
Healthy and Osteoarthritis-Affected Joints Facing the Cellular Crosstalk


Abstract
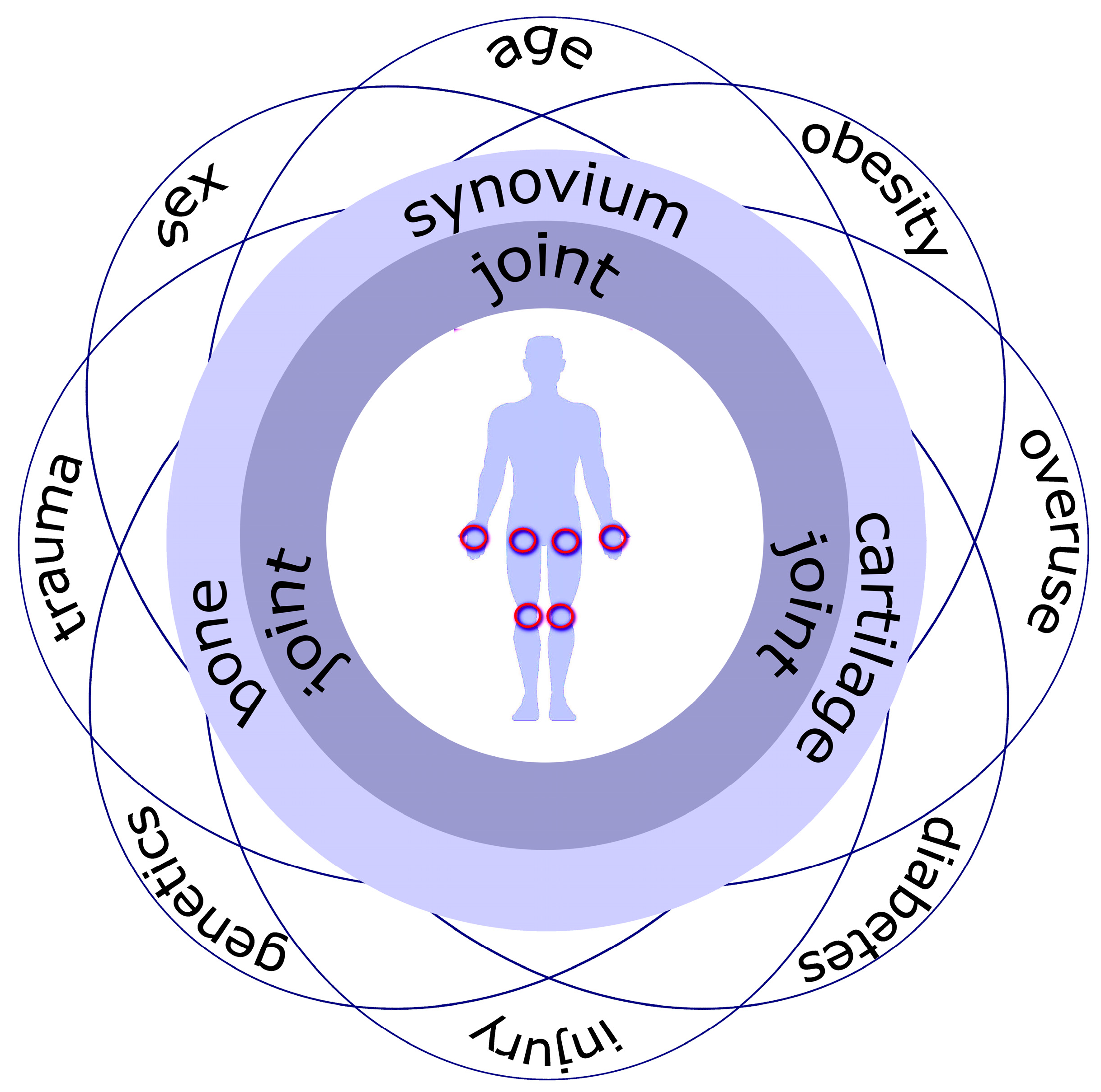
1. Introduction
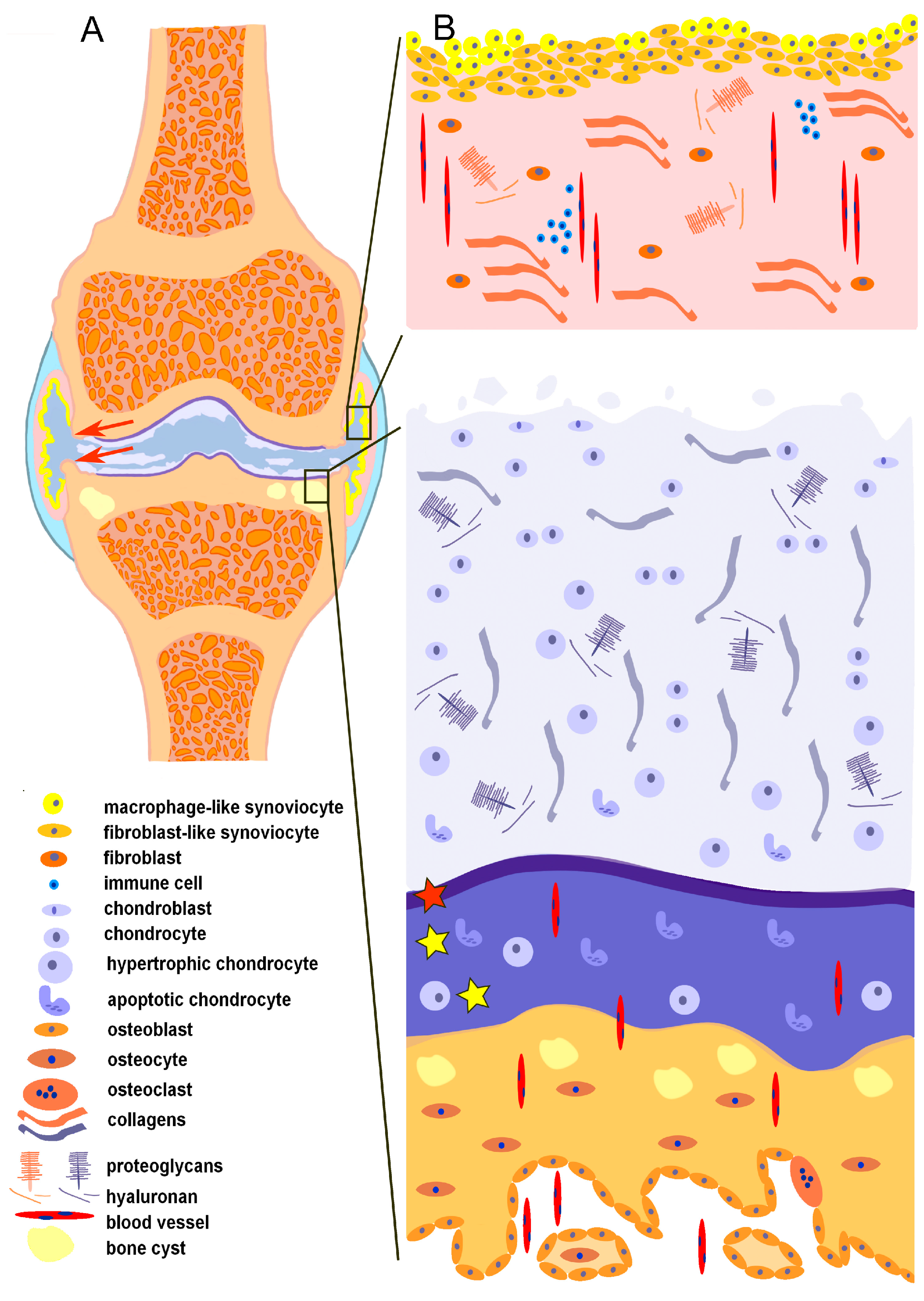
2. Joint as an Organ in a Healthy State
2.1. Synovial Membrane
2.1.1. Synovial Fluid
2.1.2. Cellularity of the Synovial Membrane
2.1.3. Synovial Populations of Macrophages
2.1.4. Synovial Populations of Fibroblasts
2.1.5. Synovial Populations of Non-Resident Cells
2.1.6. Extracellular Matrix of the Synovial Lining
2.2. Articular Cartilage
2.2.1. The Composition of Articular Cartilage
2.2.2. Chondrocytes and Their Metabolic State
2.2.3. Extracellular Matrix of Articular Cartilage
2.3. Microenvironment in the Subchondral Bone
2.3.1. Architecture of the Subchondral Bone
2.3.2. Osteoblast Lineage Cells
2.3.3. Macrophage Lineage Cells
2.4. Cellular and Molecular Interactions in the Healthy Joints
3. Pathological Changes in OA-Affected Joint Tissues
3.1. OA-Affected Synovial Membrane Triggers and Perpetuates the Pathological Process
3.1.1. Changes in the Biochemical Profile of Synovial Fluid in OA
3.1.2. Alterations in of the Synovial Membrane Reflecting Inflammatory and Fibrotic Changes
3.1.3. Insights into Synovial Macrophage Diversity Confirmed in OA Patients
3.1.4. Distinct Subsets of Synovial Fibroblasts Confirmed in OA Patients
3.1.5. Non-Resident Cell Role in the Progression of OA
3.1.6. Danger-Associated Molecular Associated Patterns Characteristic of OA-Caused Damage
3.2. Degradation of the Articular Cartilage in OA
3.2.1. Chondrocyte Phenotypes in Cartilage Affected by OA
3.2.2. Remodeling of Extracellular Matrix during the OA
3.2.3. Cartilage Destruction-Associated Proteases Activated in OA
3.2.4. Cartilage Destruction-Associated Cytokines Contributing to OA Development
3.3. Alterations in the Subchondral Bone Affected by OA
3.3.1. Microstructure of Subchondral Bone during the Early Stage OA
3.3.2. Microstructure of Subchondral Bone during the Late-Stage OA
3.3.3. Changes in the Cellular Composition of the Subchondral Bone
4. OA-Affected Impaired Interaction between Joint Compartments
4.1. Altered Interaction between the Synovium and Cartilage
4.2. Impaired Osteochondral Communication during the Development of OA
4.3. Molecular Signaling in OA-Affected Joints
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Safiri, S.; Kolahi, A.-A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, Regional and National Burden of Osteoarthritis 1990-2017: A Systematic Analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; Lin, J.; Guo, A. Prevalence Trends of Site-Specific Osteoarthritis From 1990 to 2019: Findings From the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; March, L.; Chew, M. Osteoarthritis in 2020 and beyond: A Lancet Commission. Lancet 2020, 396, 1711–1712. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-T.; Ni, G.-X. Depression in Osteoarthritis: Current Understanding. Neuropsychiatr. Dis. Treat. 2022, 18, 375–389. [Google Scholar] [CrossRef]
- Zheng, S.; Tu, L.; Cicuttini, F.; Zhu, Z.; Han, W.; Antony, B.; Wluka, A.E.; Winzenberg, T.; Aitken, D.; Blizzard, L.; et al. Depression in Patients with Knee Osteoarthritis: Risk Factors and Associations with Joint Symptoms. BMC Musculoskelet. Disord. 2021, 22, 40. [Google Scholar] [CrossRef]
- Lu, H.; Wang, L.; Zhou, W.; Jin, S.; Chen, H.; Su, Y.; Li, N.; Shang, S. Bidirectional Association between Knee Osteoarthritis and Depressive Symptoms: Evidence from a Nationwide Population-Based Cohort. BMC Musculoskelet. Disord. 2022, 23, 213. [Google Scholar] [CrossRef]
- Grässel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. J. Clin. Med. 2021, 10, 1938. [Google Scholar] [CrossRef]
- Allen, K.D.; Golightly, Y.M. State of the Evidence. Curr. Opin. Rheumatol. 2015, 27, 276–283. [Google Scholar] [CrossRef]
- Xie, H.; Ma, Y.; Shao, M.; Kong, J.; Zhou, T.; Wang, F.; Cai, G.; Xu, S.; Pan, F. Telomere Length in Patients with Osteoarthritis: A Systematic Review and Meta-Analysis. Aging Clin. Exp. Res. 2022, 34, 495–503. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Gao, S.-G.; Zeng, C.; Li, L.-J.; Luo, W.; Zhang, F.-J.; Tian, J.; Cheng, C.; Tu, M.; Xiong, Y.-L.; Jiang, W.; et al. Correlation between Senescence-Associated Beta-Galactosidase Expression in Articular Cartilage and Disease Severity of Patients with Knee Osteoarthritis. Int. J. Rheum. Dis. 2016, 19, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Tschon, M.; Contartese, D.; Pagani, S.; Borsari, V.; Fini, M. Gender and Sex Are Key Determinants in Osteoarthritis Not Only Confounding Variables. A Systematic Review of Clinical Data. J. Clin. Med. 2021, 10, 3178. [Google Scholar] [CrossRef] [PubMed]
- Joseph, G.B.; McCulloch, C.E.; Nevitt, M.C.; Lynch, J.; Lane, N.E.; Link, T.M. Effects of Weight Change on Knee and Hip Radiographic Measurements and Pain Over Four Years: Data From the Osteoarthritis Initiative. Arthritis Care Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.H.; Lenz, K.L.; Pollitt, E.N.; Ferguson, D.; Hutson, I.; Springer, L.E.; Oestreich, A.K.; Tang, R.; Choi, Y.-R.; Meyer, G.A.; et al. Adipose Tissue Is a Critical Regulator of Osteoarthritis. Proc. Natl. Acad. Sci. USA 2021, 118, e2021096118. [Google Scholar] [CrossRef]
- Belluzzi, E.; Macchi, V.; Fontanella, C.G.; Carniel, E.L.; Olivotto, E.; Filardo, G.; Sarasin, G.; Porzionato, A.; Granzotto, M.; Pozzuoli, A.; et al. Infrapatellar Fat Pad Gene Expression and Protein Production in Patients with and without Osteoarthritis. Int. J. Mol. Sci. 2020, 21, 6016. [Google Scholar] [CrossRef]
- Onuma, H.; Tsuji, K.; Hoshino, T.; Inomata, K.; Udo, M.; Nakagawa, Y.; Katagiri, H.; Miyatake, K.; Watanabe, T.; Sekiya, I.; et al. Fibrotic Changes in the Infrapatellar Fat Pad Induce New Vessel Formation and Sensory Nerve Fiber Endings That Associate Prolonged Pain. J. Orthop. Res. 2020, 38, 1296–1306. [Google Scholar] [CrossRef]
- Wu, J.; Xu, J.; Wang, K.; Zhu, Q.; Cai, J.; Ren, J.; Zheng, S.; Ding, C. Associations between Circulating Adipokines and Bone Mineral Density in Patients with Knee Osteoarthritis: A Cross-Sectional Study. BMC Musculoskelet. Disord. 2018, 19, 16. [Google Scholar] [CrossRef]
- Cannata, F.; Vadalà, G.; Ambrosio, L.; Napoli, N.; Papalia, R.; Denaro, V.; Pozzilli, P. Osteoarthritis and Type 2 Diabetes: From Pathogenetic Factors to Therapeutic Intervention. Diabetes Metab. Res. Rev. 2020, 36, e3254. [Google Scholar] [CrossRef]
- Vincent, T.L. Mechanoflammation in Osteoarthritis Pathogenesis. Semin. Arthritis Rheum. 2019, 49, S36–S38. [Google Scholar] [CrossRef]
- Whittaker, J.L.; Roos, E.M. A Pragmatic Approach to Prevent Post-Traumatic Osteoarthritis after Sport or Exercise-Related Joint Injury. Best Pract. Res. Clin. Rheumatol. 2019, 33, 158–171. [Google Scholar] [CrossRef]
- Rice, S.J.; Beier, F.; Young, D.A.; Loughlin, J. Interplay between Genetics and Epigenetics in Osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Grässel, S.; Muschter, D. Recent Advances in the Treatment of Osteoarthritis. F1000Res 2020, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Goldring, M.B. Changes in the Osteochondral Unit during Osteoarthritis: Structure, Function and Cartilage–Bone Crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Badlani, J.T.; Borrero, C.; Golla, S.; Harner, C.D.; Irrgang, J.J. The Effects of Meniscus Injury on the Development of Knee Osteoarthritis: Data From the Osteoarthritis Initiative. Am. J. Sport. Med. 2013, 41, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Trisolino, G.; Belluzzi, E.; Lazzaro, A.; Strazzari, A.; Pozzuoli, A.; Cigolotti, A.; Ruggieri, P.; Evangelista, A.; Ometto, F.; et al. Macroscopic Synovial Inflammation Correlates with Symptoms and Cartilage Lesions in Patients Undergoing Arthroscopic Partial Meniscectomy: A Clinical Study. J. Clin. Med. 2022, 11, 4330. [Google Scholar] [CrossRef] [PubMed]
- Tarasovs, M.; Skuja, S.; Semenistaja, S.; Murovska, M.; Groma, V. Synovitis in Osteoarthritic Patients: Morphological and Virological Evidence of Its Contribution to Development of the Disease. Proc. Latv. Acad. Sci. Sect. B Nat. Exact Appl. Sci. 2019, 73, 317–324. [Google Scholar] [CrossRef]
- Groma, V.; Tarasovs, M.; Skuja, S.; Semenistaja, S.; Nora-Krukle, Z.; Svirskis, S.; Murovska, M. Inflammatory Cytokine-Producing Cells and Inflammation Markers in the Synovium of Osteoarthritis Patients Evidenced in Human Herpesvirus 7 Infection. Int. J. Mol. Sci. 2020, 21, 6004. [Google Scholar] [CrossRef]
