. Dissociation of the Contractile and Hypertrophic Effects of Vasoconstrictor Prostanoids in Vascular Smooth Muscle. J. Biol. Chem. 1992, 267, 24897–24905. [Google Scholar] [CrossRef]
- Adams, J.W.; Migita, D.S.; Yu, M.K.; Young, R.; Hellickson, M.S.; Castro-Vargas, F.E.; Domingo, J.D.; Lee, P.H.; Bui, J.S.; Henderson, S.A. Prostaglandin F2 Alpha Stimulates Hypertrophic Growth of Cultured Neonatal Rat Ventricular Myocytes. J. Biol. Chem. 1996, 271, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Jin, H.; Yang, R.; Winer, J.; Li, W.; Yen, R.; King, K.L.; Zeigler, F.; Ko, A.; Cheng, J.; et al. Prostaglandin F2 Alpha Induces Cardiac Myocyte Hypertrophy in Vitro and Cardiac Growth in Vivo. Am. J. Physiol. 1996, 271, H2197–H2208. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Katsuyama, M.; Wei, H.; Xia, Q.; Liu, W.; Yabe-Nishimura, C. Molecular Mechanisms Underlying PGF2alpha-Induced Hypertrophy of Vascular Smooth Muscle Cells. Yakugaku Zasshi 2010, 130, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Ti, Y.; Wang, J.; Wang, Z.; Xie, G.-L.; Shang, Y.; Tang, M.; Zhang, Y.; Zhang, W.; Zhong, M. Prostaglandin F2α Facilitates Collagen Synthesis in Cardiac Fibroblasts via an F-Prostanoid Receptor/Protein Kinase C/Rho Kinase Pathway Independent of Transforming Growth Factor Β1. Int. J. Biochem. Cell Biol. 2012, 44, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Liu, L.; Wang, Z.; Tang, M.; Ti, Y.; Han, L.; Zhang, L.; Zhang, Y.; Zhong, M.; Zhang, W. FP-Receptor Gene Silencing Ameliorates Myocardial Fibrosis and Protects from Diabetic Cardiomyopathy. J. Mol. Med. 2014, 92, 629–640. [Google Scholar] [CrossRef]
- Hébert, R.L.; Carmosino, M.; Saito, O.; Yang, G.; Jackson, C.A.; Qi, Z.; Breyer, R.M.; Natarajan, C.; Hata, A.N.; Zhang, Y.; et al. Characterization of a Rabbit Kidney Prostaglandin F(2{alpha}) Receptor Exhibiting G(i)-Restricted Signaling That Inhibits Water Absorption in the Collecting Duct. J. Biol. Chem. 2005, 280, 35028–35037. [Google Scholar] [CrossRef]
- Hao, C.-M.; Breyer, M.D. Physiological Regulation of Prostaglandins in the Kidney. Annu. Rev. Physiol. 2008, 70, 357–377. [Google Scholar] [CrossRef]
- Yu, Y.; Lucitt, M.B.; Stubbe, J.; Cheng, Y.; Friis, U.G.; Hansen, P.B.; Jensen, B.L.; Smyth, E.M.; FitzGerald, G.A. Prostaglandin F2alpha Elevates Blood Pressure and Promotes Atherosclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7985–7990. [Google Scholar] [CrossRef]
- Hu, B.-A.; Sai, W.-W.; Yuan, J.; Lan, H.-T.; Qi, J.; Wang, D.; Zhang, W.; Wang, Z.-H.; Zhong, M.; Shang, Y.-Y. PGF2α-FP Receptor Ameliorates Senescence of VSMCs in Vascular Remodeling by Src/PAI-1 Signal Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 2908261. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Chen, Y.; Liu, Y.; Feng, F.; Liu, W.; Guo, Q.; Zhao, L.; Sun, H. Overview of AKR1C3: Inhibitor Achievements and Disease Insights. J. Med. Chem. 2020, 63, 11305–11329. [Google Scholar] [CrossRef]
- Liang, J.; Cao, Y.; He, M.; Li, W.; Huang, G.; Ma, T.; Li, M.; Huang, Y.; Huang, X.; Hu, Y. AKR1C3 and Its Transcription Factor HOXB4 Are Promising Diagnostic Biomarkers for Acute Myocardial Infarction. Front. Cardiovasc. Med. 2021, 8, 694238. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, R.; Hu, H.; Ding, X.; Ji, Y.; Li, G.; Wang, Y.; Xie, S.; Liu, X.; Ding, Z. Potential Biomarkers of Acute Myocardial Infarction Based on Co-Expression Network Analysis. Exp. Ther. Med. 2022, 23, 162. [Google Scholar] [CrossRef] [PubMed]
- Su, E.J.; Ernst, L.; Abdallah, N.; Chatterton, R.; Xin, H.; Monsivais, D.; Coon, J.; Bulun, S.E. Estrogen Receptor-β and Fetoplacental Endothelial Prostanoid Biosynthesis: A Link to Clinically Demonstrated Fetal Growth Restriction. J. Clin. Endocrinol. Metab. 2011, 96, E1558–E1567. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-J.; Zhang, L.; Zhang, W.-Y. Gene Expression Profiling of Maternal Blood in Early Onset Severe Preeclampsia: Identification of Novel Biomarkers. J. Perinat. Med. 2009, 37, 609–616. [Google Scholar] [CrossRef]
- Sun, C.-J.; Li, L.; Li, X.; Zhang, W.-Y.; Liu, X.-W. Novel SNPs of WNK1 and AKR1C3 Are Associated with Preeclampsia. Gene 2018, 668, 27–32. [Google Scholar] [CrossRef]
- Gonzalez-Covarrubias, V.; Kalabus, J.L.; Blanco, J.G. Inhibition of Polymorphic Human Carbonyl Reductase 1 (CBR1) by the Cardioprotectant Flavonoid 7-Monohydroxyethyl Rutoside (MonoHER). Pharm. Res. 2008, 25, 1730–1734. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, W.; Lv, T.; Wang, Y.; Liu, T.; Chen, Y.; Jin, Y.; Huang, J.; Zheng, L.; Huang, Y.; et al. Aidi Injection Reduces Doxorubicin-Induced Cardiotoxicity by Inhibiting Carbonyl Reductase 1 Expression. Pharm. Biol. 2022, 60, 1616–1624. [Google Scholar] [CrossRef]
- Gonzalez-Covarrubias, V.; Ghosh, D.; Lakhman, S.S.; Pendyala, L.; Blanco, J.G. A Functional Genetic Polymorphism on Human Carbonyl Reductase 1 (CBR1 V88I) Impacts on Catalytic Activity and NADPH Binding Affinity. Drug Metab. Dispos. 2007, 35, 973–980. [Google Scholar] [CrossRef]
- Wei, X.; Wu, Y.; Pan, H.; Zhang, Q.; He, K.; Xia, G.; Xia, H.; Lin, S.; Shang, H.-C. Proteomics Revealed That Mitochondrial Function Contributed to the Protective Effect of Herba Siegesbeckiae Against Cardiac Ischemia/Reperfusion Injury. Front. Cardiovasc. Med. 2022, 9, 895797. [Google Scholar] [CrossRef]
- Zhou, L.; Zhan, W.; Wei, X. Clinical Pharmacology and Pharmacogenetics of Prostaglandin Analogues in Glaucoma. Front. Pharmacol. 2022, 13, 1015338. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Gu, S.-M.; Li, M.J.; Li, J.; Tao, B.; Wang, Y.; Wang, Y.; Zuo, S.; Shen, Y.; Yu, Y.; et al. Rare SNP Rs12731181 in the MiR-590-3p Target Site of the Prostaglandin F2α Receptor Gene Confers Risk for Essential Hypertension in the Han Chinese Population. Arter. Thromb. Vasc. Biol. 2015, 35, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-Induced Inflammation: Relevance of Prostaglandin E Receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, K.; Inazumi, T.; Shimamoto, A.; Sugimoto, Y. Molecular Mechanisms Underlying Prostaglandin E2-Exacerbated Inflammation and Immune Diseases. Int. Immunol. 2019, 31, 597–606. [Google Scholar] [CrossRef]
- Sun, C.-C.; Zhou, Z.-Q.; Yang, D.; Chen, Z.-L.; Zhou, Y.-Y.; Wen, W.; Feng, C.; Zheng, L.; Peng, X.-Y.; Tang, C.-F. Recent Advances in Studies of 15-PGDH as a Key Enzyme for the Degradation of Prostaglandins. Int. Immunopharmacol. 2021, 101, 108176. [Google Scholar] [CrossRef]
