
Methylphenidate Promotes Premature Growth Plate Closure: In Vitro Evidence

 , ,
, ,  , , and
, , and 
Abstract
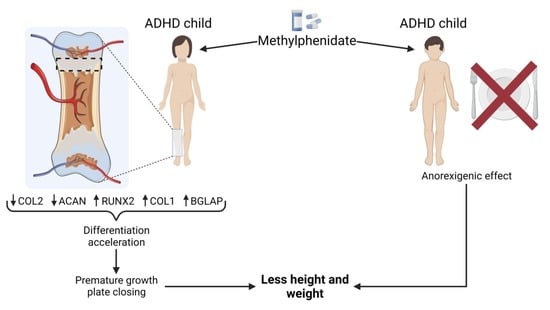
1. Introduction
2. Results
2.1. MPH Does Not Affect Chondrocyte Viability and Proliferation
2.2. MPH Treatment Alters In Vitro Chondrocyte Differentiation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. MTT Assay
4.4. RT-PCR
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Danielson, M.L.; Bitsko, R.H.; Ghandour, R.M.; Holbrook, J.R.; Kogan, M.D.; Blumberg, S.J. Prevalence of parent-reported ADHD diagnosis and associated treatment Among U.S. children and adolescents 2016. J. Clin. Child Adolesc. Psychol. 2018, 47, 199–212. [Google Scholar] [CrossRef]
- Martella, D.; Aldunate, N.; Fuentes, L.J.; Sánchez-Pérez, N. Arousal and executive alterations in attention deficit hyperactivity disorder (ADHD). Front. Psychol. 2020, 11, 1991. [Google Scholar] [CrossRef] [PubMed]
- Groenman, A.P.; Schweren, L.J.S.; Dietrich, A.; Hoekstra, P.J. An update on the safety of psychostimulants for the treatment of attention-deficit/hyperactivity disorder. Expert Opin. Drug Saf. 2017, 16, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Comim, C.M.; Gomes, K.M.; Réus, G.Z.; Petronilho, F.; Ferreira, G.K.; Streck, E.L.; Dal-Pizzol, F.; Quevedo, J. Methylphenidate treatment causes oxidative stress and alters energetic metabolism in an animal model of attention-deficit hyperactivity disorder. Acta Neuropsychiatr. 2014, 26, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Samuel, S.; Patel, D.R. Pharmacologic management of attention deficit hyperactivity disorder in children and adolescents: A review for practitioners. Transl. Pediatr. 2018, 7, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.J.; Philipsen, A.; Hoffmann, F. ADHD in Germany: Trends in Diagnosis and pharmacotherapy-a country-wide analysis of health insurance data on attention-deficit/ hyperactivity disorder (ADHD) in children, adolescents and adults from 2009–2014. Dtsch. Arztebl. Int. 2017, 114, 141–148. [Google Scholar] [CrossRef]
- Schweren, L.J.S.; de Zeeuw, P.; Durston, S. MR Imaging of the effects of methylphenidate on brain structure and function in attention-deficit/hyperactivity disorder. Eur. Neuropsychopharmacol. 2013, 23, 1151–1164. [Google Scholar] [CrossRef]
- Díez-Suárez, A.; Vallejo-Valdivielso, M.; Marín-Méndez, J.J.; De Castro-Manglano, P.; Soutullo, C.A. Weight, height, and body mass index in patients with attention-deficit/hyperactivity disorder treated with methylphenidate. J. Child Adolesc. Psychopharmacol. 2017, 27, 723–730. [Google Scholar] [CrossRef]
- Greenhill, L.L.; Swanson, J.M.; Hechtman, L.; Waxmonsky, J.; Arnold, L.E.; Molina, B.S.G.; Hinshaw, S.P.; Jensen, P.S.; Abikoff, H.B.; Wigal, T.; et al. Trajectories of growth associated with long-term stimulant medication in the multimodal treatment study of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 2020, 59, 978–989. [Google Scholar] [CrossRef]
- Waxmonsky, J.G.; Pelham, W.E.; Campa, A.; Waschbusch, D.A.; Li, T.; Marshall, R.; Babocsai, L.; Humphery, H.; Gnagy, E.; Swanson, J.; et al. A Randomized controlled trial of interventions for growth suppression in children with attention-deficit/hyperactivity disorder treated with central nervous system stimulants. J. Am. Acad. Child Adolesc. Psychiatry 2020, 59, 1330–1341. [Google Scholar] [CrossRef]