- Masoumi, M.; Bashiri, H.; Khorramdelazad, H.; Barzaman, K.; Hashemi, N.; Sereshki, H.A.; Sahebkar, A.; Karami, J. Destructive Roles of Fibroblast-like Synoviocytes in Chronic Inflammation and Joint Damage in Rheumatoid Arthritis. Inflammation 2021, 44, 466–479. [Google Scholar] [CrossRef]
- Bijlsma, J.W.J.; Berenbaum, F.; Lafeber, F.P.J.G. Osteoarthritis: An Update with Relevance for Clinical Practice. Lancet 2011, 377, 2115–2126. [Google Scholar] [CrossRef]
- De Lange-Brokaar, B.J.E.; Ioan-Facsinay, A.; van Osch, G.J.V.M.; Zuurmond, A.-M.; Schoones, J.; Toes, R.E.M.; Huizinga, T.W.J.; Kloppenburg, M. Synovial Inflammation, Immune Cells and Their Cytokines in Osteoarthritis: A Review. Osteoarthr. Cartil. 2012, 20, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Blüml, S.; Redlich, K.; Smolen, J.S. Mechanisms of Tissue Damage in Arthritis. Semin. Immunopathol. 2014, 36, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.F.; Cheverud, J.M.; Schmidt, E.J.; Sandell, L.J. Genetic Correlations between Cartilage Regeneration and Degeneration Reveal an Inverse Relationship. Osteoarthr. Cartil. 2020, 28, 1111–1120. [Google Scholar] [CrossRef]
- Chou, C.-H.; Jain, V.; Gibson, J.; Attarian, D.E.; Haraden, C.A.; Yohn, C.B.; Laberge, R.-M.; Gregory, S.; Kraus, V.B. Synovial Cell Cross-Talk with Cartilage Plays a Major Role in the Pathogenesis of Osteoarthritis. Sci. Rep. 2020, 10, 10868. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory Mediators in Osteoarthritis: A Critical Review of the State-of-the-Art, Current Prospects, and Future Challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Siebuhr, A.S.; Bay-Jensen, A.C.; Jordan, J.M.; Kjelgaard-Petersen, C.F.; Christiansen, C.; Abramson, S.B.; Attur, M.; Berenbaum, F.; Kraus, V.; Karsdal, M.A. Inflammation (or Synovitis)-Driven Osteoarthritis: An Opportunity for Personalizing Prognosis and Treatment? Scand. J. Rheumatol. 2016, 45, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.M.; Scanzello, C.R. Innate Inflammation and Synovial Macrophages in Osteoarthritis Pathophysiology. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 120), 57–63. [Google Scholar]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Zappia, J.; Sanchez, C.; Florin, A.; Dubuc, J.-E.; Henrotin, Y. The Damage-Associated Molecular Patterns (DAMPs) as Potential Targets to Treat Osteoarthritis: Perspectives From a Review of the Literature. Front. Med. 2021, 7, 607186. [Google Scholar] [CrossRef]
- Hamasaki, M.; Terkawi, M.A.; Onodera, T.; Tian, Y.; Ebata, T.; Matsumae, G.; Alhasan, H.; Takahashi, D.; Iwasaki, N. Transcriptional Profiling of Murine Macrophages Stimulated with Cartilage Fragments Revealed a Strategy for Treatment of Progressive Osteoarthritis. Sci. Rep. 2020, 10, 7558. [Google Scholar] [CrossRef]
- Berenbaum, F. Osteoarthritis as an Inflammatory Disease (Osteoarthritis Is Not Osteoarthrosis!). Osteoarthr. Cartil. 2013, 21, 16–21. [Google Scholar] [CrossRef]
- Li, Y.-S.; Luo, W.; Zhu, S.-A.; Lei, G.-H. T Cells in Osteoarthritis: Alterations and Beyond. Front. Immunol. 2017, 8, 356. [Google Scholar] [CrossRef] [PubMed]
- Benito, M.J. Synovial Tissue Inflammation in Early and Late Osteoarthritis. Ann. Rheum. Dis. 2005, 64, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Rosshirt, N.; Trauth, R.; Platzer, H.; Tripel, E.; Nees, T.A.; Lorenz, H.-M.; Tretter, T.; Moradi, B. Proinflammatory T Cell Polarization Is Already Present in Patients with Early Knee Osteoarthritis. Arthritis Res. Ther. 2021, 23, 37. [Google Scholar] [CrossRef] [PubMed]
- Haynes, M.K.; Hume, E.L.; Smith, J.B. Phenotypic Characterization of Inflammatory Cells from Osteoarthritic Synovium and Synovial Fluids. Clin. Immunol. 2002, 105, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.-P. Osteoarthritis. Nat. Rev. Dis. Prim. 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A Disease of the Joint as an Organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D. The Normal Synovium. Open Rheumatol. J. 2011, 5, 100–106. [Google Scholar] [CrossRef]
- Hügle, T.; Geurts, J. What Drives Osteoarthritis?—Synovial versus Subchondral Bone Pathology. Rheumatology 2017, 56, 1461–1471. [Google Scholar] [CrossRef]
- Kulkarni, P.; Martson, A.; Vidya, R.; Chitnavis, S.; Harsulkar, A. Pathophysiological Landscape of Osteoarthritis. Adv. Clin. Chem. 2021, 100, 37–90. [Google Scholar] [CrossRef]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.Á.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally Renewing Resident Synovial Macrophages Provide a Protective Barrier for the Joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef]
- Carpintero-Fernandez, P.; Gago-Fuentes, R.; Wang, H.Z.; Fonseca, E.; Caeiro, J.R.; Valiunas, V.; Brink, P.R.; Mayan, M.D. Intercellular Communication via Gap Junction Channels between Chondrocytes and Bone Cells. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2018, 1860, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Marsh, L.; Kemble, S.; Reis Nisa, P.; Singh, R.; Croft, A.P. Fibroblast Pathology in Inflammatory Joint Disease. Immunol. Rev. 2021, 302, 163–183. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like Synoviocytes: Key Effector Cells in Rheumatoid Arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Morawietz, L.; Burmester, G.-R.; Kinne, R.W.; Mueller-Ladner, U.; Muller, B.; Haupl, T. Synovitis Score: Discrimination between Chronic Low-Grade and High-Grade Synovitis. Histopathology 2006, 49, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct Synovial Tissue Macrophage Subsets Regulate Inflammation and Remission in Rheumatoid Arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, N.; Apparailly, F.; Courties, G. Synovial Macrophages: From Ordinary Eaters to Extraordinary Multitaskers. Trends Immunol. 2021, 42, 368–371. [Google Scholar] [CrossRef]
- Thomson, A.; Hilkens, C.M.U. Synovial Macrophages in Osteoarthritis: The Key to Understanding Pathogenesis? Front. Immunol. 2021, 12, 678757. [Google Scholar] [CrossRef]
- Knab, K.; Chambers, D.; Krönke, G. Synovial Macrophage and Fibroblast Heterogeneity in Joint Homeostasis and Inflammation. Front. Med. 2022, 9, 862161. [Google Scholar] [CrossRef]
- Mushenkova, N.V.; Nikiforov, N.G.; Shakhpazyan, N.K.; Orekhova, V.A.; Sadykhov, N.K.; Orekhov, A.N. Phenotype Diversity of Macrophages in Osteoarthritis: Implications for Development of Macrophage Modulating Therapies. Int. J. Mol. Sci. 2022, 23, 8381. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-Tissue Organization of the Fibroblast Lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Guilak, F.; Nims, R.J.; Dicks, A.; Wu, C.-L.; Meulenbelt, I. Osteoarthritis as a Disease of the Cartilage Pericellular Matrix. Matrix Biol. 2018, 71–72, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Arayssi, T.; Duray, P.; Schumacher, H.R. Immunohistochemistry of Normal Human Knee Synovium: A Quantitative Study. Ann. Rheum. Dis. 2004, 63, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Muntz, I.; Fenu, M.; van Osch, G.J.V.M.; Koenderink, G.H. The Role of Cell–Matrix Interactions in Connective Tissue Mechanics. Phys. Biol. 2022, 19, 021001. [Google Scholar] [CrossRef] [PubMed]
- Charlier, E.; Deroyer, C.; Ciregia, F.; Malaise, O.; Neuville, S.; Plener, Z.; Malaise, M.; de Seny, D. Chondrocyte Dedifferentiation and Osteoarthritis (OA). Biochem. Pharmacol. 2019, 165, 49–65. [Google Scholar] [CrossRef]
- Primorac, D.; Molnar, V.; Rod, E.; Jeleč, Ž.; Čukelj, F.; Matišić, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Borić, I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, L.; Zeng, L.; He, D.; Wei, X. Nutrition and Degeneration of Articular Cartilage. Knee Surg. Sport. Traumatol. Arthrosc. 2013, 21, 1751–1762. [Google Scholar] [CrossRef]
- Luo, Y.; Sinkeviciute, D.; He, Y.; Karsdal, M.; Henrotin, Y.; Mobasheri, A.; Önnerfjord, P.; Bay-Jensen, A. The Minor Collagens in Articular Cartilage. Protein Cell 2017, 8, 560–572. [Google Scholar] [CrossRef]
- Hall, A.C. The Role of Chondrocyte Morphology and Volume in Controlling Phenotype—Implications for Osteoarthritis, Cartilage Repair, and Cartilage Engineering. Curr. Rheumatol. Rep. 2019, 21, 38. [Google Scholar] [CrossRef]
- Wang, X.; Ning, Y.; Zhang, P.; Poulet, B.; Huang, R.; Gong, Y.; Hu, M.; Li, C.; Zhou, R.; Lammi, M.J.; et al. Comparison of the Major Cell Populations among Osteoarthritis, Kashin–Beck Disease and Healthy Chondrocytes by Single-Cell RNA-Seq Analysis. Cell Death Dis. 2021, 12, 551. [Google Scholar] [CrossRef]
- Basso, P.R.; Carava’, E.; Protasoni, M.; Reguzzoni, M.; Raspanti, M. The Synovial Surface of the Articular Cartilage. Eur. J. Histochem. 2020, 64, 3146. [Google Scholar] [CrossRef]