- Bryson, T.D.; Harding, P. Prostaglandin E2 EP Receptors in Cardiovascular Disease: An Update. Biochem. Pharmacol. 2022, 195, 114858. [Google Scholar] [CrossRef]
- Perretti, M.; Ahluwalia, A. The Microcirculation and Inflammation: Site of Action for Glucocorticoids. Microcirculation 2000, 7, 147–161. [Google Scholar] [CrossRef]
- Martínez-Colón, G.J.; Moore, B.B. Prostaglandin E2 as a Regulator of Immunity to Pathogens. Pharmacol. Ther. 2018, 185, 135–146. [Google Scholar] [CrossRef]
- Morimoto, K.; Shirata, N.; Taketomi, Y.; Tsuchiya, S.; Segi-Nishida, E.; Inazumi, T.; Kabashima, K.; Tanaka, S.; Murakami, M.; Narumiya, S.; et al. Prostaglandin E2-EP3 Signaling Induces Inflammatory Swelling by Mast Cell Activation. J. Immunol. 2014, 192, 1130–1137. [Google Scholar] [CrossRef]
- Omori, K.; Kida, T.; Hori, M.; Ozaki, H.; Murata, T. Multiple Roles of the PGE2 -EP Receptor Signal in Vascular Permeability. Br. J. Pharmacol. 2014, 171, 4879–4889. [Google Scholar] [CrossRef]
- Takaishi, O.; Arakawa, T.; Fujiwara, Y.; Fukuda, T.; Otani, K.; Yamasaki, K.; Higuchi, K.; Kuroki, T. Inhibition by 16,16-Dimethyl Prostaglandin E2 of Tumor Necrosis Factor-Alpha and Interleukin-1beta Production and Messenger RNA Expression in Human Monocytes Stimulated by Helicobacter Pylori. Dig. Dis. Sci. 1999, 44, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, M.; Chen, L.-Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.-Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J. Immunol. 2015, 194, 5472–5487. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 Inflammasome Inhibition Is Disrupted in a Group of Auto-Inflammatory Disease CAPS Mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.-F.; Chou, J.-C.; Ka, S.-M.; Tasi, Y.-L.; Chen, A.; Wu, S.-H.; Chiu, H.-W.; Wong, W.-T.; Wang, Y.-F.; Tsai, C.-L.; et al. Cyclooxygenase-2 Regulates NLRP3 Inflammasome-Derived IL-1β Production. J. Cell. Physiol. 2015, 230, 863–874. [Google Scholar] [CrossRef]
- Martínez-Colón, G.J.; Taylor, Q.M.; Wilke, C.A.; Podsiad, A.B.; Moore, B.B. Elevated Prostaglandin E2 Post-Bone Marrow Transplant Mediates Interleukin-1β-Related Lung Injury. Mucosal Immunol. 2018, 11, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Zasłona, Z.; Pålsson-McDermott, E.M.; Menon, D.; Haneklaus, M.; Flis, E.; Prendeville, H.; Corcoran, S.E.; Peters-Golden, M.; O’Neill, L.A.J. The Induction of Pro-IL-1β by Lipopolysaccharide Requires Endogenous Prostaglandin E2 Production. J. Immunol. 2017, 198, 3558–3564. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Aoki, T.; Narumiya, S. Prostaglandins and Chronic Inflammation. Trends Pharmacol. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive Lipids, Inflammation and Chronic Diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef]
- Aoki, T.; Nishimura, M.; Matsuoka, T.; Yamamoto, K.; Furuyashiki, T.; Kataoka, H.; Kitaoka, S.; Ishibashi, R.; Ishibazawa, A.; Miyamoto, S.; et al. PGE(2) -EP(2) Signalling in Endothelium Is Activated by Haemodynamic Stress and Induces Cerebral Aneurysm through an Amplifying Loop via NF-ΚB. Br. J. Pharmacol. 2011, 163, 1237–1249. [Google Scholar] [CrossRef]
- Yao, C.; Sakata, D.; Esaki, Y.; Li, Y.; Matsuoka, T.; Kuroiwa, K.; Sugimoto, Y.; Narumiya, S. Prostaglandin E2-EP4 Signaling Promotes Immune Inflammation through Th1 Cell Differentiation and Th17 Cell Expansion. Nat. Med. 2009, 15, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and Unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef]
- Cipollone, F.; Prontera, C.; Pini, B.; Marini, M.; Fazia, M.; De Cesare, D.; Iezzi, A.; Ucchino, S.; Boccoli, G.; Saba, V.; et al. Overexpression of Functionally Coupled Cyclooxygenase-2 and Prostaglandin E Synthase in Symptomatic Atherosclerotic Plaques as a Basis of Prostaglandin E(2)-Dependent Plaque Instability. Circulation 2001, 104, 921–927. [Google Scholar] [CrossRef]
- Cipollone, F.; Fazia, M.L.; Iezzi, A.; Cuccurullo, C.; De Cesare, D.; Ucchino, S.; Spigonardo, F.; Marchetti, A.; Buttitta, F.; Paloscia, L.; et al. Association between Prostaglandin E Receptor Subtype EP4 Overexpression and Unstable Phenotype in Atherosclerotic Plaques in Human. Arter. Thromb. Vasc. Biol. 2005, 25, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; García-Cardena, G.; Sukhova, G.K.; Comander, J.; Gimbrone, M.A.; Libby, P. Prostaglandin E2 Suppresses Chemokine Production in Human Macrophages through the EP4 Receptor. J. Biol. Chem. 2002, 277, 44147–44154. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Shimizu, K.; Okamoto, Y.; Folco, E.; Ilasaca, M.-L.; Feinberg, M.W.; Aikawa, M.; Libby, P. Prostaglandin E Receptor Type 4-Associated Protein Interacts Directly with NF-KappaB1 and Attenuates Macrophage Activation. J. Biol. Chem. 2008, 283, 9692–9703. [Google Scholar] [CrossRef]
- Wang, W.; Liang, M.; Wang, L.; Bei, W.; Rong, X.; Xu, J.; Guo, J. Role of Prostaglandin E2 in Macrophage Polarization: Insights into Atherosclerosis. Biochem. Pharmacol. 2023, 207, 115357. [Google Scholar] [CrossRef]
- Fabre, J.E.; Nguyen, M.; Athirakul, K.; Coggins, K.; McNeish, J.D.; Austin, S.; Parise, L.K.; FitzGerald, G.A.; Coffman, T.M.; Koller, B.H. Activation of the Murine EP3 Receptor for PGE2 Inhibits CAMP Production and Promotes Platelet Aggregation. J. Clin. Investig. 2001, 107, 603–610. [Google Scholar] [CrossRef]
- Gross, S.; Tilly, P.; Hentsch, D.; Vonesch, J.-L.; Fabre, J.-E. Vascular Wall-Produced Prostaglandin E2 Exacerbates Arterial Thrombosis and Atherothrombosis through Platelet EP3 Receptors. J. Exp. Med. 2007, 204, 311–320. [Google Scholar] [CrossRef]
- Tilly, P.; Charles, A.-L.; Ludwig, S.; Slimani, F.; Gross, S.; Meilhac, O.; Geny, B.; Stefansson, K.; Gurney, M.E.; Fabre, J.-E. Blocking the EP3 Receptor for PGE2 with DG-041 Decreases Thrombosis without Impairing Haemostatic Competence. Cardiovasc. Res. 2014, 101, 482–491. [Google Scholar] [CrossRef]
- Smith, J.P.; Haddad, E.V.; Downey, J.D.; Breyer, R.M.; Boutaud, O. PGE2 Decreases Reactivity of Human Platelets by Activating EP2 and EP4. Thromb. Res. 2010, 126, e23–e29. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.; Foudi, N.; Longrois, D.; Norel, X. The Role of Prostaglandin E2 in Human Vascular Inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Jia, Z.; Yu, J.; Huang, S. Roles of EP Receptors in the Regulation of Fluid Balance and Blood Pressure. Front. Endocrinol. 2022, 13, 875425. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Breyer, R.M.; Fowler, B.; Davis, L.; Hébert, R.L.; Breyer, M.D. Prostaglandin E2 Inhibits Renal Collecting Duct Na+ Absorption by Activating the EP1 Receptor. J. Clin. Investig. 1998, 102, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, R.; Zimpelmann, J.; Eckert, D.; Ghossein, J.; Geddes, S.; Beique, J.-C.; Thibodeau, J.-F.; Kennedy, C.R.J.; Burns, K.D.; Hébert, R.L. PGE2 EP1 Receptor Inhibits Vasopressin-Dependent Water Reabsorption and Sodium Transport in Mouse Collecting Duct. Lab. Investig. 2018, 98, 360–370. [Google Scholar] [CrossRef]