- Bou Khalil, R.; Fares, N.; Saliba, Y.; Tamraz, J.; Richa, S. L’effet de la méthylphénidate sur l’appétit et le poids. Encephale 2017, 43, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Poulton, A.S.; Bui, Q.; Melzer, E.; Evans, R. Stimulant Medication Effects on growth and bone age in children with attention-deficit/hyperactivity disorder: A prospective cohort study. Int. Clin. Psychopharmacol. 2016, 31, 93–99. [Google Scholar] [CrossRef]
- Chirokikh, A.A.; Uddin, S.M.Z.; Areikat, N.; Jones, R.; Duque, E.; Connor, C.; Hadjiargyrou, M.; Thanos, P.K.; Komatsu, D.E. Combined methylphenidate and fluoxetine treatment in adolescent rats significantly impairs weight gain with minimal effects on skeletal development. Bone 2023, 167, 116637. [Google Scholar] [CrossRef]
- Uddin, S.M.Z.; Robison, L.S.; Fricke, D.; Chernoff, E.; Hadjiargyrou, M.; Thanos, P.K.; Komatsu, D.E. Methylphenidate regulation of osteoclasts in a dose- a nd sex-dependent manner adversely affects skeletal mechanical integrity. Sci. Rep. 2018, 8, 1515. [Google Scholar] [CrossRef] [PubMed]
- Gumustas, F.; Yilmaz, I.; Sirin, D.Y.; Gumustas, S.A.; Batmaz, A.G.; Isyar, M.; Akkaya, S.; Mahirogullari, M. Chondrocyte proliferation, viability and differentiation is declined following administration of methylphenidate utilized for the treatment of attention-deficit/hyperactivity disorder. Hum. Exp. Toxicol. 2017, 36, 981–992. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Kronenberg, developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Tian, Y.; Yang, C.; Ma, X.; Wang, X.; Pei, J.; Qian, A. Mesenchymal stem cell migration during bone formation and bone diseases therapy. Int. J. Mol. Sci. 2018, 19, 2343. [Google Scholar] [CrossRef]
- Ağirdil, Y. The growth plate: A physiologic overview. EFORT Open Rev. 2020, 5, 498–507. [Google Scholar] [CrossRef]
- Kodama, J.; Wilkinson, K.J.; Iwamoto, M.; Otsuru, S.; Enomoto-Iwamoto, M. The role of hypertrophic chondrocytes in regulation of the cartilage-to-bone transition in fracture healing. Bone Rep. 2022, 17, 101616. [Google Scholar] [CrossRef]
- Hallett, S.A.; Ono, W.; Ono, N. Growth plate chondrocytes: Skeletal development, growth and beyond. Int. J. Mol. Sci. 2019, 20, 6009. [Google Scholar] [CrossRef]
- Kvist, O.; Luiza Dallora, A.; Nilsson, O.; Anderberg, P.; Sanmartin Berglund, J.; Flodmark, C.E.; Diaz, S. A cross-sectional magnetic resonance imaging study of factors influencing growth plate closure in adolescents and young adults. Acta Paediatr. 2021, 110, 1249–1256. [Google Scholar] [CrossRef]
- Koyama, E.; Mundy, C.; Saunders, C.; Chung, J.; Catheline, S.E.; Rux, D.; Iwamoto, M.; Pacifici, M. Premature growth plate closure caused by a hedgehog cancer drug is preventable by co-administration of a retinoid antagonist in mice. J. Bone Miner. Res. 2021, 36, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.G.; Schaz, U.; Storch, A.; Liebau, S.; Fegert, J.M.; Boeckers, T.M. Methylphenidate exerts no neurotoxic, but neuroprotective effects in vitro. J. Neural Transm. 2006, 113, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Balcioglu, A.; Ren, J.Q.; McCarthy, D.; Spencer, T.J.; Biederman, J.; Bhide, P.G. Plasma and brain concentrations of oral therapeutic doses of methylphenidate and their impact on brain monoamine content in mice. Neuropharmacology 2009, 57, 687. [Google Scholar] [CrossRef] [PubMed]