- Pan, J.; Wang, B.; Li, W.; Zhou, X.; Scherr, T.; Yang, Y.; Price, C.; Wang, L. Elevated Cross-Talk between Subchondral Bone and Cartilage in Osteoarthritic Joints. Bone 2012, 51, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Zecca, P.A.; Reguzzoni, M.; Protasoni, M.; Raspanti, M. The Chondro-Osseous Junction Of Articular Cartilage. Tissue Cell 2022, 80, 101993. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.J.; Stoddart, R.W.; McClure, S.F.; McClure, J. The Tidemark of the Chondro-Osseous Junction of the Normal Human Knee Joint. J. Mol. Hist. 2005, 36, 207–215. [Google Scholar] [CrossRef]
- Boyde, A. The Bone Cartilage Interface and Osteoarthritis. Calcif. Tissue Int. 2021, 109, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Chery, D.R.; Han, B.; Zhou, Y.; Wang, C.; Adams, S.M.; Chandrasekaran, P.; Kwok, B.; Heo, S.-J.; Enomoto-Iwamoto, M.; Lu, X.L.; et al. Decorin Regulates Cartilage Pericellular Matrix Micromechanobiology. Matrix Biol. 2021, 96, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Sun, H.; Bunpetch, V.; Koh, Y.; Wen, Y.; Wu, D.; Ouyang, H. The Regulation of Cartilage Extracellular Matrix Homeostasis in Joint Cartilage Degeneration and Regeneration. Biomaterials 2021, 268, 120555. [Google Scholar] [CrossRef]
- Brandt, K.D. Yet More Evidence That Osteoarthritis Is Not a Cartilage Disease. Ann. Rheum. Dis. 2006, 65, 1261–1264. [Google Scholar] [CrossRef]
- Danalache, M.; Kleinert, R.; Schneider, J.; Erler, A.L.; Schwitalle, M.; Riester, R.; Traub, F.; Hofmann, U.K. Changes in Stiffness and Biochemical Composition of the Pericellular Matrix as a Function of Spatial Chondrocyte Organisation in Osteoarthritic Cartilage. Osteoarthr. Cartil. 2019, 27, 823–832. [Google Scholar] [CrossRef]
- Wang, C.; Brisson, B.K.; Terajima, M.; Li, Q.; Hoxha, K.; Han, B.; Goldberg, A.M.; Sherry Liu, X.; Marcolongo, M.S.; Enomoto-Iwamoto, M.; et al. Type III Collagen Is a Key Regulator of the Collagen Fibrillar Structure and Biomechanics of Articular Cartilage and Meniscus. Matrix Biol. 2020, 85–86, 47–67. [Google Scholar] [CrossRef]
- Eyre, D. Articular Cartilage and Changes in Arthritis: Collagen of Articular Cartilage. Arthritis Res. Ther. 2001, 4, 30. [Google Scholar] [CrossRef]
- Sandell, L.J.; Aigner, T. Articular Cartilage and Changes in Arthritis. An Introduction: Cell Biology of Osteoarthritis. Arthritis Res. Ther. 2001, 3, 107–113. [Google Scholar] [CrossRef]
- Pullig, O.; Weseloh, G.; Klatt, A.R.; Wagener, R.; Swoboda, B. Matrilin-3 in Human Articular Cartilage: Increased Expression in Osteoarthritis. Osteoarthr. Cartil. 2002, 10, 253–263. [Google Scholar] [CrossRef]
- Roseti, L.; Desando, G.; Cavallo, C.; Petretta, M.; Grigolo, B. Articular Cartilage Regeneration in Osteoarthritis. Cells 2019, 8, 1305. [Google Scholar] [CrossRef]
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral Bone in Osteoarthritis: Insight into Risk Factors and Microstructural Changes. Arthritis Res. Ther. 2013, 15, 223. [Google Scholar] [CrossRef]
- Pan, J.; Zhou, X.; Li, W.; Novotny, J.E.; Doty, S.B.; Wang, L. In Situ Measurement of Transport between Subchondral Bone and Articular Cartilage: TRANSPORT BETWEEN BONE AND CARTILAGE. J. Orthop. Res. 2009, 27, 1347–1352. [Google Scholar] [CrossRef]
- Mountcastle, S.E.; Allen, P.; Mellors, B.O.L.; Lawless, B.M.; Cooke, M.E.; Lavecchia, C.E.; Fell, N.L.A.; Espino, D.M.; Jones, S.W.; Cox, S.C. Dynamic Viscoelastic Characterisation of Human Osteochondral Tissue: Understanding the Effect of the Cartilage-Bone Interface. BMC Musculoskelet. Disord. 2019, 20, 575. [Google Scholar] [CrossRef]
- Hu, W.; Chen, Y.; Dou, C.; Dong, S. Microenvironment in Subchondral Bone: Predominant Regulator for the Treatment of Osteoarthritis. Ann. Rheum. Dis. 2021, 80, 413–422. [Google Scholar] [CrossRef]
- Burr, D.B.; Gallant, M.A. Bone Remodelling in Osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Donell, S. Subchondral Bone Remodelling in Osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Zhao, X.; Ma, L.; Guo, H.; Wang, J.; Zhang, S.; Yang, X.; Yang, L.; Jin, Q. Osteoclasts Secrete Leukemia Inhibitory Factor to Promote Abnormal Bone Remodeling of Subchondral Bone in Osteoarthritis. BMC Musculoskelet. Disord. 2022, 23, 87. [Google Scholar] [CrossRef]
- Sharma, A.; Jagga, S.; Lee, S.-S.; Nam, J.-S. Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19805–19830. [Google Scholar] [CrossRef]
- Lajeunesse, D.; Reboul, P. Subchondral Bone in Osteoarthritis: A Biologic Link with Articular Cartilage Leading to Abnormal Remodeling. Curr. Opin. Rheumatol. 2003, 15, 628–633. [Google Scholar] [CrossRef]
- Wu, X.; Crawford, R.; Xiao, Y.; Mao, X.; Prasadam, I. Osteoarthritic Subchondral Bone Release Exosomes That Promote Cartilage Degeneration. Cells 2021, 10, 251. [Google Scholar] [CrossRef]
- Buenzli, P.R.; Sims, N.A. Quantifying the Osteocyte Network in the Human Skeleton. Bone 2015, 75, 144–150. [Google Scholar] [CrossRef]
- Schurman, C.A.; Verbruggen, S.W.; Alliston, T. Disrupted Osteocyte Connectivity and Pericellular Fluid Flow in Bone with Aging and Defective TGF-β Signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2023999118. [Google Scholar] [CrossRef]
- Zhang, L.; Wen, C. Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 6522. [Google Scholar] [CrossRef]
- Morrell, A.E.; Brown, G.N.; Robinson, S.T.; Sattler, R.L.; Baik, A.D.; Zhen, G.; Cao, X.; Bonewald, L.F.; Jin, W.; Kam, L.C.; et al. Mechanically Induced Ca2+ Oscillations in Osteocytes Release Extracellular Vesicles and Enhance Bone Formation. Bone Res. 2018, 6, 6. [Google Scholar] [CrossRef]
- Chen, J.; Tu, X.; Esen, E.; Joeng, K.S.; Lin, C.; Arbeit, J.M.; Rüegg, M.A.; Hall, M.N.; Ma, L.; Long, F. WNT7B Promotes Bone Formation in Part through MTORC1. PLoS Genet. 2014, 10, e1004145. [Google Scholar] [CrossRef]
- Song, D.; He, G.; Song, F.; Wang, Z.; Liu, X.; Liao, L.; Ni, J.; Silva, M.J.; Long, F. Inducible Expression of Wnt7b Promotes Bone Formation in Aged Mice and Enhances Fracture Healing. Bone Res. 2020, 8, 4. [Google Scholar] [CrossRef]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical Stimulation of Bone in Vivo Reduces Osteocyte Expression of Sost/Sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, A.; Pennisi, P.; Bratengeier, C.; Torrisi, V.; Lindner, B.; Mangiafico, R.A.; Pulvirenti, I.; Hawa, G.; Tringali, G.; Fiore, C.E. Increased Sclerostin Serum Levels Associated with Bone Formation and Resorption Markers in Patients with Immobilization-Induced Bone Loss. J. Clin. Endocrinol. Metab. 2010, 95, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.S.; Manilay, J.O.; Chang, J.C.; Hum, N.R.; Murugesh, D.K.; Bajwa, J.; Mendez, M.E.; Economides, A.E.; Horan, D.J.; Robling, A.G.; et al. Conditional Deletion of Sost in MSC-Derived Lineages Identifies Specific Cell-Type Contributions to Bone Mass and B-Cell Development. J. Bone Min. Res. 2018, 33, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Van Bezooijen, R.L.; Roelen, B.A.J.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; ten Dijke, P.; Löwik, C.W.G.M. Sclerostin Is an Osteocyte-Expressed Negative Regulator of Bone Formation, But Not a Classical BMP Antagonist. J. Exp. Med. 2004, 199, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.A.; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt Signaling in Differentiated Osteoblasts Controls Osteoclast Differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef]
- Zimmerman, S.M.; Heard-Lipsmeyer, M.E.; Dimori, M.; Thostenson, J.D.; Mannen, E.M.; O’Brien, C.A.; Morello, R. Loss of RANKL in Osteocytes Dramatically Increases Cancellous Bone Mass in the Osteogenesis Imperfecta Mouse (Oim). Bone Rep. 2018, 9, 61–73. [Google Scholar] [CrossRef]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast Differentiation by RANKL and OPG Signaling Pathways. J. Bone Min. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Pellicore, M.J.; Gangi, L.R.; Murphy, L.A.; Lee, A.J.; Jacobsen, T.; Kenawy, H.M.; Shah, R.P.; Chahine, N.O.; Ateshian, G.A.; Hung, C.T. Toward Defining the Role of the Synovium in Mitigating Normal Articular Cartilage Wear and Tear. J. Biomech. 2023, 148, 111472. [Google Scholar] [CrossRef]
- Dy, P.; Wang, W.; Bhattaram, P.; Wang, Q.; Wang, L.; Ballock, R.T.; Lefebvre, V. Sox9 Directs Hypertrophic Maturation and Blocks Osteoblast Differentiation of Growth Plate Chondrocytes. Dev. Cell 2012, 22, 597–609. [Google Scholar] [CrossRef]
- Ouyang, Y.; Wang, W.; Tu, B.; Zhu, Y.; Fan, C.; Li, Y. Overexpression of SOX9 Alleviates the Progression of Human Osteoarthritis in Vitro and in Vivo. Drug Des. Devel. 2019, 13, 2833–2842. [Google Scholar] [CrossRef]