- Bartlett, C.S.; Boyd, K.L.; Harris, R.C.; Zent, R.; Breyer, R.M. EP1 Disruption Attenuates End-Organ Damage in a Mouse Model of Hypertension. Hypertension 2012, 60, 1184–1191. [Google Scholar] [CrossRef]
- Rutkai, I.; Feher, A.; Erdei, N.; Henrion, D.; Papp, Z.; Edes, I.; Koller, A.; Kaley, G.; Bagi, Z. Activation of Prostaglandin E2 EP1 Receptor Increases Arteriolar Tone and Blood Pressure in Mice with Type 2 Diabetes. Cardiovasc. Res. 2009, 83, 148–154. [Google Scholar] [CrossRef]
- Hassouneh, R.; Nasrallah, R.; Zimpelmann, J.; Gutsol, A.; Eckert, D.; Ghossein, J.; Burns, K.D.; Hébert, R.L. PGE2 Receptor EP3 Inhibits Water Reabsorption and Contributes to Polyuria and Kidney Injury in a Streptozotocin-Induced Mouse Model of Diabetes. Diabetologia 2016, 59, 1318–1328. [Google Scholar] [CrossRef]
- Chen, L.; Miao, Y.; Zhang, Y.; Dou, D.; Liu, L.; Tian, X.; Yang, G.; Pu, D.; Zhang, X.; Kang, J.; et al. Inactivation of the E-Prostanoid 3 Receptor Attenuates the Angiotensin II Pressor Response via Decreasing Arterial Contractility. Arter. Thromb. Vasc. Biol. 2012, 32, 3024–3032. [Google Scholar] [CrossRef]
- Olesen, E.T.B.; Rützler, M.R.; Moeller, H.B.; Praetorius, H.A.; Fenton, R.A. Vasopressin-Independent Targeting of Aquaporin-2 by Selective E-Prostanoid Receptor Agonists Alleviates Nephrogenic Diabetes Insipidus. Proc. Natl. Acad. Sci. USA 2011, 108, 12949–12954. [Google Scholar] [CrossRef]
- Gao, M.; Cao, R.; Du, S.; Jia, X.; Zheng, S.; Huang, S.; Han, Q.; Liu, J.; Zhang, X.; Miao, Y.; et al. Disruption of Prostaglandin E2 Receptor EP4 Impairs Urinary Concentration via Decreasing Aquaporin 2 in Renal Collecting Ducts. Proc. Natl. Acad. Sci. USA 2015, 112, 8397–8402. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The Prostanoid EP4 Receptor and Its Signaling Pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Du, S.; Fang, B.; Li, C.; Jia, X.; Zheng, S.; Wang, S.; Li, Q.; Su, W.; Wang, N.; et al. VSMC-Specific EP4 Deletion Exacerbates Angiotensin II-Induced Aortic Dissection by Increasing Vascular Inflammation and Blood Pressure. Proc. Natl. Acad. Sci. USA 2019, 116, 8457–8462. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, M.; He, W.; Milne, G.L.; Howard, J.R.H.; Morrow, J.; Hébert, R.L.; Breyer, R.M.; Chen, J.; Hao, C.-M. Increased Dietary NaCl Induces Renal Medullary PGE2 Production and Natriuresis via the EP2 Receptor. Am. J. Physiol. Renal. Physiol. 2008, 295, F818–F825. [Google Scholar] [CrossRef]
- Levy, D.; Larson, M.G.; Vasan, R.S.; Kannel, W.B.; Ho, K.K. The Progression from Hypertension to Congestive Heart Failure. JAMA 1996, 275, 1557–1562. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Kim, S.; Fu, Q.; Parikh, D.; Sridhar, B.; Shi, Q.; Zhang, X.; Guan, Y.; Chen, X.; et al. Phosphodiesterases Coordinate CAMP Propagation Induced by Two Stimulatory G Protein-Coupled Receptors in Hearts. Proc. Natl. Acad. Sci. USA 2012, 109, 6578–6583. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What Is the Role of Beta-Adrenergic Signaling in Heart Failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Fu, J.; Li, L.; Chen, L.; Su, C.; Feng, X.; Huang, K.; Zhang, L.; Yang, X.; Fu, Q. PGE2 Protects against Heart Failure through Inhibiting TGF-Β1 Synthesis in Cardiomyocytes and Crosstalk between TGF-Β1 and GRK2. J. Mol. Cell Cardiol. 2022, 172, 63–77. [Google Scholar] [CrossRef]
- Bryson, T.D.; Gu, X.; Khalil, R.M.; Khan, S.; Zhu, L.; Xu, J.; Peterson, E.; Yang, X.-P.; Harding, P. Overexpression of Prostaglandin E2 EP4 Receptor Improves Cardiac Function after Myocardial Infarction. J. Mol. Cell. Cardiol. 2018, 118, 1–12. [Google Scholar] [CrossRef]
- Mendez, M.; LaPointe, M.C. PGE2-Induced Hypertrophy of Cardiac Myocytes Involves EP4 Receptor-Dependent Activation of P42/44 MAPK and EGFR Transactivation. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2111–H2117. [Google Scholar] [CrossRef]
- Qian, J.-Y.; Harding, P.; Liu, Y.; Shesely, E.; Yang, X.-P.; LaPointe, M.C. Reduced Cardiac Remodeling and Function in Cardiac-Specific EP4 Receptor Knockout Mice with Myocardial Infarction. Hypertension 2008, 51, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-Y.; Yuhki, K.; Hara, A.; Fujino, T.; Kuriyama, S.; Yamada, T.; Takayama, K.; Takahata, O.; Karibe, H.; Taniguchi, T.; et al. Prostaglandin E2 Protects the Heart from Ischemia-Reperfusion Injury via Its Receptor Subtype EP4. Circulation 2004, 109, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xu, C.; Huo, X.; Hao, H.; Wan, Q.; Chen, H.; Zhang, X.; Breyer, R.M.; Huang, Y.; Cao, X.; et al. The Cyclooxygenase-1/MPGES-1/Endothelial Prostaglandin EP4 Receptor Pathway Constrains Myocardial Ischemia-Reperfusion Injury. Nat. Commun. 2019, 10, 1888. [Google Scholar] [CrossRef] [PubMed]
- Zacharowski, K.; Olbrich, A.; Otto, M.; Hafner, G.; Thiemermann, C. Effects of the Prostanoid EP3-Receptor Agonists M&B 28767 and GR 63799X on Infarct Size Caused by Regional Myocardial Ischaemia in the Anaesthetized Rat. Br. J. Pharmacol. 1999, 126, 849–858. [Google Scholar] [CrossRef]
- Zacharowski, K.; Olbrich, A.; Piper, J.; Hafner, G.; Kondo, K.; Thiemermann, C. Selective Activation of the Prostanoid EP(3) Receptor Reduces Myocardial Infarct Size in Rodents. Arter. Thromb. Vasc. Biol. 1999, 19, 2141–2147. [Google Scholar] [CrossRef]
- Hohlfeld, T.; Meyer-Kirchrath, J.; Vogel, Y.C.; Schrör, K. Reduction of Infarct Size by Selective Stimulation of Prostaglandin EP(3)Receptors in the Reperfused Ischemic Pig Heart. J. Mol. Cell. Cardiol. 2000, 32, 285–296. [Google Scholar] [CrossRef]
- Martin, M.; Meyer-Kirchrath, J.; Kaber, G.; Jacoby, C.; Flögel, U.; Schrader, J.; Rüther, U.; Schrör, K.; Hohlfeld, T. Cardiospecific Overexpression of the Prostaglandin EP3 Receptor Attenuates Ischemia-Induced Myocardial Injury. Circulation 2005, 112, 400–406. [Google Scholar] [CrossRef]
- Gu, X.; Xu, J.; Zhu, L.; Bryson, T.; Yang, X.-P.; Peterson, E.; Harding, P. Prostaglandin E2 Reduces Cardiac Contractility via EP3 Receptor. Circ. Heart Fail. 2016, 9, e003291. [Google Scholar] [CrossRef]