- Shukunami, C.; Ishizeki, K.; Atsumi, T.; Ohta, Y.; Suzuki, F.; Hiraki, Y. Cellular hypertrophy and calcification of embryonal carcinoma-derived chondrogenic cell line ATDC5 in vitro. J. Bone Miner. Res. 1997, 12, 1174–1188. [Google Scholar] [CrossRef] [PubMed]
- Newton, P.T.; Staines, K.A.; Spevak, L.; Boskey, A.L.; Teixeira, C.C.; Macrae, V.E.; Canfield, A.E.; Farquharson, C. Chondrogenic ATDC5 cells: An optimised model for rapid and physiological matrix mineralisation. Int. J. Mol. Med. 2012, 30, 1187–1193. [Google Scholar] [CrossRef]
- Yan, J.; Li, J.; Hu, J.; Zhang, L.; Wei, C.; Sultana, N.; Cai, X.; Zhang, W.; Cai, C.L. Smad4 deficiency impairs chondrocyte hypertrophy via the runx2 transcription factor in mouse skeletal development. J. Biol. Chem. 2018, 293, 9162–9175. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Bruderer, M.; Richards, R.G.; Alini, M.; Stoddart, M.J. Role and regulation of RUNX2 in osteogenesis. Eur. Cell. Mater. 2014, 28, 269–286. [Google Scholar] [CrossRef]
- Lin, X.; Patil, S.; Gao, Y.G.; Qian, A. The Bone Extracellular matrix in bone formation and regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef]
- Carvalho, M.S.; Cabral, J.M.S.; da Silva, C.L.; Vashishth, D. Synergistic Effect of extracellularly supplemented osteopontin and osteocalcin on stem cell proliferation, osteogenic differentiation, and angiogenic properties. J. Cell. Biochem. 2019, 120, 6555–6569. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.; Wang, X.; Qiu, X.; Wu, Z.; Gao, B.; Liu, L.; Liang, G.; Zhou, H.; Yang, X.; Peng, Y.; et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin Β1−SMAD1 interaction. Bone Res. 2019, 7, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.J.; Janssen, M.P.F.; Peeters, L.; Haudenschild, D.R.; Cremers, A.; Surtel, D.A.M.; van Rhijn, L.W.; Emans, P.J.; Welting, T.J.M. Aggrecan and COMP improve periosteal chondrogenesis by delaying chondrocyte hypertrophic maturation. Front. Bioeng. Biotechnol. 2020, 8, 1036. [Google Scholar] [CrossRef]
- Domowicz, M.S.; Cortes, M.; Henry, J.G.; Schwartz, N.B. Aggrecan modulation of growth plate morphogenesis. Dev. Biol. 2009, 329, 242. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.F.; Angelozzi, M.; Haseeb, A.; Lefebvre, V. SOX9 is dispensable for the initiation of epigenetic remodeling and the activation of marker genes at the onset of chondrogenesis. Development 2018, 145, dev164459. [Google Scholar] [CrossRef] [PubMed]
- Grünblatt, E.; Bartl, J.; Walitza, S. Methylphenidate Enhances neuronal differentiation and reduces proliferation concomitant to activation of wnt signal transduction pathways. Transl. Psychiatry 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Schizas, N.P.; Zafeiris, C.; Neri, A.A.; Anastasopoulos, P.P.; Papaioannou, N.A.; Dontas, I.A. Inhibition versus activation of canonical wnt-signaling, to promote chondrogenic differentiation of mesenchymal stem cells. A review. Orthop. Rev. 2021, 13, 27098. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible Growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295. [Google Scholar] [CrossRef]