- Hossain, M.A.; Adithan, A.; Alam, M.J.; Kopalli, S.R.; Kim, B.; Kang, C.-W.; Hwang, K.-C.; Kim, J.-H. IGF-1 Facilitates Cartilage Reconstruction by Regulating PI3K/AKT, MAPK, and NF-KB Signaling in Rabbit Osteoarthritis. J. Inflamm. Res. 2021, 14, 3555–3568. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Xu, L.; Xu, X.; Wang, D.; Liang, Y.; Duan, L. Insulin-like Growth Factor-1 in Articular Cartilage Repair for Osteoarthritis Treatment. Arthritis Res. Ther. 2021, 23, 277. [Google Scholar] [CrossRef] [PubMed]
- Cherifi, C.; Monteagudo, S.; Lories, R.J. Promising Targets for Therapy of Osteoarthritis: A Review on the Wnt and TGF-β Signalling Pathways. Ther. Adv. Musculoskelet. 2021, 13, 1759720X211006959. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Maldonado, M.; Nam, J. The Role of Changes in Extracellular Matrix of Cartilage in the Presence of Inflammation on the Pathology of Osteoarthritis. BioMed Res. Int. 2013, 2013, 284873. [Google Scholar] [CrossRef]
- Berenbaum, F.; Walker, C. Osteoarthritis and Inflammation: A Serious Disease with Overlapping Phenotypic Patterns. Postgrad. Med. 2020, 132, 377–384. [Google Scholar] [CrossRef]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-Grade Inflammation as a Key Mediator of the Pathogenesis of Osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, X.; Jiang, Y.; Liu, X.; Huang, L.; Wei, Q.; Huang, Y.; Wu, W.; Gu, J. Alterations in Peripheral T Cell and B Cell Subsets in Patients with Osteoarthritis. Clin. Rheumatol. 2020, 39, 523–532. [Google Scholar] [CrossRef]
- Hayashi, D.; Roemer, F.W.; Katur, A.; Felson, D.T.; Yang, S.-O.; Alomran, F.; Guermazi, A. Imaging of Synovitis in Osteoarthritis: Current Status and Outlook. Semin. Arthritis Rheum. 2011, 41, 116–130. [Google Scholar] [CrossRef]
- Atukorala, I.; Kwoh, C.K.; Guermazi, A.; Roemer, F.W.; Boudreau, R.M.; Hannon, M.J.; Hunter, D.J. Synovitis in Knee Osteoarthritis: A Precursor of Disease? Ann. Rheum. Dis. 2016, 75, 390–395. [Google Scholar] [CrossRef]
- Boffa, A.; Merli, G.; Andriolo, L.; Lattermann, C.; Salzmann, G.M.; Filardo, G. Synovial Fluid Biomarkers in Knee Osteoarthritis: A Systematic Review and Quantitative Evaluation Using BIPEDs Criteria. Cartilage 2021, 13, 82S–103S. [Google Scholar] [CrossRef] [PubMed]
- Manferdini, C.; Paolella, F.; Gabusi, E.; Silvestri, Y.; Gambari, L.; Cattini, L.; Filardo, G.; Fleury-Cappellesso, S.; Lisignoli, G. From Osteoarthritic Synovium to Synovial-Derived Cells Characterization: Synovial Macrophages Are Key Effector Cells. Arthritis Res. Ther. 2016, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Thoenen, J.; MacKay, J.W.; Sandford, H.J.C.; Gold, G.E.; Kogan, F. Imaging of Synovial Inflammation in Osteoarthritis, From the AJR Special Series on Inflammation. AJR Am. J. Roentgenol. 2022, 218, 405–417. [Google Scholar] [CrossRef]
- Baker, K.; Grainger, A.; Niu, J.; Clancy, M.; Guermazi, A.; Crema, M.; Hughes, L.; Buckwalter, J.; Wooley, A.; Nevitt, M.; et al. Relation of Synovitis to Knee Pain Using Contrast-Enhanced MRIs. Ann. Rheum. Dis. 2010, 69, 1779–1783. [Google Scholar] [CrossRef] [PubMed]
- Roemer, F.W.; Guermazi, A.; Felson, D.T.; Niu, J.; Nevitt, M.C.; Crema, M.D.; Lynch, J.A.; Lewis, C.E.; Torner, J.; Zhang, Y. Presence of MRI-Detected Joint Effusion and Synovitis Increases the Risk of Cartilage Loss in Knees without Osteoarthritis at 30-Month Follow-up: The MOST Study. Ann. Rheum. Dis. 2011, 70, 1804–1809. [Google Scholar] [CrossRef]
- Wang, X.; Hunter, D.J.; Jin, X.; Ding, C. The Importance of Synovial Inflammation in Osteoarthritis: Current Evidence from Imaging Assessments and Clinical Trials. Osteoarthr. Cartil. 2018, 26, 165–174. [Google Scholar] [CrossRef]
- Felson, D.T.; Niu, J.; Neogi, T.; Goggins, J.; Nevitt, M.C.; Roemer, F.; Torner, J.; Lewis, C.E.; Guermazi, A. Synovitis and the Risk of Knee Osteoarthritis: The MOST Study. Osteoarthr. Cartil. 2016, 24, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Ayral, X.; Pickering, E.H.; Woodworth, T.G.; Mackillop, N.; Dougados, M. Synovitis: A Potential Predictive Factor of Structural Progression of Medial Tibiofemoral Knee Osteoarthritis-Results of a 1 Year Longitudinal Arthroscopic Study in 422 Patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef]
- Riis, R.G.C.; Gudbergsen, H.; Simonsen, O.; Henriksen, M.; Al-Mashkur, N.; Eld, M.; Petersen, K.K.; Kubassova, O.; Bay Jensen, A.C.; Damm, J.; et al. The Association between Histological, Macroscopic and Magnetic Resonance Imaging Assessed Synovitis in End-Stage Knee Osteoarthritis: A Cross-Sectional Study. Osteoarthr. Cartil. 2017, 25, 272–280. [Google Scholar] [CrossRef]
- Abbasi, B.; Pezeshki-Rad, M.; Akhavan, R.; Sahebari, M. Association between Clinical and Sonographic Synovitis in Patients with Painful Knee Osteoarthritis. Int. J. Rheum. Dis. 2017, 20, 561–566. [Google Scholar] [CrossRef]
- Zhang, Y.; Nevitt, M.; Niu, J.; Lewis, C.; Torner, J.; Guermazi, A.; Roemer, F.; McCulloch, C.; Felson, D. Fluctuation of Knee Pain and Changes in Bone Marrow Lesions, Effusions and Synovitis on MRI: The MOST Study. Arthritis Rheum. 2011, 63, 573–862. [Google Scholar] [CrossRef] [PubMed]
- Bacon, K.; LaValley, M.P.; Jafarzadeh, S.R.; Felson, D. Does Cartilage Loss Cause Pain in Osteoarthritis and If so, How Much? Ann. Rheum. Dis. 2020, 79, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lin, L.; Guo, Y.; Zou, R.; Wang, Z.; Shi, Z.; Lin, F. Matrix Metalloproteinase-13, NF-κB P65 and Interleukin-1β Are Associated with the Severity of Knee Osteoarthritis. Exp. Ther. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stannus, O.; Jones, G.; Cicuttini, F.; Parameswaran, V.; Quinn, S.; Burgess, J.; Ding, C. Circulating Levels of IL-6 and TNF-α Are Associated with Knee Radiographic Osteoarthritis and Knee Cartilage Loss in Older Adults. Osteoarthr. Cartil. 2010, 18, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Hoff, P.; Buttgereit, F.; Burmester, G.-R.; Jakstadt, M.; Gaber, T.; Andreas, K.; Matziolis, G.; Perka, C.; Röhner, E. Osteoarthritis Synovial Fluid Activates Pro-Inflammatory Cytokines in Primary Human Chondrocytes. Int. Orthop. (SICOT) 2013, 37, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mai, Y.; Cao, P.; Wen, X.; Fan, T.; Wang, X.; Ruan, G.; Tang, S.; Ding, C.; Zhu, Z. Relative Efficacy and Safety of Anti-Inflammatory Biologic Agents for Osteoarthritis: A Conventional and Network Meta-Analysis. J. Clin. Med. 2022, 11, 3958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, M.; Li, X.; Liao, T.; Ma, Z.; Zhang, L.; Xing, R.; Wang, P.; Mao, J. Characteristics of Sensory Innervation in Synovium of Rats within Different Knee Osteoarthritis Models and the Correlation between Synovial Fibrosis and Hyperalgesia. J. Adv. Res. 2022, 35, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Nanus, D.E.; Badoume, A.; Wijesinghe, S.N.; Halsey, A.M.; Hurley, P.; Ahmed, Z.; Botchu, R.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Synovial Tissue from Sites of Joint Pain in Knee Osteoarthritis Patients Exhibits a Differential Phenotype with Distinct Fibroblast Subsets. eBioMedicine 2021, 72, 103618. [Google Scholar] [CrossRef]
- Wang, M.; Lessard, S.G.; Singh, P.; Pannellini, T.; Chen, T.; Rourke, B.J.; Chowdhury, L.; Craveiro, V.; Sculco, P.K.; van der Meulen, M.C.H.; et al. Knee Fibrosis Is Associated with the Development of Osteoarthritis in a Murine Model of Tibial Compression. J. Orthop. Res. 2021, 39, 1030–1040. [Google Scholar] [CrossRef]
- Favero, M.; El-Hadi, H.; Belluzzi, E.; Granzotto, M.; Porzionato, A.; Sarasin, G.; Rambaldo, A.; Iacobellis, C.; Cigolotti, A.; Fontanella, C.G.; et al. Infrapatellar Fat Pad Features in Osteoarthritis: A Histopathological and Molecular Study. Rheumatology 2017, 56, 1784–1793. [Google Scholar] [CrossRef]
- Macchi, V.; Stocco, E.; Stecco, C.; Belluzzi, E.; Favero, M.; Porzionato, A.; De Caro, R. The infrapatellar fat pad and the synovial membrane: An anatomo-functional unit. J. Anat. 2018, 233, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, E.; Stocco, E.; Pozzuoli, A.; Granzotto, M.; Porzionato, A.; Vettor, R.; De Caro, R.; Ruggieri, P.; Ramonda, R.; Rossato, M.; et al. Contribution of Infrapatellar Fat Pad and Synovial Membrane to Knee Osteoarthritis Pain. BioMed Res. Int. 2019, 2019, e6390182. [Google Scholar] [CrossRef] [PubMed]