- Meyer-Kirchrath, J.; Martin, M.; Schooss, C.; Jacoby, C.; Flögel, U.; Marzoll, A.; Fischer, J.W.; Schrader, J.; Schrör, K.; Hohlfeld, T. Overexpression of Prostaglandin EP3 Receptors Activates Calcineurin and Promotes Hypertrophy in the Murine Heart. Cardiovasc. Res. 2009, 81, 310–318. [Google Scholar] [CrossRef]
- Tóth, A.D.; Schell, R.; Lévay, M.; Vettel, C.; Theis, P.; Haslinger, C.; Alban, F.; Werhahn, S.; Frischbier, L.; Krebs-Haupenthal, J.; et al. Inflammation Leads through PGE/EP3 Signaling to HDAC5/MEF2-Dependent Transcription in Cardiac Myocytes. EMBO Mol. Med. 2018, 10, e8536. [Google Scholar] [CrossRef]
- Maxwell, D.L.; Bryson, T.D.; Taube, D.; Xu, J.; Peterson, E.; Harding, P. Deleterious Effects of Cardiomyocyte-Specific Prostaglandin E2 EP3 Receptor Overexpression on Cardiac Function after Myocardial Infarction. Life Sci. 2023, 313, 121277. [Google Scholar] [CrossRef] [PubMed]
- Harding, P.; LaPointe, M.C. Prostaglandin E2 Increases Cardiac Fibroblast Proliferation and Increases Cyclin D Expression via EP1 Receptor. Prostaglandins. Leukot. Essent. Fat. Acids 2011, 84, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Cao, X.; Zou, J.; Ding, X.; Shen, B.; Lv, W. Prostaglandin E2 Induced Cardiac Hypertrophy through EP2 Receptor-Dependent Activation of β-Catenin in 5/6 Nephrectomy Rats. ESC Heart Fail. 2021, 8, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, P.J.; Thorén, S.; Morgenstern, R.; Samuelsson, B. Identification of Human Prostaglandin E Synthase: A Microsomal, Glutathione-Dependent, Inducible Enzyme, Constituting a Potential Novel Drug Target. Proc. Natl. Acad. Sci. USA 1999, 96, 7220–7225. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 Synthesis and Secretion: The Role of PGE2 Synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Murakami, M.; Naraba, H.; Tanioka, T.; Semmyo, N.; Nakatani, Y.; Kojima, F.; Ikeda, T.; Fueki, M.; Ueno, A.; Oh, S.; et al. Regulation of Prostaglandin E2 Biosynthesis by Inducible Membrane-Associated Prostaglandin E2 Synthase That Acts in Concert with Cyclooxygenase-2. J. Biol. Chem. 2000, 275, 32783–32792. [Google Scholar] [CrossRef]
- Uematsu, S.; Matsumoto, M.; Takeda, K.; Akira, S. Lipopolysaccharide-Dependent Prostaglandin E(2) Production Is Regulated by the Glutathione-Dependent Prostaglandin E(2) Synthase Gene Induced by the Toll-like Receptor 4/MyD88/NF-IL6 Pathway. J. Immunol. 2002, 168, 5811–5816. [Google Scholar] [CrossRef]
- Yang, G.; Chen, L. An Update of Microsomal Prostaglandin E Synthase-1 and PGE2 Receptors in Cardiovascular Health and Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 5249086. [Google Scholar] [CrossRef]
- Gómez-Hernández, A.; Martín-Ventura, J.L.; Sánchez-Galán, E.; Vidal, C.; Ortego, M.; Blanco-Colio, L.M.; Ortega, L.; Tuñón, J.; Egido, J. Overexpression of COX-2, Prostaglandin E Synthase-1 and Prostaglandin E Receptors in Blood Mononuclear Cells and Plaque of Patients with Carotid Atherosclerosis: Regulation by Nuclear Factor-KappaB. Atherosclerosis 2006, 187, 139–149. [Google Scholar] [CrossRef]
- Wang, M.; Zukas, A.M.; Hui, Y.; Ricciotti, E.; Puré, E.; FitzGerald, G.A. Deletion of Microsomal Prostaglandin E Synthase-1 Augments Prostacyclin and Retards Atherogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 14507–14512. [Google Scholar] [CrossRef]
- Chen, L.; Yang, G.; Monslow, J.; Todd, L.; Cormode, D.P.; Tang, J.; Grant, G.R.; DeLong, J.H.; Tang, S.Y.; Lawson, J.A.; et al. Myeloid Cell Microsomal Prostaglandin E Synthase-1 Fosters Atherogenesis in Mice. Proc. Natl. Acad. Sci. USA 2014, 111, 6828–6833. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, M.S.; García-Redondo, A.B.; Zalba, G.; González-Amor, M.; Aguado, A.; Martínez-Revelles, S.; Beltrán, L.M.; Camacho, M.; Cachofeiro, V.; Alonso, M.J.; et al. MPGES-1 (Microsomal Prostaglandin E Synthase-1) Mediates Vascular Dysfunction in Hypertension Through Oxidative Stress. Hypertension 2018, 72, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Afif, H.; Martel-Pelletier, J.; Pelletier, J.-P.; Li, X.; Farrajota, K.; Lavigne, M.; Fahmi, H. Activation of Peroxisome Proliferator-Activated Receptor Gamma Inhibits Interleukin-1beta-Induced Membrane-Associated Prostaglandin E2 Synthase-1 Expression in Human Synovial Fibroblasts by Interfering with Egr-1. J. Biol. Chem. 2004, 279, 22057–22065. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Moulin, D.; Sebillaud, S.; Koufany, M.; Galteau, M.-M.; Netter, P.; Terlain, B.; Jouzeau, J.-Y. Contrasting Effects of Peroxisome-Proliferator-Activated Receptor (PPAR)Gamma Agonists on Membrane-Associated Prostaglandin E2 Synthase-1 in IL-1beta-Stimulated Rat Chondrocytes: Evidence for PPARgamma-Independent Inhibition by 15-Deoxy-Delta12,14prostaglandin J2. Arthritis Res. Ther. 2005, 7, R1325–R1337. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakashima, K.; Kamei, D.; Masuda, S.; Ishikawa, Y.; Ishii, T.; Ohmiya, Y.; Watanabe, K.; Kudo, I. Cellular Prostaglandin E2 Production by Membrane-Bound Prostaglandin E Synthase-2 via Both Cyclooxygenases-1 and -2. J. Biol. Chem. 2003, 278, 37937–37947. [Google Scholar] [CrossRef]
- Jania, L.A.; Chandrasekharan, S.; Backlund, M.G.; Foley, N.A.; Snouwaert, J.; Wang, I.-M.; Clark, P.; Audoly, L.P.; Koller, B.H. Microsomal Prostaglandin E Synthase-2 Is Not Essential for in Vivo Prostaglandin E2 Biosynthesis. Prostaglandins Other Lipid Mediat. 2009, 88, 73–81. [Google Scholar] [CrossRef]
- Tanioka, T.; Nakatani, Y.; Semmyo, N.; Murakami, M.; Kudo, I. Molecular Identification of Cytosolic Prostaglandin E2 Synthase That Is Functionally Coupled with Cyclooxygenase-1 in Immediate Prostaglandin E2 Biosynthesis. J. Biol. Chem. 2000, 275, 32775–32782. [Google Scholar] [CrossRef]
- Young, W.J.; Lahrouchi, N.; Isaacs, A.; Duong, T.; Foco, L.; Ahmed, F.; Brody, J.A.; Salman, R.; Noordam, R.; Benjamins, J.-W.; et al. Genetic Analyses of the Electrocardiographic QT Interval and Its Components Identify Additional Loci and Pathways. Nat. Commun. 2022, 13, 5144. [Google Scholar] [CrossRef]
- Cárcel-Márquez, J.; Muiño, E.; Gallego-Fabrega, C.; Cullell, N.; Lledós, M.; Llucià-Carol, L.; Sobrino, T.; Campos, F.; Castillo, J.; Freijo, M.; et al. A Polygenic Risk Score Based on a Cardioembolic Stroke Multitrait Analysis Improves a Clinical Prediction Model for This Stroke Subtype. Front. Cardiovasc. Med. 2022, 9, 940696. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A Cross-Population Atlas of Genetic Associations for 220 Human Phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Roselli, C.; Chaffin, M.D.; Weng, L.-C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-Ethnic Genome-Wide Association Study for Atrial Fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef]