- Bliziotes, M.; McLoughlin, S.; Gunness, M.; Fumagalli, F.; Jones, S.R.; Caron, M.G. Bone Histomorphometric and biomechanical abnormalities in mice homozygous for deletion of the dopamine transporter gene. Bone 2000, 26, 15–19. [Google Scholar] [CrossRef]
- Bliziotes, M.M.; Eshleman, A.J.; Zhang, X.W.; Wiren, K.M. Neurotransmitter action in osteoblasts: Expression of a functional system for serotonin receptor activation and reuptake. Bone 2001, 29, 477–486. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Symbol | Forward Primer | Reverse Primer |
|---|---|---|---|
| Hypoxanthine Phosphoribosyltransferase 1 | HPRT | AGGGATTTGAATCACGTTTG | TTTACTGGCAACATCAACAG |
| SRY-Box Transcription Factor 9 | SOX9 | CTCTGGAGACTTCTGAACG | AGATGTGCGTCTGCTC |
| Collagen Type II | COL2 | GAAGAGTGGAGACTACTGG | CAGATGTGTTTCTTCTCCTTG |
| Aggrecan | ACAN | CACCCCATGCAATTTGAG | AGATCATCACCACACAGTC |
| Collagen Type X | COLX | GCTAGTATCCTTGAACTTGG | CCTTTACTCTTTATGGTGTAGG |
| RUNX Family Transcription Factor 2 | RUNX2 | AAGCTTGATGACTCTAAACC | TCTGTAATCTGACTCTGTCC |
| Collagen Type I | COL1 | GCTATGATGAGAAATCAACCG | TCATCTCCATTCTTTCCAGG |
| Alkaline Phosphatase | ALPL | TCTTCACATTTGGTGGATAC | ATGGAGACATTCTCTCGTTC |
| Glycoprotein Nmb | GPNMB | CAGATCAGATTCCTGTGTTTG | ACAGTATGATTGGTGGAAAC |
| Bone Gamma-Carboxyglutamate Protein | BGLAP | TTCTTTCCTCTTCCCCTTG | CCTCTTCTGGAGTTTATTTGG |
| Secreted Phosphoprotein 1 | SPP1 | GACCAAGGAAAACTCACTAC | CTGTTTAACTGGTATGGCAC |
| Bone Morphogenetic Protein 2 | BMP2 | CAAGAGACACCCTTTGTATG | GAATTCACAGAGTTCACCAG |
| Parathyroid Hormone 1 Receptor | PTHR | AGAGAAGAAGTATCTGTGGG | GATAAAGAGGATGAAGTTGAGC |
| Dickkopf WNT Signaling Pathway Inhibitor 1 | DKK1 | ATATCACACCAAAGGACAAC | CCTTCTTTAAGGACAGGTTTAC |
| Axis Inhibition Protein 2 | AXIN2 | AAGATCACAAAGAGCCAAAG | GAAAAAGTAGGTGACAACCAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pazos-Pérez, A.; Piñeiro-Ramil, M.; Franco-Trepat, E.; Guillán-Fresco, M.; López-López, V.; Jorge-Mora, A.; Alonso-Pérez, A.; Gómez, R. Methylphenidate Promotes Premature Growth Plate Closure: In Vitro Evidence. Int. J. Mol. Sci. 2023, 24, 4175. https://doi.org/10.3390/ijms24044175
Pazos-Pérez A, Piñeiro-Ramil M, Franco-Trepat E, Guillán-Fresco M, López-López V, Jorge-Mora A, Alonso-Pérez A, Gómez R. Methylphenidate Promotes Premature Growth Plate Closure: In Vitro Evidence. International Journal of Molecular Sciences. 2023; 24(4):4175. https://doi.org/10.3390/ijms24044175
Chicago/Turabian StylePazos-Pérez, Andrés, María Piñeiro-Ramil, Eloi Franco-Trepat, María Guillán-Fresco, Verónica López-López, Alberto Jorge-Mora, Ana Alonso-Pérez, and Rodolfo Gómez. 2023. "Methylphenidate Promotes Premature Growth Plate Closure: In Vitro Evidence" International Journal of Molecular Sciences 24, no. 4: 4175. https://doi.org/10.3390/ijms24044175
APA StylePazos-Pérez, A., Piñeiro-Ramil, M., Franco-Trepat, E., Guillán-Fresco, M., López-López, V., Jorge-Mora, A., Alonso-Pérez, A., & Gómez, R. (2023). Methylphenidate Promotes Premature Growth Plate Closure: In Vitro Evidence. International Journal of Molecular Sciences, 24(4), 4175. https://doi.org/10.3390/ijms24044175