- Emmi, A.; Stocco, E.; Boscolo-Berto, R.; Contran, M.; Belluzzi, E.; Favero, M.; Ramonda, R.; Porzionato, A.; Ruggieri, P.; De Caro, R.; et al. Infrapatellar Fat Pad-Synovial Membrane Anatomo-Fuctional Unit: Microscopic Basis for Piezo1/2 Mechanosensors Involvement in Osteoarthritis Pain. Front. Cell. Dev. Biol. 2022, 10, 886604. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.B.; van Lent, P.L.; Libregts, S.; Holthuysen, A.E.; van der Kraan, P.M.; van Rooijen, N.; van den Berg, W.B. Crucial Role of Macrophages in Matrix Metalloproteinase–Mediated Cartilage Destruction during Experimental Osteoarthritis: Involvement of Matrix Metalloproteinase 3. Arthritis Rheum. 2007, 56, 147–157. [Google Scholar] [CrossRef]
- Kraus, V.B.; McDaniel, G.; Huebner, J.L.; Stabler, T.V.; Pieper, C.F.; Shipes, S.W.; Petry, N.A.; Low, P.S.; Shen, J.; McNearney, T.A.; et al. Direct in Vivo Evidence of Activated Macrophages in Human Osteoarthritis. Osteoarthr. Cartil. 2016, 24, 1613–1621. [Google Scholar] [CrossRef]
- Huo, L.W.; Ye, Y.L.; Wang, G.W.; Ye, Y.G. Fractalkine (CX3CL1): A Biomarker Reflecting Symptomatic Severity in Patients With Knee Osteoarthritis. J. Investig. Med. 2015, 63, 626–631. [Google Scholar] [CrossRef]
- Ancuta, P.; Wang, J.; Gabuzda, D. CD16+ Monocytes Produce IL-6, CCL2, and Matrix Metalloproteinase-9 upon Interaction with CX3CL1-Expressing Endothelial Cells. J. Leukoc. Biol. 2006, 80, 1156–1164. [Google Scholar] [CrossRef]
- Raghu, H.; Lepus, C.M.; Wang, Q.; Wong, H.H.; Lingampalli, N.; Oliviero, F.; Punzi, L.; Giori, N.J.; Goodman, S.B.; Chu, C.R.; et al. CCL2/CCR2, but Not CCL5/CCR5, Mediates Monocyte Recruitment, Inflammation and Cartilage Destruction in Osteoarthritis. Ann. Rheum. Dis. 2017, 76, 914–922. [Google Scholar] [CrossRef]
- Daghestani, H.N.; Pieper, C.F.; Kraus, V.B. Soluble Macrophage Biomarkers Indicate Inflammatory Phenotypes in Patients With Knee Osteoarthritis. Arthritis Rheumatol. 2015, 67, 956–965. [Google Scholar] [CrossRef]
- Blom, A.B.; van Lent, P.L.E.M.; Holthuysen, A.E.M.; van der Kraan, P.M.; Roth, J.; van Rooijen, N.; van den Berg, W.B. Synovial Lining Macrophages Mediate Osteophyte Formation during Experimental Osteoarthritis. Osteoarthr. Cartil. 2004, 12, 627–635. [Google Scholar] [CrossRef]
- Jamal, J.; Roebuck, M.M.; Lee, S.-Y.; Frostick, S.P.; Abbas, A.A.; Merican, A.M.; Teo, S.-H.; Wood, A.; Tan, S.-L.; Bou-Gharios, G.; et al. Modulation of the Mechanical Responses of Synovial Fibroblasts by Osteoarthritis-Associated Inflammatory Stressors. Int. J. Biochem. Cell Biol. 2020, 126, 105800. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.M.; Leonard, C.; Regmi, C.S.; De Rantere, D.; Tailor, P.; Ren, G.; Ishida, H.; Hsu, C.y.; Abubacker, S.; Pang, D.S.J.; et al. Lubricin/Proteoglycan 4 Binds to and Regulates the Activity of Toll-Like Receptors In Vitro. Sci. Rep. 2016, 6, 18910. [Google Scholar] [CrossRef]
- Chavda, S.; Rabbani, S.A.; Wadhwa, T. Role and Effectiveness of Intra-Articular Injection of Hyaluronic Acid in the Treatment of Knee Osteoarthritis: A Systematic Review. Cureus 2022, 14, e24503. [Google Scholar] [CrossRef]
- Avenoso, A.; Bruschetta, G.; D’Ascola, A.; Scuruchi, M.; Mandraffino, G.; Saitta, A.; Campo, S.; Campo, G.M. Hyaluronan Fragmentation During Inflammatory Pathologies: A Signal That Empowers Tissue Damage. Mini Rev. Med. Chem. 2020, 20, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally Distinct Disease-Associated Fibroblast Subsets in Rheumatoid Arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, G.; Firestein, G.S. Restoring Synovial Homeostasis in Rheumatoid Arthritis by Targeting Fibroblast-like Synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Miao, K.; Zhou, X.; Xu, N.; Li, C.; Zhu, R.; Sun, R.; Wang, Y. The Involvement of Follistatin-like Protein 1 in Osteoarthritis by Elevating NF-ΚB-Mediated Inflammatory Cytokines and Enhancing Fibroblast like Synoviocyte Proliferation. Arthritis Res. Ther. 2015, 17, 91. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Richendrfer, H.; Schmidt, T.A.; Elsaid, K.A. Proteoglycan-4 Regulates Fibroblast to Myofibroblast Transition and Expression of Fibrotic Genes in the Synovium. Arthritis Res. Ther. 2020, 22, 113. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Kotela, A.; Deszczyński, J.; Kotela, I.; Szukiewicz, D. The Chemokine CX3CL1 (Fractalkine) and Its Receptor CX3CR1: Occurrence and Potential Role in Osteoarthritis. Arch. Immunol. Et Ther. Exp. (Warsz) 2014, 62, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.-M.; Hou, C.-H.; Liu, J.-F. CX3CL1 Promotes MMP-3 Production via the CX3CR1, c-Raf, MEK, ERK, and NF-ΚB Signaling Pathway in Osteoarthritis Synovial Fibroblasts. Arthritis Res. Ther. 2017, 19, 282. [Google Scholar] [CrossRef] [PubMed]
- Frank-Bertoncelj, M.; Trenkmann, M.; Klein, K.; Karouzakis, E.; Rehrauer, H.; Bratus, A.; Kolling, C.; Armaka, M.; Filer, A.; Michel, B.A.; et al. Epigenetically-Driven Anatomical Diversity of Synovial Fibroblasts Guides Joint-Specific Fibroblast Functions. Nat. Commun. 2017, 8, 14852. [Google Scholar] [CrossRef] [PubMed]
- Trajerova, M.; Kriegova, E.; Mikulkova, Z.; Savara, J.; Kudelka, M.; Gallo, J. Knee Osteoarthritis Phenotypes Based on Synovial Fluid Immune Cells Correlate with Clinical Outcome Trajectories. Osteoarthr. Cartil. 2022, 30, 1583–1592. [Google Scholar] [CrossRef]
- Rosshirt, N.; Hagmann, S.; Tripel, E.; Gotterbarm, T.; Kirsch, J.; Zeifang, F.; Lorenz, H.-M.; Tretter, T.; Moradi, B. A Predominant Th1 Polarization Is Present in Synovial Fluid of End-Stage Osteoarthritic Knee Joints: Analysis of Peripheral Blood, Synovial Fluid and Synovial Membrane. Clin. Exp. Immunol. 2019, 195, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.; Manninen, M.; Eklund, K.K. Osteoarthritis and Toll-Like Receptors: When Innate Immunity Meets Chondrocyte Apoptosis. Biology 2020, 9, 65. [Google Scholar] [CrossRef]
- Schedel, J.; Wenglen, C.; Distler, O.; Muller-Ladner, U.; Scholmerich, J.; Heinegard, D.; Krenn, V. Differential Adherence of Osteoarthritis and Rheumatoid Arthritis Synovial Fibroblasts to Cartilage and Bone Matrix Proteins and Its Implication for Osteoarthritis Pathogenesis. Scand. J. Immunol. 2004, 60, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The Role of Metabolism in the Pathogenesis of Osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Woodell-May, J.E.; Sommerfeld, S.D. Role of Inflammation and the Immune System in the Progression of Osteoarthritis. J. Orthop. Res. 2020, 38, 253–257. [Google Scholar] [CrossRef]
- Mobasheri, A.; Fonseca, J.E.; Gualillo, O.; Henrotin, Y.; Largo, R.; Herrero-Beaumont, G.; Rocha, F.A.C. Editorial: Inflammation and Biomarkers in Osteoarthritis. Front. Med. 2021, 8, 727700. [Google Scholar] [CrossRef]
- Orlowsky, E.W.; Kraus, V.B. The Role of Innate Immunity in Osteoarthritis: When Our First Line of Defense Goes On the Offensive. J. Rheumatol. 2015, 42, 363–371. [Google Scholar] [CrossRef]
- Millerand, M.; Berenbaum, F.; Jacques, C. Danger Signals and Inflammaging in Osteoarthritis. Clin. Exp. Rheumatol. 2019, 37, 48–56. [Google Scholar] [PubMed]
- Avenoso, A.; Bruschetta, G.; D’Ascola, A.; Scuruchi, M.; Mandraffino, G.; Gullace, R.; Saitta, A.; Campo, S.; Campo, G.M. Hyaluronan Fragments Produced during Tissue Injury: A Signal Amplifying the Inflammatory Response. Arch. Biochem. Biophys. 2019, 663, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Deroyer, C.; Poulet, C.; Paulissen, G.; Ciregia, F.; Malaise, O.; Plener, Z.; Cobraiville, G.; Daniel, C.; Gillet, P.; Malaise, M.G.; et al. CEMIP (KIAA1199) Regulates Inflammation, Hyperplasia and Fibrosis in Osteoarthritis Synovial Membrane. Cell. Mol. Life Sci. 2022, 79, 260. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.; Belluzzi, E.; Frallonardo, P.; Peruzzo, L.; Tauro, L.; Oliviero, F.; Ramonda, R.; Punzi, L. Synovial Fluid Fetuin-A Levels in Patients Affected by Osteoarthritis with or without Evidence of Calcium Crystals. Rheumatology 2019, 58, 729–730. [Google Scholar] [CrossRef] [PubMed]
- Stücker, S.; Bollmann, M.; Garbers, C.; Bertrand, J. The Role of Calcium Crystals and Their Effect on Osteoarthritis Pathogenesis. Best Pract. Res. Clin. Rheumatol. 2021, 35, 101722. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, F.; Eymard, F.; Houard, X. Osteoarthritis, Inflammation and Obesity. Curr. Opin. Rheumatol. 2013, 25, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Stolberg-Stolberg, J.; Boettcher, A.; Sambale, M.; Stuecker, S.; Sherwood, J.; Raschke, M.; Pap, T.; Bertrand, J. Toll-like Receptor 3 Activation Promotes Joint Degeneration in Osteoarthritis. Cell Death Dis. 2022, 13, 224. [Google Scholar] [CrossRef]