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Genetic Analysis of over 1 Million People Identifies 535 New Loci Associated with Blood Pressure Traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef] [PubMed]
- Gieger, C.; Radhakrishnan, A.; Cvejic, A.; Tang, W.; Porcu, E.; Pistis, G.; Serbanovic-Canic, J.; Elling, U.; Goodall, A.H.; Labrune, Y.; et al. New Gene Functions in Megakaryopoiesis and Platelet Formation. Nature 2011, 480, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Eicher, J.D.; Chami, N.; Kacprowski, T.; Nomura, A.; Chen, M.-H.; Yanek, L.R.; Tajuddin, S.M.; Schick, U.M.; Slater, A.J.; Pankratz, N.; et al. Platelet-Related Variants Identified by Exomechip Meta-Analysis in 157,293 Individuals. Am. J. Hum. Genet. 2016, 99, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.C.; Fedirko, V.; Baron, J.A.; Barry, E.L.; Flanders, W.D.; McCullough, M.L.; Yacoub, R.; Raavi, T.; Rutherford, R.E.; Seabrook, M.E.; et al. Inflammation Modulation by Vitamin D and Calcium in the Morphologically Normal Colorectal Mucosa of Patients with Colorectal Adenoma in a Clinical Trial. Cancer Prev. Res. 2021, 14, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.; Travers, J.G.; Headrick, A.L.; Enyart, B.T.; Lemieux, M.E.; Cavasin, M.A.; Schwisow, J.A.; Hardy, E.J.; Kaltenbacher, K.J.; Felisbino, M.B.; et al. Inhibition of Eicosanoid Degradation Mitigates Fibrosis of the Heart. Circ. Res. 2023, 132, 10–29. [Google Scholar] [CrossRef]
- Kakavandi, N.; Rezaee, S.; Hosseini-Fard, S.R.; Ghasempour, G.; Khosravi, M.; Shabani, M.; Najafi, M. Prostaglandin E2 (PGE2) Synthesis Pathway Is Involved in Coronary Artery Stenosis and Restenosis. Gene 2021, 765, 145131. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, Y.-K.; Park, H.-W.; Jee, Y.-K.; Kim, S.-H.; Bahn, J.-W.; Chang, Y.-S.; Kim, S.-H.; Ye, Y.-M.; Shin, E.-S.; et al. Association between Polymorphisms in Prostanoid Receptor Genes and Aspirin-Intolerant Asthma. Pharm. Genom. 2007, 17, 295–304. [Google Scholar] [CrossRef]
- Plaza-Serón, M.D.C.; Ayuso, P.; Pérez-Sánchez, N.; Doña, I.; Blanca-Lopez, N.; Flores, C.; Galindo, L.; Molina, A.; Perkins, J.R.; Cornejo-García, J.A.; et al. Copy Number Variation in ALOX5 and PTGER1 Is Associated with NSAIDs-Induced Urticaria and/or Angioedema. Pharm. Genom. 2016, 26, 280–287. [Google Scholar] [CrossRef]
- Park, B.-L.; Park, S.-M.; Park, J.-S.; Uh, S.-T.; Choi, J.-S.; Kim, Y.-H.; Kim, M.-K.; Choi, I.S.; Choi, B.-W.; Cho, S.-H.; et al. Association of PTGER Gene Family Polymorphisms with Aspirin Intolerant Asthma in Korean Asthmatics. BMB Rep. 2010, 43, 445–449. [Google Scholar] [CrossRef]
- Palikhe, N.S.; Sin, H.J.; Kim, S.H.; Sin, H.J.; Hwang, E.K.; Ye, Y.M.; Park, H.-S. Genetic Variability of Prostaglandin E2 Receptor Subtype EP4 Gene in Aspirin-Intolerant Chronic Urticaria. J. Hum. Genet. 2012, 57, 494–499. [Google Scholar] [CrossRef]
- González, L.M.; Robles, N.R.; Mota-Zamorano, S.; Valdivielso, J.M.; López-Gómez, J.; Gervasini, G. Genetic Variants in PGE2 Receptors Modulate the Risk of Nephrosclerosis and Clinical Outcomes in These Patients. J. Pers. Med. 2021, 11, 772. [Google Scholar] [CrossRef] [PubMed]
- Sõber, S.; Org, E.; Kepp, K.; Juhanson, P.; Eyheramendy, S.; Gieger, C.; Lichtner, P.; Klopp, N.; Veldre, G.; Viigimaa, M.; et al. Targeting 160 Candidate Genes for Blood Pressure Regulation with a Genome-Wide Genotyping Array. PLoS ONE 2009, 4, e6034. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Sanak, M.; Rostoff, P.; Piwowarska, W.; Jakiela, B.; Szczeklik, A. Common Polymorphisms of Cyclooxygenase-2 and Prostaglandin E2 Receptor and Increased Risk for Acute Coronary Syndrome in Coronary Artery Disease. Thromb. Haemost. 2008, 100, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, N. Thromboxane A2: Physiology/Pathophysiology, Cellular Signal Transduction and Pharmacology. Pharmacol. Ther. 2008, 118, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Rocca, B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front. Pharmacol. 2019, 10, 1244. [Google Scholar] [CrossRef]
- Rucker, D.; Dhamoon, A.S. Physiology, Thromboxane A2. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Raychowdhury, M.K.; Yukawa, M.; Collins, L.J.; McGrail, S.H.; Kent, K.C.; Ware, J.A. Alternative Splicing Produces a Divergent Cytoplasmic Tail in the Human Endothelial Thromboxane A2 Receptor. J. Biol. Chem. 1994, 269, 19256–19261. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Activation of Platelet Function through G Protein-Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef]
- Scridon, A. Platelets and Their Role in Hemostasis and Thrombosis-From Physiology to Pathophysiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 12772. [Google Scholar] [CrossRef]
- Ozen, G.; Norel, X. Prostanoids in the Pathophysiology of Human Coronary Artery. Prostaglandins Other Lipid Mediat. 2017, 133, 20–28. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet Activation and Prothrombotic Mediators at the Nexus of Inflammation and Atherosclerosis: Potential Role of Antiplatelet Agents. Blood Rev. 2021, 45, 100694. [Google Scholar] [CrossRef] [PubMed]
- Martin, W. The Combined Role of Atheroma, Cholesterol, Platelets, the Endothelium and Fibrin in Heart Attacks and Strokes. Med. Hypotheses 1984, 15, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Liel, N.; Trask, J.L.; Mais, D.E.; Assey, M.E.E.; Halushka, P.V. Increased Platelet Thromboxane A2/Prostaglandin H2 Receptors in Patients with Acute Myocardial Infarction. Circulation 1990, 81, 212–218. [Google Scholar] [CrossRef]
- Gabrielsen, A.; Qiu, H.; Bäck, M.; Hamberg, M.; Hemdahl, A.-L.; Agardh, H.; Folkersen, L.; Swedenborg, J.; Hedin, U.; Paulsson-Berne, G.; et al. Thromboxane Synthase Expression and Thromboxane A2 Production in the Atherosclerotic Lesion. J. Mol. Med. 2010, 88, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Kawakami, M.; Hidaka, T.; Matsuki, Y.; Takamizawa, M.; Suzuki, K.; Kurita, A.; Nakamura, H. Stimulation with Thromboxane A2 (TXA2) Receptor Agonist Enhances ICAM-1, VCAM-1 or ELAM-1 Expression by Human Vascular Endothelial Cells. Clin. Exp. Immunol. 1998, 112, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tahara, Y.; Matsumoto, M.; Iguchi, M.; Sano, H.; Murayama, T.; Arai, H.; Oida, H.; Yurugi-Kobayashi, T.; Yamashita, J.K.; et al. Roles of Thromboxane A(2) and Prostacyclin in the Development of Atherosclerosis in ApoE-Deficient Mice. J. Clin. Investig. 2004, 114, 784–794. [Google Scholar] [CrossRef]