- Miller, R.E.; Ishihara, S.; Tran, P.B.; Golub, S.B.; Last, K.; Miller, R.J.; Fosang, A.J.; Malfait, A.-M. An Aggrecan Fragment Drives Osteoarthritis Pain through Toll-like Receptor 2. JCI Insight 2018, 3, e95704. [Google Scholar] [CrossRef]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a Glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Zelenski, N.A.; Leddy, H.A.; Sanchez-Adams, J.; Zhang, J.; Bonaldo, P.; Liedtke, W.; Guilak, F. Type VI Collagen Regulates Pericellular Matrix Properties, Chondrocyte Swelling, and Mechanotransduction in Mouse Articular Cartilage. Arthritis Rheumatol. 2015, 67, 1286–1294. [Google Scholar] [CrossRef]
- Rim, Y.A.; Ju, J.H. The Role of Fibrosis in Osteoarthritis Progression. Life 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Bush, P.G.; Hodkinson, P.D.; Hamilton, G.L.; Hall, A.C. Viability and Volume of in Situ Bovine Articular Chondrocytes-Changes Following a Single Impact and Effects of Medium Osmolarity. Osteoarthr. Cartil. 2005, 13, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Zheng, Y.; Zhang, G.; Hu, Y.; Fan, X.; Hou, Y.; Wen, L.; Li, L.; Xu, Y.; Wang, Y.; et al. Single-Cell RNA-Seq Analysis Reveals the Progression of Human Osteoarthritis. Ann. Rheum. Dis. 2019, 78, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Styczynska-Soczka, K.; Amin, A.K.; Hall, A.C. Cell-associated Type I Collagen in Nondegenerate and Degenerate Human Articular Cartilage. J. Cell Physiol. 2021, 236, 7672–7681. [Google Scholar] [CrossRef]
- Haseeb, A.; Kc, R.; Angelozzi, M.; de Charleroy, C.; Rux, D.; Tower, R.J.; Yao, L.; Pellegrino da Silva, R.; Pacifici, M.; Qin, L.; et al. SOX9 Keeps Growth Plates and Articular Cartilage Healthy by Inhibiting Chondrocyte Dedifferentiation/Osteoblastic Redifferentiation. Proc. Natl. Acad. Sci. USA 2021, 118, e2019152118. [Google Scholar] [CrossRef]
- Schminke, B.; Frese, J.; Bode, C.; Goldring, M.B.; Miosge, N. Laminins and Nidogens in the Pericellular Matrix of Chondrocytes: Their Role in Osteoarthritis and Chondrogenic Differentiation. Am. J. Pathol. 2016, 186, 410–418. [Google Scholar] [CrossRef]
- Gómez, R.; Scotece, M.; Conde, J.; Gómez-Reino, J.J.; Lago, F.; Gualillo, O. Adiponectin and Leptin Increase IL-8 Production in Human Chondrocytes. Ann. Rheum. Dis. 2011, 70, 2052–2054. [Google Scholar] [CrossRef]
- Salucci, S.; Falcieri, E.; Battistelli, M. Chondrocyte Death Involvement in Osteoarthritis. Cell Tissue Res. 2022, 389, 159–170. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent Cells and Osteoarthritis: A Painful Connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Shane Anderson, A.; Loeser, R.F. Why Is Osteoarthritis an Age-Related Disease? Best Pract. Res. Clin. Rheumatol. 2010, 24, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Aggarwal, A.; Aggarwal, A.; Bhattacharyya, S.; Kumar, V.; Sharma, V.; Sahni, D. Senescent Chondrogenic Progenitor Cells Derived from Articular Cartilage of Knee Osteoarthritis Patients Contributes to Senescence-Associated Secretory Phenotype via Release of IL-6 and IL-8. Acta Histochem. 2022, 124, 151867. [Google Scholar] [CrossRef] [PubMed]
- Malaise, O.; Tachikart, Y.; Constantinides, M.; Mumme, M.; Ferreira-Lopez, R.; Noack, S.; Krettek, C.; Noël, D.; Wang, J.; Jorgensen, C.; et al. Mesenchymal Stem Cell Senescence Alleviates Their Intrinsic and Seno-Suppressive Paracrine Properties Contributing to Osteoarthritis Development. Aging 2019, 11, 9128–9146. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Nagase, H. Proteases Involved in Cartilage Matrix Degradation in Osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef]
- Calvo, E.; Palacios, I.; Delgado, E.; Sánchez-Pernaute, O.; Largo, R.; Egido, J.; Herrero-Beaumont, G. Histopathological Correlation of Cartilage Swelling Detected by Magnetic Resonance Imaging in Early Experimental Osteoarthritis. Osteoarthr. Cartil. 2004, 12, 878–886. [Google Scholar] [CrossRef]
- Bank, R.A.; Soudry, M.; Maroudas, A.; Mizrahi, J.; TeKoppele, J.M. The Increased Swelling and Instantaneous Deformation of Osteoarthritic Cartilage Is Highly Correlated with Collagen Degradation. Arthritis Rheum. 2000, 43, 2202–2210. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Turunen, M.J.; Finnilä, M.A.; Joukainen, A.; Kröger, H.; Saarakkala, S.; Korhonen, R.K.; Tanska, P. Structure–Function Relationships of Healthy and Osteoarthritic Human Tibial Cartilage: Experimental and Numerical Investigation. Ann. Biomed. Eng. 2020, 48, 2887–2900. [Google Scholar] [CrossRef]
- Saarakkala, S.; Julkunen, P.; Kiviranta, P.; Mäkitalo, J.; Jurvelin, J.S.; Korhonen, R.K. Depth-Wise Progression of Osteoarthritis in Human Articular Cartilage: Investigation of Composition, Structure and Biomechanics. Osteoarthr. Cartil. 2010, 18, 73–81. [Google Scholar] [CrossRef]
- Mort, J.S.; Geng, Y.; Fisher, W.D.; Roughley, P.J. Aggrecan Heterogeneity in Articular Cartilage from Patients with Osteoarthritis. BMC Musculoskelet. Disord. 2016, 17, 89. [Google Scholar] [CrossRef]
- Lorenzo, P.; Bayliss, M.T.; Heinegård, D. Altered Patterns and Synthesis of Extracellular Matrix Macromolecules in Early Osteoarthritis. Matrix Biol. 2004, 23, 381–391. [Google Scholar] [CrossRef]
- Wilusz, R.E.; Sanchez-Adams, J.; Guilak, F. The Structure and Function of the Pericellular Matrix of Articular Cartilage. Matrix Biol. 2014, 39, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Wang, M.; Zhao, S.; Zhao, Z.; Fang, J. Mechanotransduction Pathways in the Regulation of Cartilage Chondrocyte Homoeostasis. J. Cell. Mol. Med. 2020, 24, 5408–5419. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F. Integrins and Chondrocyte–Matrix Interactions in Articular Cartilage. Matrix Biol. 2014, 39, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Reappraising Metalloproteinases in Rheumatoid Arthritis and Osteoarthritis: Destruction or Repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.F.; Smookler, D.S.; Khokha, R. Metalloproteinases, Inflammation, and Rheumatoid Arthritis. Ann. Rheum. Dis. 2003, 62 (Suppl. 2), ii43–ii47. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM Metalloproteinases in Osteoarthritis–Looking beyond the ‘Usual Suspects’. Osteoarthr. Cartil. 2017, 25, 1000–1009. [Google Scholar] [CrossRef]
- Yamamoto, K.; Troeberg, L.; Scilabra, S.D.; Pelosi, M.; Murphy, C.L.; Strickland, D.K.; Nagase, H. LRP-1-mediated Endocytosis Regulates Extracellular Activity of ADAMTS-5 in Articular Cartilage. FASEB J. 2013, 27, 511–521. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yang, H.; Xu, Z.; Li, J.; Chen, G.; Jiang, L.; Wu, L.; Zhou, X. A Functional Polymorphism in the Paired Basic Amino Acid-Cleaving Enzyme 4 Gene Confers Osteoarthritis Risk in a Population of Eastern China. Genet. Mol. Biol. 2020, 43, e20190115. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, L.; Zhang, Y.; Zhang, J.; Zhou, D.; Wu, L.; Huang, Y.; Xu, N. Genetic Variation of Aggrecanase-2 (ADAMTS5) in Susceptibility to Osteoarthritis. Braz. J. Med. Biol. Res. 2019, 52, e8109. [Google Scholar] [CrossRef]
- Stefik, D.; Vranic, V.; Ivkovic, N.; Abazovic, D.; Maric, D.; Vojvodic, D.; Supic, G. An Insight into Osteoarthritis Susceptibility: Integration of Immunological and Genetic Background. Bosn. J. Basic Med. Sci. 2020. [Google Scholar] [CrossRef]
- Sulzbacher, I. Osteoarthritis: Histology and Pathogenesis. Wien. Med. Wochenschr. 2013, 163, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Huang, J.; Huang, D.; Lv, H.; Wang, D.; Wang, H.; Miao, H.; Wu, L.; Wang, F. Deficiency of Immune-Responsive Gene 1 Exacerbates Interleukin-1beta-Elicited the Inflammatory Response of Chondrocytes via Enhancing the Activation of NLRP3 Inflammasome. Int. Immunopharmacol. 2023, 114, 109456. [Google Scholar] [CrossRef] [PubMed]
- Aho, O.-M.; Finnilä, M.; Thevenot, J.; Saarakkala, S.; Lehenkari, P. Subchondral Bone Histology and Grading in Osteoarthritis. PLoS ONE 2017, 12, e0173726. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, X.; Wang, S.; Jing, Y.; Su, J. Subchondral Bone Microenvironment in Osteoarthritis and Pain. Bone Res. 2021, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Huebner, J.L.; Bay-Jensen, A.C.; Huffman, K.M.; He, Y.; Leeming, D.J.; McDaniel, G.E.; Karsdal, M.A.; Kraus, V.B. Alpha C-Telopeptide of Type I Collagen Is Associated With Subchondral Bone Turnover and Predicts Progression of Joint Space Narrowing and Osteophytes in Osteoarthritis: Urinary α-CTX Predicts OA Progression. Arthritis Rheumatol. 2014, 66, 2440–2449. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xiao, Q.; Hu, Z.; Pu, B.; Shu, J.; Yang, Q.; Lao, H.; Hao, J. Tissue Levels of Leukemia Inhibitory Factor Vary by Osteoarthritis Grade. Orthopedics 2014, 37, e460–e464. [Google Scholar] [CrossRef]
- Alliston, T.; Hernandez, C.J.; Findlay, D.M.; Felson, D.T.; Kennedy, O.D. Bone Marrow Lesions in Osteoarthritis: What Lies beneath: BMLs WHAT LIES BENEATH. J. Orthop. Res. 2018, 36, 1818–1825. [Google Scholar] [CrossRef]