- Keen, H.L.; Brands, M.W.; Smith, M.J.; Shek, E.W.; Hall, J.E. Thromboxane Is Required for Full Expression of Angiotensin Hypertension in Rats. Hypertension 1997, 29, 310–314. [Google Scholar] [CrossRef][Green Version]
- Francois, H.; Athirakul, K.; Mao, L.; Rockman, H.; Coffman, T.M. Role for Thromboxane Receptors in Angiotensin-II-Induced Hypertension. Hypertension 2004, 43, 364–369. [Google Scholar] [CrossRef]
- Thomas, D.W.; Mannon, R.B.; Mannon, P.J.; Latour, A.; Oliver, J.A.; Hoffman, M.; Smithies, O.; Koller, B.H.; Coffman, T.M. Coagulation Defects and Altered Hemodynamic Responses in Mice Lacking Receptors for Thromboxane A2. J. Clin. Investig. 1998, 102, 1994–2001. [Google Scholar] [CrossRef]
- Sparks, M.A.; Makhanova, N.A.; Griffiths, R.C.; Snouwaert, J.N.; Koller, B.H.; Coffman, T.M. Thromboxane Receptors in Smooth Muscle Promote Hypertension, Vascular Remodeling, and Sudden Death. Hypertension 2013, 61, 166–173. [Google Scholar] [CrossRef]
- Montalescot, G.; Drobinski, G.; Maclouf, J.; Maillet, F.; Salloum, J.; Ankri, A.; Kazatchkine, M.; Eugène, L.; Thomas, D.; Grosgogeat, Y. Evaluation of Thromboxane Production and Complement Activation during Myocardial Ischemia in Patients with Angina Pectoris. Circulation 1991, 84, 2054–2062. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Santilli, F.; Davì, G.; Basili, S.; Lattanzio, S.; Cavoni, A.; Guizzardi, G.; De Feudis, L.; Traisci, G.; Pettinella, C.; Paloscia, L.; et al. Thromboxane and Prostacyclin Biosynthesis in Heart Failure of Ischemic Origin: Effects of Disease Severity and Aspirin Treatment. J. Thromb. Haemost. 2010, 8, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.J.; Best, S.R.; Kosloski, L.M.; Stachura, C.J.; Smoot, R.L.; Porter, C.B.; Orr, J.A. Thromboxane A2-Induced Arrhythmias in the Anesthetized Rabbit. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1353–H1361. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.J.; Kosloski, L.M.; Gilbert, W.J.R.; Touchberry, C.D.; Moore, D.S.; Kelly, J.K.; Brotto, M.; Orr, J.A. Inhibition of Thromboxane A2-Induced Arrhythmias and Intracellular Calcium Changes in Cardiac Myocytes by Blockade of the Inositol Trisphosphate Pathway. J. Pharmacol. Exp. Ther. 2009, 331, 917–924. [Google Scholar] [CrossRef]
- Touchberry, C.D.; Silswal, N.; Tchikrizov, V.; Elmore, C.J.; Srinivas, S.; Akthar, A.S.; Swan, H.K.; Wetmore, L.A.; Wacker, M.J. Cardiac Thromboxane A2 Receptor Activation Does Not Directly Induce Cardiomyocyte Hypertrophy but Does Cause Cell Death That Is Prevented with Gentamicin and 2-APB. BMC Pharmacol. Toxicol. 2014, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Fuse, I.; Higuchi, W.; Aizawa, Y. Pathogenesis of a Bleeding Disorder Characterized by Platelet Unresponsiveness to Thromboxane A2. Semin. Thromb. Hemost. 2000, 26, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Mesitskaya, D.F.; Syrkin, A.L.; Aksenova, M.G.; Zhang, Y.; Zamyatnin, A.A.; Kopylov, P.Y. Thromboxane A Synthase: A New Target for the Treatment of Cardiovascular Diseases. Cardiovasc. Hematol. Agents Med. Chem. 2018, 16, 81–87. [Google Scholar] [CrossRef]
- Capra, V.; Bäck, M.; Angiolillo, D.J.; Cattaneo, M.; Sakariassen, K.S. Impact of Vascular Thromboxane Prostanoid Receptor Activation on Hemostasis, Thrombosis, Oxidative Stress, and Inflammation. J. Thromb. Haemost. 2014, 12, 126–137. [Google Scholar] [CrossRef]
- Cyrus, T.; Ding, T.; Praticò, D. Expression of Thromboxane Synthase, Prostacyclin Synthase and Thromboxane Receptor in Atherosclerotic Lesions: Correlation with Plaque Composition. Atherosclerosis 2010, 208, 376–381. [Google Scholar] [CrossRef]
- Tang, E.H.C.; Vanhoutte, P.M. Gene Expression Changes of Prostanoid Synthases in Endothelial Cells and Prostanoid Receptors in Vascular Smooth Muscle Cells Caused by Aging and Hypertension. Physiol. Genom. 2008, 32, 409–418. [Google Scholar] [CrossRef]
- Chen, H. Role of Thromboxane A2 Signaling in Endothelium-Dependent Contractions of Arteries. Prostaglandins Other Lipid Mediat. 2018, 134, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-H.; Kim, Y.-H.; Park, S.-M.; Cho, S.-H.; Park, J.-S.; Jang, A.-S.; Park, S.-W.; Uh, S.-T.; Lee, Y.-M.; Kim, M.-K.; et al. Association Analysis of Thromboxane A Synthase 1 Gene Polymorphisms with Aspirin Intolerance in Asthmatic Patients. Pharmacogenomics 2011, 12, 351–363. [Google Scholar] [CrossRef]
- Peng, J.; Lu, F.; Zhong, M.; Zhao, Y.; Wang, Z.; Zhang, W. TBXAS1 Gene Polymorphism Is Associated with the Risk of Ischemic Stroke of Metabolic Syndrome in a Chinese Han Population. Dis. Mrk. 2022, 2022, 9717510. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Z.-Y.; Wang, Y.-Z.; Liu, X.; Yuan, L.-Y. Associations between Thromboxane A Synthase 1 Gene Polymorphisms and the Risk of Ischemic Stroke in a Chinese Han Population. Neural Regen. Res. 2018, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Lin, J.; Wang, C.; Huang, R.; Liu, Y. Interactions among Variants in Eicosanoid Genes Increase Risk of Atherothrombotic Stroke in Chinese Populations. J. Stroke Cerebrovasc. Dis. 2017, 26, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Lin, J.; Luo, H.; Wang, C.; Liu, Y. Genetic Variants of PTGS2, TXA2R and TXAS1 Are Associated with Carotid Plaque Vulnerability, Platelet Activation and TXA2 Levels in Ischemic Stroke Patients. PLoS ONE 2017, 12, e0180704. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Ming, B.; Wang, C.; Chen, H.; Ma, C. Variants in COX-2, PTGIS, and TBXAS1 Are Associated with Carotid Artery or Intracranial Arterial Stenosis and Neurologic Deterioration in Ischemic Stroke Patients. J. Stroke Cerebrovasc. Dis. 2017, 26, 1128–1135. [Google Scholar] [CrossRef]
- Mundell, S.J.; Mumford, A. TBXA2R Gene Variants Associated with Bleeding. Platelets 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Peng, L.-L.; Zhao, Y.-Q.; Zhou, Z.-Y.; Jin, J.; Zhao, M.; Chen, X.-M.; Chen, L.-Y.; Cai, Y.-F.; Li, J.-L.; Huang, M. Associations of MDR1, TBXA2R, PLA2G7, and PEAR1 Genetic Polymorphisms with the Platelet Activity in Chinese Ischemic Stroke Patients Receiving Aspirin Therapy. Acta Pharmacol. Sin. 2016, 37, 1442–1448. [Google Scholar] [CrossRef]
- Liang, X.; Zhou, Y.; Li, S. Association of TBXA2R, P2Y12 and ADD1 Genes Polymorphisms with Ischemic Stroke Susceptibility: A Metaanalysis. Clin. Investig. Med. 2020, 43, E33–E43. [Google Scholar] [CrossRef]
- Palikhe, N.S.; Kim, S.-H.; Lee, H.-Y.; Kim, J.-H.; Ye, Y.-M.; Park, H.-S. Association of Thromboxane A2 Receptor (TBXA2R) Gene Polymorphism in Patients with Aspirin-Intolerant Acute Urticaria. Clin. Exp. Allergy 2011, 41, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Choi, J.-H.; Park, H.-S.; Holloway, J.W.; Lee, S.-K.; Park, C.-S.; Shin, H.-D. Association of Thromboxane A2 Receptor Gene Polymorphism with the Phenotype of Acetyl Salicylic Acid-Intolerant Asthma. Clin. Exp. Allergy 2005, 35, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, S.; Xie, X.; Li, M. Association between Thromboxane A2 Receptor Polymorphisms and Asthma Risk: A Meta-Analysis. J. Asthma 2016, 53, 576–582. [Google Scholar] [CrossRef]
- Dorris, S.L.; Peebles, R.S. PGI2 as a Regulator of Inflammatory Diseases. Mediat. Inflamm. 2012, 2012, 926968. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Kirkby, N.S. Eicosanoids, Prostacyclin and Cyclooxygenase in the Cardiovascular System. Br. J. Pharmacol. 2019, 176, 1038–1050. [Google Scholar] [CrossRef]
- Cheng, Y.; Austin, S.C.; Rocca, B.; Koller, B.H.; Coffman, T.M.; Grosser, T.; Lawson, J.A.; FitzGerald, G.A. Role of Prostacyclin in the Cardiovascular Response to Thromboxane A2. Science 2002, 296, 539–541. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Shala, F.; Elghazouli, Y.; Warner, T.D.; Gaston-Massuet, C.; Crescente, M.; Armstrong, P.C.; Herschman, H.R.; Kirkby, N.S. Cell-Specific Gene Deletion Reveals the Antithrombotic Function of COX1 and Explains the Vascular COX1/Prostacyclin Paradox. Circ. Res. 2019, 125, 847–854. [Google Scholar] [CrossRef]
- Luttmann, W.; Herzog, V.; Virchow, J.C.; Matthys, H.; Thierauch, K.H.; Kroegel, C. Prostacyclin Modulates Granulocyte/Macrophage Colony-Stimulating Factor Release by Human Blood Mononuclear Cells. Pulm. Pharmacol. 1996, 9, 43–48. [Google Scholar] [CrossRef]
- Ipseiz, N.; Pickering, R.J.; Rosas, M.; Tyrrell, V.J.; Davies, L.C.; Orr, S.J.; Czubala, M.A.; Fathalla, D.; Robertson, A.A.; Bryant, C.E.; et al. Tissue-Resident Macrophages Actively Suppress IL-1beta Release via a Reactive Prostanoid/IL-10 Pathway. EMBO J. 2020, 39, e103454. [Google Scholar] [CrossRef]
- Zhou, W.; Blackwell, T.S.; Goleniewska, K.; O’Neal, J.F.; Fitzgerald, G.A.; Lucitt, M.; Breyer, R.M.; Peebles, R.S. Prostaglandin I2 Analogs Inhibit Th1 and Th2 Effector Cytokine Production by CD4 T Cells. J. Leukoc. Biol. 2007, 81, 809–817. [Google Scholar] [CrossRef]
- Zhou, W.; Hashimoto, K.; Goleniewska, K.; O’Neal, J.F.; Ji, S.; Blackwell, T.S.; Fitzgerald, G.A.; Egan, K.M.; Geraci, M.W.; Peebles, R.S. Prostaglandin I2 Analogs Inhibit Proinflammatory Cytokine Production and T Cell Stimulatory Function of Dendritic Cells. J. Immunol. 2007, 178, 702–710. [Google Scholar] [CrossRef]
- Di Francesco, L.; Totani, L.; Dovizio, M.; Piccoli, A.; Di Francesco, A.; Salvatore, T.; Pandolfi, A.; Evangelista, V.; Dercho, R.A.; Seta, F.; et al. Induction of Prostacyclin by Steady Laminar Shear Stress Suppresses Tumor Necrosis Factor-Alpha Biosynthesis via Heme Oxygenase-1 in Human Endothelial Cells. Circ. Res. 2009, 104, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Luttmann, W.; Herzog, V.; Matthys, H.; Thierauch, K.H.; Virchow, J.C.; Kroegel, C. Modulation of Cytokine Release from Mononuclear Cells by Prostacyclin, IL-4 and IL-13. Cytokine 1999, 11, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Ushikubi, F.; Matsuoka, T.; Hirata, M.; Yamasaki, A.; Sugimoto, Y.; Ichikawa, A.; Aze, Y.; Tanaka, T.; Yoshida, N.; et al. Altered Pain Perception and Inflammatory Response in Mice Lacking Prostacyclin Receptor. Nature 1997, 388, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Francois, H.; Athirakul, K.; Howell, D.; Dash, R.; Mao, L.; Kim, H.-S.; Rockman, H.A.; Fitzgerald, G.A.; Koller, B.H.; Coffman, T.M. Prostacyclin Protects against Elevated Blood Pressure and Cardiac Fibrosis. Cell Metab. 2005, 2, 201–207. [Google Scholar] [CrossRef]
- Hara, A.; Yuhki, K.; Fujino, T.; Yamada, T.; Takayama, K.; Kuriyama, S.; Takahata, O.; Karibe, H.; Okada, Y.; Xiao, C.-Y.; et al. Augmented Cardiac Hypertrophy in Response to Pressure Overload in Mice Lacking the Prostaglandin I2 Receptor. Circulation 2005, 112, 84–92. [Google Scholar] [CrossRef]
- Chen, H.-H.; Chen, T.-W.; Lin, H. Prostacyclin-Induced Peroxisome Proliferator-Activated Receptor-Alpha Translocation Attenuates NF-KappaB and TNF-Alpha Activation after Renal Ischemia-Reperfusion Injury. Am. J. Physiol. Renal. Physiol. 2009, 297, F1109–F1118. [Google Scholar] [CrossRef]
- Chen, R.; Xue, J.; Xie, M.-L. Reduction of Isoprenaline-Induced Myocardial TGF-Β1 Expression and Fibrosis in Osthole-Treated Mice. Toxicol. Appl. Pharmacol. 2011, 256, 168–173. [Google Scholar] [CrossRef]
- Yu, H.; Gallagher, A.M.; Garfin, P.M.; Printz, M.P. Prostacyclin Release by Rat Cardiac Fibroblasts: Inhibition of Collagen Expression. Hypertension 1997, 30, 1047–1053. [Google Scholar] [CrossRef]
- Li, Y.; Connolly, M.; Nagaraj, C.; Tang, B.; Bálint, Z.; Popper, H.; Smolle-Juettner, F.M.; Lindenmann, J.; Kwapiszewska, G.; Aaronson, P.I.; et al. Peroxisome Proliferator-Activated Receptor-β/δ, the Acute Signaling Factor in Prostacyclin-Induced Pulmonary Vasodilation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 372–379. [Google Scholar] [CrossRef]
- Miyata, A.; Hara, S.; Yokoyama, C.; Inoue, H.; Ullrich, V.; Tanabe, T. Molecular Cloning and Expression of Human Prostacyclin Synthase. Biochem. Biophys. Res. Commun. 1994, 200, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Honsawa, T.; Sasaki, Y.; Hara, S. Prostacyclin Synthase as an Ambivalent Regulator of Inflammatory Reactions. Biol. Pharm. Bull. 2022, 45, 979–984. [Google Scholar] [CrossRef]
- Yokoyama, C.; Yabuki, T.; Shimonishi, M.; Wada, M.; Hatae, T.; Ohkawara, S.; Takeda, J.; Kinoshita, T.; Okabe, M.; Tanabe, T. Prostacyclin-Deficient Mice Develop Ischemic Renal Disorders, Including Nephrosclerosis and Renal Infarction. Circulation 2002, 106, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Todaka, T.; Yokoyama, C.; Yanamoto, H.; Hashimoto, N.; Nagata, I.; Tsukahara, T.; Hara, S.; Hatae, T.; Morishita, R.; Aoki, M.; et al. Gene Transfer of Human Prostacyclin Synthase Prevents Neointimal Formation after Carotid Balloon Injury in Rats. Stroke 1999, 30, 419–426. [Google Scholar] [CrossRef]