- Klement, M.R.; Sharkey, P.F. The Significance of Osteoarthritis-Associated Bone Marrow Lesions in the Knee. J. Am. Acad. Orthop. Surg. 2019, 27, 752–759. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for Osteocyte Regulation of Bone Homeostasis through RANKL Expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Jaiprakash, A.; Prasadam, I.; Feng, J.Q.; Liu, Y.; Crawford, R.; Xiao, Y. Phenotypic Characterization of Osteoarthritic Osteocytes from the Sclerotic Zones: A Possible Pathological Role in Subchondral Bone Sclerosis. Int. J. Biol. Sci. 2012, 8, 406–417. [Google Scholar] [CrossRef]
- Yee, C.S.; Schurman, C.A.; White, C.R.; Alliston, T. Investigating Osteocytic Perilacunar/Canalicular Remodeling. Curr. Osteoporos. Rep. 2019, 17, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Mazur, C.M.; Woo, J.J.; Yee, C.S.; Fields, A.J.; Acevedo, C.; Bailey, K.N.; Kaya, S.; Fowler, T.W.; Lotz, J.C.; Dang, A.; et al. Osteocyte Dysfunction Promotes Osteoarthritis through MMP13-Dependent Suppression of Subchondral Bone Homeostasis. Bone Res. 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.W.; Acevedo, C.; Mazur, C.M.; Hall-Glenn, F.; Fields, A.J.; Bale, H.A.; Ritchie, R.O.; Lotz, J.C.; Vail, T.P.; Alliston, T. Glucocorticoid Suppression of Osteocyte Perilacunar Remodeling Is Associated with Subchondral Bone Degeneration in Osteonecrosis. Sci. Rep. 2017, 7, 44618. [Google Scholar] [CrossRef] [PubMed]
- Hemmatian, H.; Jalali, R.; Semeins, C.M.; Hogervorst, J.M.A.; van Lenthe, G.H.; Klein-Nulend, J.; Bakker, A.D. Mechanical Loading Differentially Affects Osteocytes in Fibulae from Lactating Mice Compared to Osteocytes in Virgin Mice: Possible Role for Lacuna Size. Calcif. Tissue Int. 2018, 103, 675–685. [Google Scholar] [CrossRef]
- Busse, B.; Djonic, D.; Milovanovic, P.; Hahn, M.; Püschel, K.; Ritchie, R.O.; Djuric, M.; Amling, M. Decrease in the Osteocyte Lacunar Density Accompanied by Hypermineralized Lacunar Occlusion Reveals Failure and Delay of Remodeling in Aged Human Bone. Aging Cell 2010, 9, 1065–1075. [Google Scholar] [CrossRef]
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone Remodeling: An Operational Process Ensuring Survival and Bone Mechanical Competence. Bone Res. 2022, 10, 48. [Google Scholar] [CrossRef]
- Andreev, D.; Liu, M.; Weidner, D.; Kachler, K.; Faas, M.; Grüneboom, A.; Schlötzer-Schrehardt, U.; Muñoz, L.E.; Steffen, U.; Grötsch, B.; et al. Osteocyte Necrosis Triggers Osteoclast-Mediated Bone Loss through Macrophage-Inducible C-Type Lectin. J. Clin. Investig. 2020, 130, 4811–4830. [Google Scholar] [CrossRef]
- Bouaziz, W.; Funck-Brentano, T.; Lin, H.; Marty, C.; Ea, H.-K.; Hay, E.; Cohen-Solal, M. Loss of Sclerostin Promotes Osteoarthritis in Mice via β-Catenin-Dependent and -Independent Wnt Pathways. Arthritis Res. Ther. 2015, 17, 24. [Google Scholar] [CrossRef]
- Sanchez, C.; Deberg, M.A.; Bellahcène, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic Characterization of Osteoblasts from the Sclerotic Zones of Osteoarthritic Subchondral Bone. Arthritis Rheum. 2008, 58, 442–455. [Google Scholar] [CrossRef]
- Couchourel, D.; Aubry, I.; Delalandre, A.; Lavigne, M.; Martel-Pelletier, J.; Pelletier, J.-P.; Lajeunesse, D. Altered Mineralization of Human Osteoarthritic Osteoblasts Is Attributable to Abnormal Type I Collagen Production. Arthritis Rheum. 2009, 60, 1438–1450. [Google Scholar] [CrossRef]
- Housmans, B.A.; van den Akker, G.G.; Neefjes, M.; Surtel, D.A.; Cremers, A.; van der Kraan, P.; van Rhijn, L.; Welting, T. OA Synovial Fluid Induces Chondrocyte Fibrosis and Proliferation via MAPK and RHOGTPASE Signaling Routes. Osteoarthr. Cartil. 2021, 29, S136. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Z.; Guo, H.; Wang, Z.; Sun, K.; Yang, X.; Zhao, X.; Ma, L.; Wang, J.; Meng, Z.; et al. Extensive Cytokine Analysis in Synovial Fluid of Osteoarthritis Patients. Cytokine 2021, 143, 155546. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M.; Stefani, R.M.; Sobczak, E.; Tong, E.L.; Attur, M.G.; Shah, R.P.; Bulinski, J.C.; Ateshian, G.A.; Hung, C.T. Toward Understanding the Role of Cartilage Particulates in Synovial Inflammation. Osteoarthr. Cartil. 2017, 25, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhou, J.; Ruan, A.; Guan, H.; Xie, J.; Zeng, L.; Liu, J.; Wang, Q. Synovial Tissue-Derived Extracellular Vesicles Induce Chondrocyte Inflammation and Degradation via NF-ΚB Signalling Pathway: An in Vitro Study. J. Cell. Mol. Med. 2022, 26, 2038–2048. [Google Scholar] [CrossRef]
- Zreiqat, H.; Belluoccio, D.; Smith, M.M.; Wilson, R.; Rowley, L.A.; Jones, K.; Ramaswamy, Y.; Vogl, T.; Roth, J.; Bateman, J.F.; et al. S100A8 and S100A9 in Experimental Osteoarthritis. Arthritis Res. Ther. 2010, 12, R16. [Google Scholar] [CrossRef]
- Nakashima, M.; Sakai, T.; Hiraiwa, H.; Hamada, T.; Omachi, T.; Ono, Y.; Inukai, N.; Ishizuka, S.; Matsukawa, T.; Oda, T.; et al. Role of S100A12 in the Pathogenesis of Osteoarthritis. Biochem. Biophys. Res. Commun. 2012, 422, 508–514. [Google Scholar] [CrossRef]
- Chan, M.W.Y.; Gomez-Aristizábal, A.; Mahomed, N.; Gandhi, R.; Viswanathan, S. A Tool for Evaluating Novel Osteoarthritis Therapies Using Multivariate Analyses of Human Cartilage-Synovium Explant Co-Culture. Osteoarthr. Cartil. 2022, 30, 147–159. [Google Scholar] [CrossRef]
- Fahy, N.; Melle, M.L.d.V.; Lehmann, J.; Wei, W.; Grotenhuis, N.; Farrell, E.; van der Kraan, P.M.; Murphy, J.M.; Bastiaansen-Jenniskens, Y.M.; Osch, G.J.V.M. van Human Osteoarthritic Synovium Impacts Chondrogenic Differentiation of Mesenchymal Stem Cells via Macrophage Polarisation State. Osteoarthr. Cartil. 2014, 22, 1167–1175. [Google Scholar] [CrossRef]
- Intema, F.; Hazewinkel, H.A.W.; Gouwens, D.; Bijlsma, J.W.J.; Weinans, H.; Lafeber, F.P.J.G.; Mastbergen, S.C. In Early OA, Thinning of the Subchondral Plate Is Directly Related to Cartilage Damage: Results from a Canine ACLT-Meniscectomy Model. Osteoarthr. Cartil. 2010, 18, 691–698. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Y.-C.; Yan, C.H.; Chiu, K.Y.; Wei, Q.; Zhao, J.; Guo, X.E.; Leung, F.; Lu, W.W. Abnormal Subchondral Bone Remodeling and Its Association with Articular Cartilage Degradation in Knees of Type 2 Diabetes Patients. Bone Res. 2017, 5, 17034. [Google Scholar] [CrossRef]
- Fell, N.L.A.; Lawless, B.M.; Cox, S.C.; Cooke, M.E.; Eisenstein, N.M.; Shepherd, D.E.T.; Espino, D.M. The Role of Subchondral Bone, and Its Histomorphology, on the Dynamic Viscoelasticity of Cartilage, Bone and Osteochondral Cores. Osteoarthr. Cartil. 2019, 27, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Bellido, M.; Lugo, L.; Roman-Blas, J.A.; Castañeda, S.; Calvo, E.; Largo, R.; Herrero-Beaumont, G. Improving Subchondral Bone Integrity Reduces Progression of Cartilage Damage in Experimental Osteoarthritis Preceded by Osteoporosis. Osteoarthr. Cartil. 2011, 19, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Neogi, T.; Felson, D.; Niu, J.; Lynch, J.; Nevitt, M.; Guermazi, A.; Roemer, F.; Lewis, C.E.; Wallace, B.; Zhang, Y. Cartilage Loss Occurs in the Same Subregions as Subchondral Bone Attrition: A within-Knee Subregion-Matched Approach from the Multicenter Osteoarthritis Study. Arthritis Rheum. 2009, 61, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Liu, G.; Liu, X.; Zhou, Y.; Sun, Q.; Zhen, G.; Wang, X.; Hu, Y.; Gao, P.; Demehri, S.; et al. Angiogenesis Stimulated by Elevated PDGF-BB in Subchondral Bone Contributes to Osteoarthritis Development. JCI Insight 2020, 5, e135446. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Bonnet, C.S.; Turner, E.L.; Wilson, D.; Situ, M.; McWilliams, D.F. Angiogenesis in the Synovium and at the Osteochondral Junction in Osteoarthritis. Osteoarthr. Cartil. 2007, 15, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Aso, K.; Shahtaheri, S.M.; Hill, R.; Wilson, D.; McWilliams, D.F.; Nwosu, L.N.; Chapman, V.; Walsh, D.A. Contribution of Nerves within Osteochondral Channels to Osteoarthritis Knee Pain in Humans and Rats. Osteoarthr. Cartil. 2020, 28, 1245–1254. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-Inflammatory Cytokines: The Link between Obesity and Osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Yu, C.D.; Miao, W.H.; Zhang, Y.Y.; Zou, M.J.; Yan, X.F. Inhibition of MiR-126 Protects Chondrocytes from IL-1β Induced Inflammation via Upregulation of Bcl-2. Bone Jt. Res. 2018, 7, 414–421. [Google Scholar] [CrossRef]
- Mehana, E.-S.E.; Khafaga, A.F.; El-Blehi, S.S. The Role of Matrix Metalloproteinases in Osteoarthritis Pathogenesis: An Updated Review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.-K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic Inhibition of IL-6/Stat3 Signalling Protects against Experimental Osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 Functions as an Osteoclastogenic Helper T Cell Subset That Links T Cell Activation and Bone Destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Sinkeviciute, D.; Aspberg, A.; He, Y.; Bay-Jensen, A.-C.; Önnerfjord, P. Characterization of the Interleukin-17 Effect on Articular Cartilage in a Translational Model: An Explorative Study. BMC Rheumatol. 2020, 4, 30. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, P.; Yang, D.; Wang, F.; Yuan, L.; Lin, Z.; Jiang, J. Interleukin-18-Induced Inflammatory Responses in Synoviocytes and Chondrocytes from Osteoarthritic Patients. Int. J. Mol. Med. 2012, 30, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, J.; Liu, Q.; Luo, A. Tumor Necrosis Factor-α Induces ADAMTS-4 Expression in Human Osteoarthritis Chondrocytes. Mol. Med. Rep. 2013, 8, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Hore, Z.; Pattison, L.A.; Lalnunhlimi, S.; Bhebhe, C.N.; Callejo, G.; Bulmer, D.C.; Taams, L.S.; Denk, F.; Smith, E.S.T.J. Sensitization of Knee-Innervating Sensory Neurons by Tumor Necrosis Factor-α-Activated Fibroblast-like Synoviocytes: An in Vitro, Coculture Model of Inflammatory Pain. Pain 2020, 161, 2129–2141. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, K.A.; Papavassiliou, A.G. Redox and NF-ΚB Signaling in Osteoarthritis. Free Radic. Biol. Med. 2019, 132, 90–100. [Google Scholar] [CrossRef]
- Conde, J.; Otero, M.; Scotece, M.; Abella, V.; López, V.; Pino, J.; Gómez, R.; Lago, F.; Goldring, M.B.; Gualillo, O. E74-like Factor 3 and Nuclear Factor-ΚB Regulate Lipocalin-2 Expression in Chondrocytes. J. Physiol. 2016, 594, 6133–6146. [Google Scholar] [CrossRef]
- Choi, M.-C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-ΚB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Chemical Messenger | Signaling Factor | Cells | Effect | References |
|---|---|---|---|---|
| Transcription factor | SOX-9 1 | Synoviocytes, chondrocytes, osteoblast lineage cells | Maintains the chondrogenic phenotype and functions of the chondrocyte, prevents hypertrophy, antagonizing with RUNX2 Inhibits IL-1β-induced inflammatory response and chondrocyte apoptosis Inhibits the production of ADAMTS in cartilage tissue | [97,109,110] |
| Growth factor | IGF-1 2 | Synoviocytes, chondrocytes, osteoblast lineage cells | Exhibits anti-inflammatory and anti-catabolic effects in chondrocytes Maintains articular cartilage anabolism, stimulating ECM production and chondrogenesis | [111,112] |
| Growth factor | TGF-β 3 | Chondrocytes, synovial fibroblasts, and macrophages | Inhibits chondrocytes phenotype switch to hypertrophic chondrocytes and collagen type X production Induces proteoglycan synthesis by chondrocytes Inhibits IL-1β effects | [113] |
| Chemical Messenger | Signaling Factor | Cells | Effect | References |
|---|---|---|---|---|
| Cytokine | IL-1β 1 | Synovial macrophages and fibroblasts, chondrocytes, osteoblasts | Induces cartilage degradation and inhibits its repair abilities Stimulates production of MMPs (MMP-1, 3, 9, 13), ADAMTSs 4, 5 by chondrocytes Suppresses the synthesis of collagen type II and aggrecan Enhances chondrocytes’ pro-inflammatory response, hypertrophy, dedifferentiation, and apoptosis, inhibits chondrogenesis Stimulates synovial inflammation and the production of pro-inflammatory cytokines (TNF-α, IL-6, IL-8, IL-17, CCL5), mediators (NO, COX-1), prostaglandins (PGE2) Induces the formation of pannus-like tissue, fibrosis, and production of pro-fibrotic factors (PDGF, TGF-β) | [126,133,212,247,248,249] |
| Cytokine | IL-6 | Synovial macrophages and fibroblasts, chondrocytes, osteoblasts | Promotes osteoclast formation and subchondral bone resorption Increases production of MMPs (MMP-1, 3, 13) and ADAMTS (ADAMTS-4) by chondrocytes Induces catabolic changes in chondrocytes, as well as promotes cellular senescence Shows synergic action with IL-1β and TNF-α, sustaining articular cartilage degradation and synovial inflammation | [192,250] |
| Cytokine | IL-17 | CD4+ T cells, macrophages, NK cells, mast cells | Induces cartilage degradation by upregulating catabolic factors (MMP-1, 3, 13; ADAMTS) and downregulating anabolic factors (SOX-9, COL2A1) in chondrocytes Promotes recruitment of inflammatory cells and release of pro-inflammatory mediators, induces angiogenesis Induces RANKL expression and osteoclastogenesis | [251,252] |
| Cytokine | IL-18 | Synovial macrophages, fibroblasts, Chondrocytes, and osteoblasts | Promotes articular cartilage degradation by upregulating MMP-1, 3, 13 and suppressing aggrecan synthesis Stimulates pro-inflammatory conditions by induction of cytokines synthesis through NF-κB and MAPK signaling pathways Stimulates both bone resorption and osteophyte formation Enhances gene expression for the synthesis of IL-6, TNF-α | [232,253,254] |
| Cytokine | TNF-α 2 | Synovial macrophages and fibroblasts, chondrocytes, osteoblasts | Levels of expression are associated with radiographic OA cartilage loss Shows action synergism with IL-1β Stimulates MMP and ADAMTS production by chondrocytes; inhibits synthesis of collagen type II and aggrecan Inhibits chondrocyte differentiation by suppressing the expression of SOX-9, and induces apoptosis Promotes pro-inflammatory signaling pathways in synoviocytes and chondrocytes, stimulating the release of IL-1β, IL-6, IL-8, IL-10 Leads to neuronal sensitization, predisposing to the development of pain in OA Promotes angiogenesis and aberrant bone formation in subchondral bone by recruiting mesenchymal stem cells | [110,134,248,253,255,256,257] |
| Enzyme | MMPs 3 | Chondrocytes, synovial macrophages, and fibroblasts | MMP-1 degrades collagen types I, II, III, and aggrecan of articular cartilage MMP-3 cleaves collagen types II, IV, IX, X, XI, and aggrecan; activates other MMPs (MMP-1, 7, 9) MMP-9 cleaves non-collagenous matric components MMP-13 levels correlate with hypertrophic chondrocytes in early stage OA; with OA severity and articular cartilage deterioration, as well as NF-κB expression; the enzyme exhibits higher activity for collagen type II cleavage and degrades aggrecan; it is associated with synovial membrane hyperplasia and cellular senescence | [88,133,249] |
| Enzyme | ADAMTSs 4 | Chondrocytes, synovial fibroblasts, and macrophages | ADAMTS-4, 5 are induced by IL-1β, TNF-α in chondrocytes and promote cleavage of aggrecan | [206] |
| Transcription factor | NF-κB 5 | All joint cells | Acts alone or in synergy with other signaling pathways Inhibits anabolic functions of chondrocytes Triggers chondrocyte hypertrophy, apoptosis, catabolic functions (MMP, ADAMTS, NO, PGE2, COX2 production), production of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-8) by chondrocytes, which augment the action of NF-κB Augments activation of other transcription factors such as ELF3, and RUNX2, that stimulate MMP13 and cartilage degradation In synovial membrane promotes inflammation, angiogenesis, production of cytokines (IL-1β, TNF-α, IL-6), enzymes (MMP-1, MMP-13, ADAMTS-4, ADAMTS-5), VEGF In subchondral bone promotes resorption | [258,259,260] |
| Transcription factor | SOX-9 6 | All joint cells | Expression is downregulated in OA-affected joint Downregulation results in calcified cartilage and osteophyte formation | [110] |
| Growth factor | TGF-β 7 | Chondrocytes, osteoblasts, osteoclasts, synovial fibroblasts, and macrophages | Leads to the osteoid formation and bone sclerosis Leads to cartilage damage and angiogenesis | [113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenistaja, S.; Skuja, S.; Kadisa, A.; Groma, V. Healthy and Osteoarthritis-Affected Joints Facing the Cellular Crosstalk. Int. J. Mol. Sci. 2023, 24, 4120. https://doi.org/10.3390/ijms24044120
Semenistaja S, Skuja S, Kadisa A, Groma V. Healthy and Osteoarthritis-Affected Joints Facing the Cellular Crosstalk. International Journal of Molecular Sciences. 2023; 24(4):4120. https://doi.org/10.3390/ijms24044120
Chicago/Turabian StyleSemenistaja, Sofija, Sandra Skuja, Anda Kadisa, and Valerija Groma. 2023. "Healthy and Osteoarthritis-Affected Joints Facing the Cellular Crosstalk" International Journal of Molecular Sciences 24, no. 4: 4120. https://doi.org/10.3390/ijms24044120
APA StyleSemenistaja, S., Skuja, S., Kadisa, A., & Groma, V. (2023). Healthy and Osteoarthritis-Affected Joints Facing the Cellular Crosstalk. International Journal of Molecular Sciences, 24(4), 4120. https://doi.org/10.3390/ijms24044120