- Liu, Q.; Xi, Y.; Terry, T.; So, S.-P.; Mohite, A.; Zhang, J.; Wu, G.; Liu, X.; Cheng, J.; Ruan, K.-H.; et al. Engineered Endothelial Progenitor Cells That Overexpress Prostacyclin Protect Vascular Cells. J. Cell Physiol. 2012, 227, 2907–2916. [Google Scholar] [CrossRef]
- Geraci, M.W.; Gao, B.; Shepherd, D.C.; Moore, M.D.; Westcott, J.Y.; Fagan, K.A.; Alger, L.A.; Tuder, R.M.; Voelkel, N.F. Pulmonary Prostacyclin Synthase Overexpression in Transgenic Mice Protects against Development of Hypoxic Pulmonary Hypertension. J. Clin. Investig. 1999, 103, 1509–1515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin Synthase Expression Is Decreased in Lungs from Patients with Severe Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.; Tatsumi, K.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Takiguchi, Y.; Kasuya, Y.; Kimura, S.; Kuriyama, T. Polymorphism of the Promoter Region of Prostacyclin Synthase Gene in Chronic Thromboembolic Pulmonary Hypertension. Respirology 2004, 9, 184–189. [Google Scholar] [CrossRef]
- Stearman, R.S.; Cornelius, A.R.; Lu, X.; Conklin, D.S.; Del Rosario, M.J.; Lowe, A.M.; Elos, M.T.; Fettig, L.M.; Wong, R.E.; Hara, N.; et al. Functional Prostacyclin Synthase Promoter Polymorphisms. Impact in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 1110–1120. [Google Scholar] [CrossRef]
- Xie, X.; Ma, Y.-T.; Fu, Z.-Y.; Yang, Y.-N.; Ma, X.; Chen, B.-D.; Wang, Y.-H.; Liu, F. Haplotype Analysis of the CYP8A1 Gene Associated with Myocardial Infarction. Clin. Appl. Thromb. Hemost. 2009, 15, 574–580. [Google Scholar] [CrossRef]
- Reid, H.M.; Kinsella, B.T. Prostacyclin Receptors: Transcriptional Regulation and Novel Signalling Mechanisms. Prostaglandins Other Lipid Mediat. 2015, 121, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Di Febbo, C.; Tacconelli, S.; Douville, K.; Guglielmi, M.D.; Horvath, R.J.; Ding, M.; Sierra, K.; Stitham, J.; Gleim, S.; et al. Differential Association between Human Prostacyclin Receptor Polymorphisms and the Development of Venous Thrombosis and Intimal Hyperplasia: A Clinical Biomarker Study. Pharm. Genom. 2008, 18, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Stitham, J.; Arehart, E.; Elderon, L.; Gleim, S.R.; Douville, K.; Kasza, Z.; Fetalvero, K.; MacKenzie, T.; Robb, J.; Martin, K.A.; et al. Comprehensive Biochemical Analysis of Rare Prostacyclin Receptor Variants: Study of Association of Signaling with Coronary Artery Obstruction. J. Biol. Chem. 2011, 286, 7060–7069. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Boeglin, W.E.; Yin, H.; Stec, D.F.; Voehler, M. Convergent Oxygenation of Arachidonic Acid by 5-Lipoxygenase and Cyclooxygenase-2. J. Am. Chem. Soc. 2006, 128, 720–721. [Google Scholar] [CrossRef][Green Version]
- Mulugeta, S.; Suzuki, T.; Hernandez, N.T.; Griesser, M.; Boeglin, W.E.; Schneider, C. Identification and Absolute Configuration of Dihydroxy-Arachidonic Acids Formed by Oxygenation of 5S-HETE by Native and Aspirin-Acetylated COX-2. J. Lipid Res. 2010, 51, 575–585. [Google Scholar] [CrossRef]
- Griesser, M.; Suzuki, T.; Tejera, N.; Mont, S.; Boeglin, W.E.; Pozzi, A.; Schneider, C. Biosynthesis of Hemiketal Eicosanoids by Cross-over of the 5-Lipoxygenase and Cyclooxygenase-2 Pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 6945–6950. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Shibata, T.; Uchida, K.; Schneider, C. Roles of 5-Lipoxygenase and Cyclooxygenase-2 in the Biosynthesis of Hemiketals E2 and D2 by Activated Human Leukocytes. FASEB J. 2017, 31, 1867–1878. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Suzuki, T.; Sprinkel, K.C.; Boeglin, W.E.; Schneider, C. Biomimetic Synthesis of Hemiketal Eicosanoids for Biological Testing. Prostaglandins Other Lipid Mediat. 2017, 132, 41–46. [Google Scholar] [CrossRef]
- Boer, R.E.; Giménez-Bastida, J.A.; Boutaud, O.; Jana, S.; Schneider, C.; Sulikowski, G.A. Total Synthesis and Biological Activity of the Arachidonic Acid Metabolite Hemiketal E2. Org. Lett. 2018, 20, 4020–4022. [Google Scholar] [CrossRef]
- Nakashima, F.; Suzuki, T.; Gordon, O.N.; Golding, D.; Okuno, T.; Giménez-Bastida, J.A.; Yokomizo, T.; Schneider, C. Biosynthetic Crossover of 5-Lipoxygenase and Cyclooxygenase-2 Yields 5-Hydroxy-PGE2 and 5-Hydroxy-PGD2. JACS Au 2021, 1, 1380–1388. [Google Scholar] [CrossRef]
- Schmid, T.; Brüne, B. Prostanoids and Resolution of Inflammation—Beyond the Lipid-Mediator Class Switch. Front. Immunol. 2021, 12, 714042. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
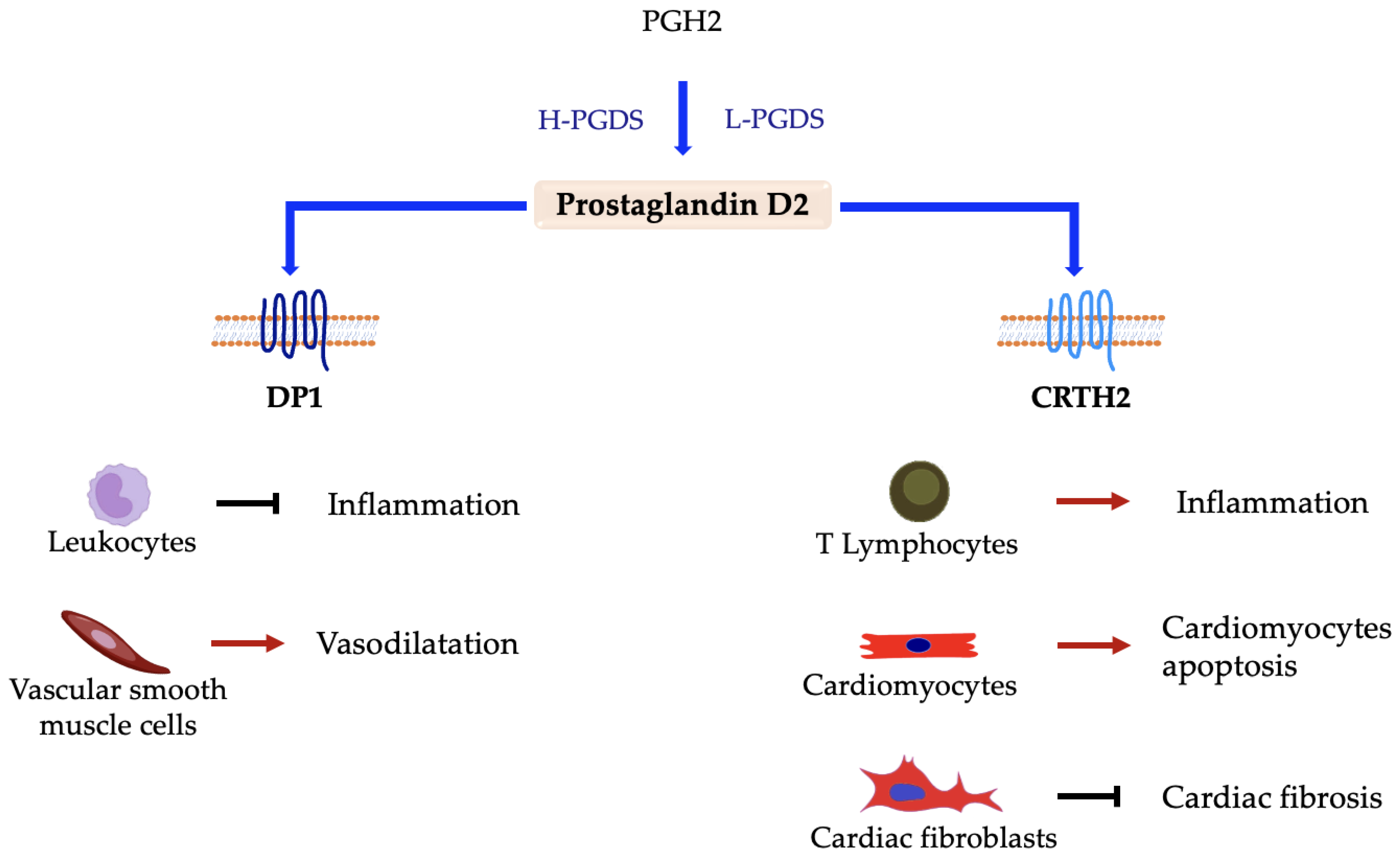{kind=link}
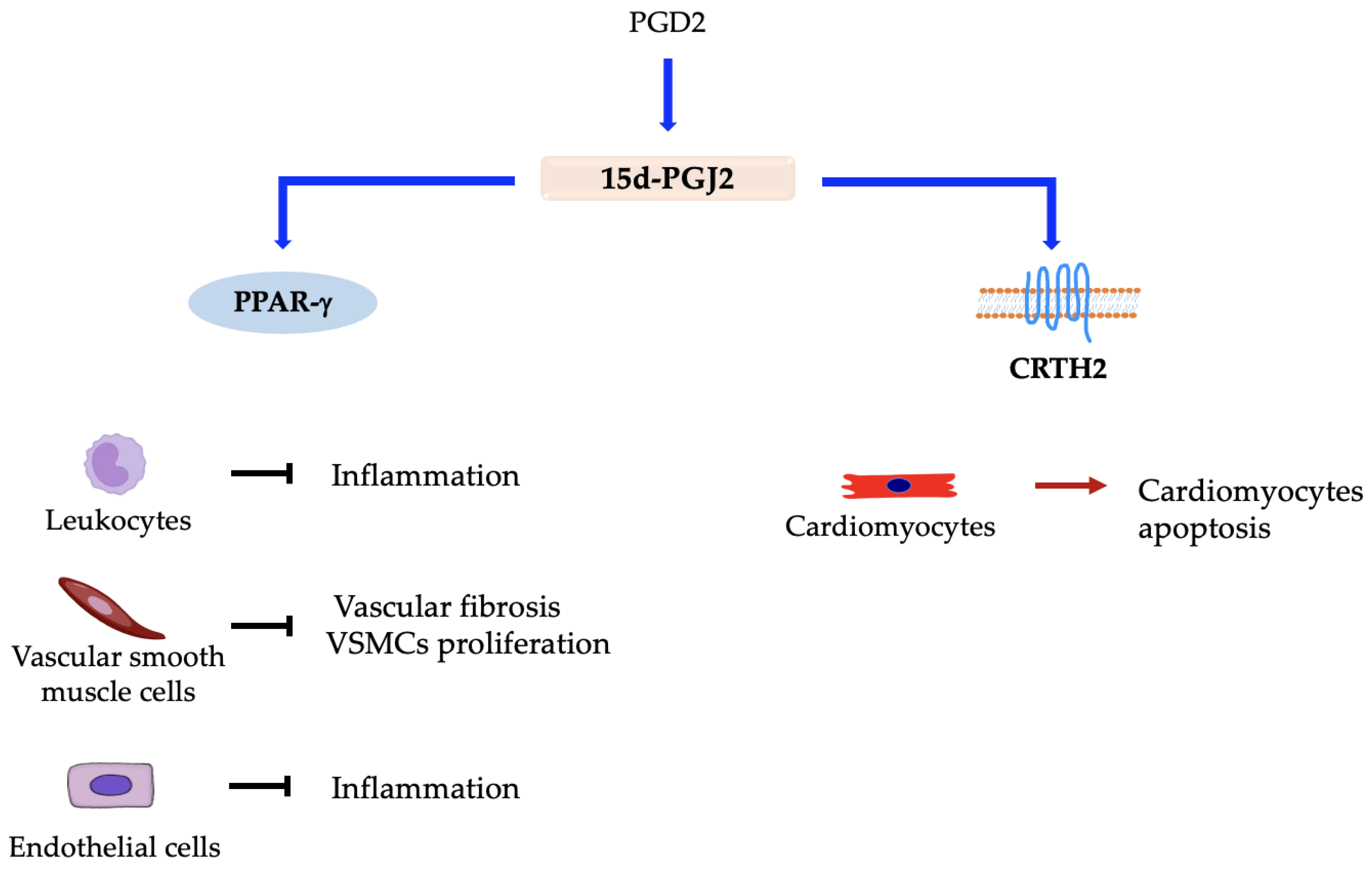{kind=link}
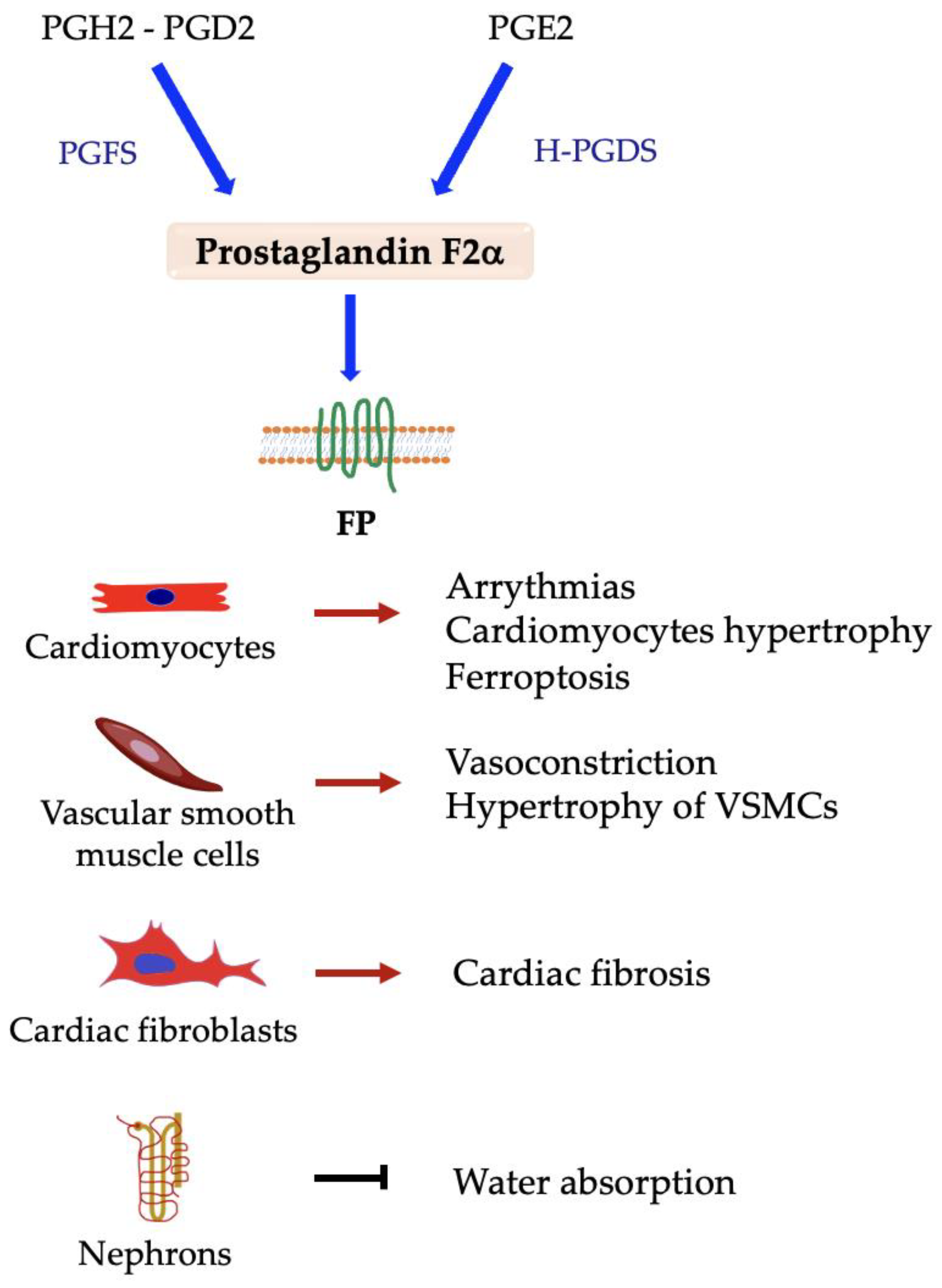{kind=link}
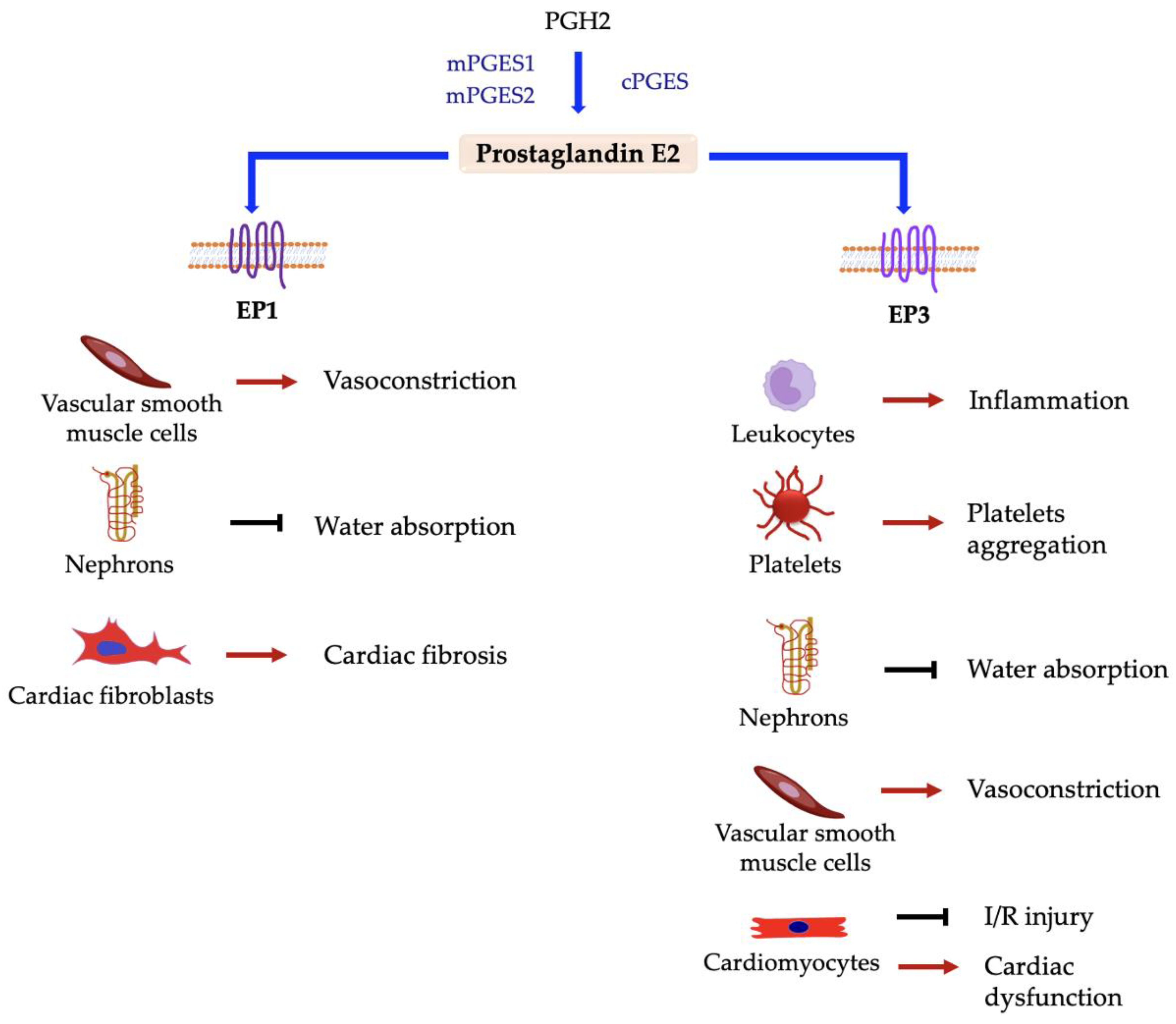{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prostanoid | Receptor | G-Protein | Signaling Pathway |
|---|---|---|---|
| PGD2 | DP1 | Gαs | AC and PKA activation—cAMP increase |
| CRTH2 | Gαi | cAMP decrease—Ca2+ increase | |
| PGF2α | FP | Gαq | IP3 and Ca2+ increase |
| Gα12/13 | Rho activation | ||
| PGE2 | EP1 | Gαq | IP3 and Ca2+ increase |
| EP2 | Gαs | AC and PKA activation—cAMP increase | |
| EP3 | Gαi or Gα12 | Ca2+ increase—Rho activation | |
| EP4 | Gαs | AC and PKA activation—cAMP increase | |
| TxA2 | TPα-TPβ | Gαq | IP3 and Ca2+ increase |
| Gα12/13 | Rho activation | ||
| PGI2 | IP | Gαs | AC and PKA activation—cAMP increase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beccacece, L.; Abondio, P.; Bini, C.; Pelotti, S.; Luiselli, D. The Link between Prostanoids and Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 4193. https://doi.org/10.3390/ijms24044193
Beccacece L, Abondio P, Bini C, Pelotti S, Luiselli D. The Link between Prostanoids and Cardiovascular Diseases. International Journal of Molecular Sciences. 2023; 24(4):4193. https://doi.org/10.3390/ijms24044193
Chicago/Turabian StyleBeccacece, Livia, Paolo Abondio, Carla Bini, Susi Pelotti, and Donata Luiselli. 2023. "The Link between Prostanoids and Cardiovascular Diseases" International Journal of Molecular Sciences 24, no. 4: 4193. https://doi.org/10.3390/ijms24044193
APA StyleBeccacece, L., Abondio, P., Bini, C., Pelotti, S., & Luiselli, D. (2023). The Link between Prostanoids and Cardiovascular Diseases. International Journal of Molecular Sciences, 24(4), 4193. https://doi.org/10.3390/ijms24044193