

Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data
 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Comparison of Genomes Structure
2.2. Comparison of Sequences on Multiple Sequence Alignment Plot
2.3. Homology Based Tools Comparison
2.4. RagTag Tool Results
2.5. BLAST Search Results
2.6. Integration of DArTseq Experiment Results
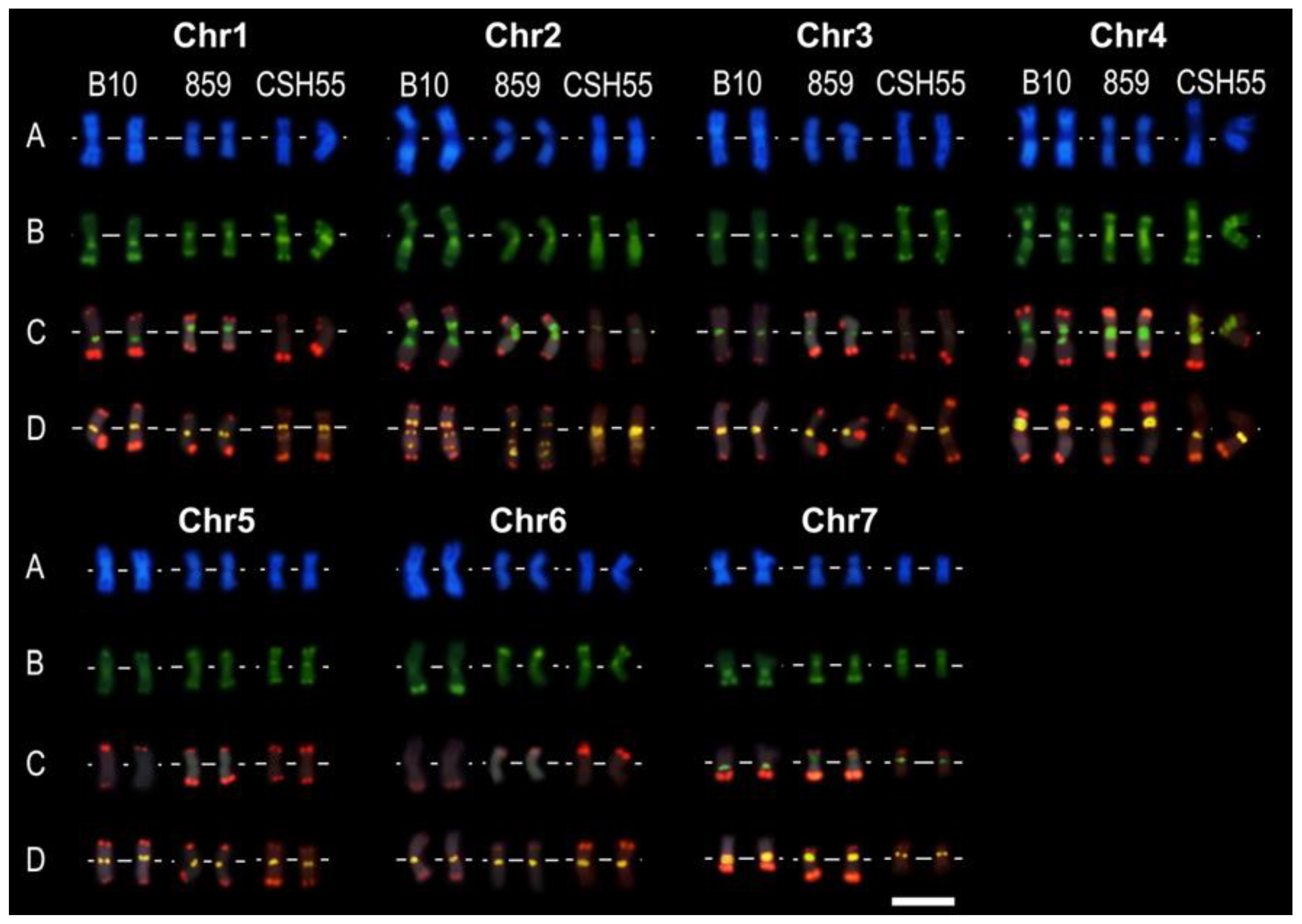
2.7. Integration of FISH Experiment Results
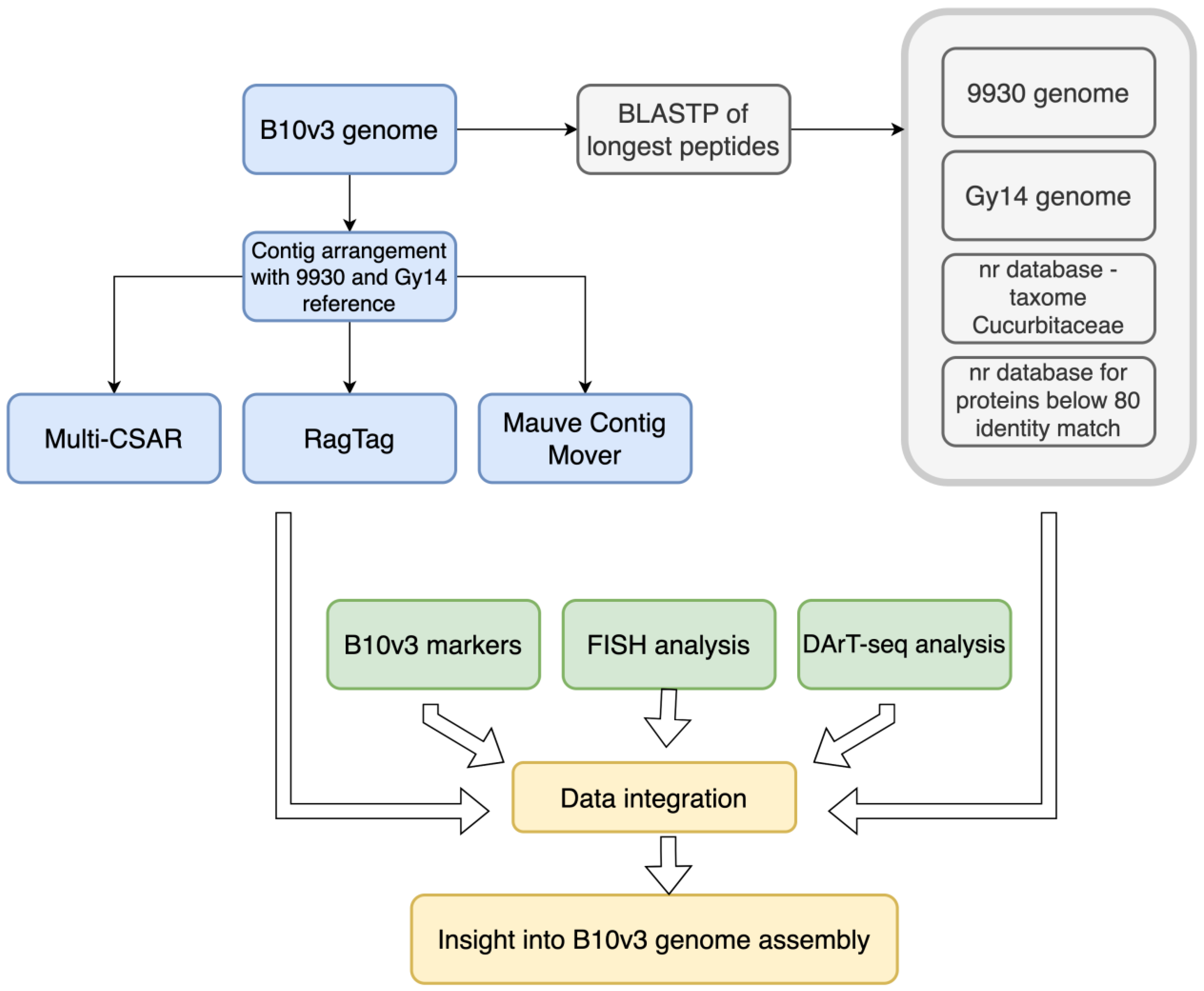
3. Materials and Methods
3.1. Sample Data Curation
3.2. Data Arrangement and Chromosome Assignment
3.3. Comparative Analysis of Coded Protein Sequences
3.4. Integration of Published Data about Cucumber B10 Genome Assembly
3.5. BAC-FISH Experiment Description
3.6. Data Consolidation
- -
- A file integrating RagTag results with BLASTP searches and data for markers used in B10v3 genome stacking together with results of DArT-seq and FISH analyses (Table S1);
- -
- Files showing the results of the BLAST program (Tables S2–S6);
- -
- A file showing the genes assigned to each contig (Table S7).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, L.; Wang, Q.; Zheng, Y.; Guo, J.; Yuan, S.; Fu, A.; Bai, C.; Zhao, X.; Zheng, S.; Wen, C.; et al. Cucurbitaceae genome evolution, gene function, and molecular breeding. Hortic. Res. 2022, 9, uhab057. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, H.; Huang, W.U.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). Gigascience 2019, 8, giz072. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Koo, D.H.; Li, Y.; Zhang, X.; Luan, F.; Havey, M.J.; Jiang, J.; Weng, Y. Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J. 2012, 71, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Osipowski, P.; Pawełkowicz, M.; Wojcieszek, M.; Skarzyńska, A.; Przybecki, Z.; Pląder, W. A high-quality cucumber genome assembly enhances computational comparative genomics. Mol. Genet. Genom. 2020, 295, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef]
- Pawełkowicz, M.; Zieliński, K.; Zielińska, D.; Pląder, W.; Yagi, K.; Wojcieszek, M.; Siedlecka, E.; Bartoszewski, G.; Skarzyńska, A.; Przybecki, Z. Next generation sequencing and omics in cucumber (Cucumis sativus L.) breeding directed research. Plant Sci. 2015, 242, 77–88. [Google Scholar] [CrossRef]
- Słomnicka, R.; Olczak-Woltman, H.; Korzeniewska, A.; Gozdowski, D.; Niemirowicz-Szczytt, K.; Bartoszewski, G. Genetic mapping of psl locus and quantitative trait loci for angular leaf spot resistance in cucumber (Cucumis sativus L.). Mol. Breed. 2018, 38, 111. [Google Scholar] [CrossRef]
- Osipowski, P.; Wojcieszek, M.; Pawełkowicz, M.; Skarzyńska, A.; Koren, S.; Lomsadze, A.; Wóycicki, R.; Pląder, W.; Yagi, K.; Borodovsky, M.; et al. Progress in assembling the cucumber (Cucumis sativus) borszczagowski B10 line genome using long single molecule, real-time reads. In Proceedings of the Cucurbitaceae 2016, XIth Eucarpia Meeting on Cucurbit Genetics & Breeding, Warsaw, Poland, 24–28 July 2016; pp. 72–74. [Google Scholar]
- Lofgren, L.A.; Ross, B.S.; Cramer, R.A.; Stajich, J.E. The pan-genome of Aspergillus fumigatus provides a high-resolution view of its population structure revealing high levels of lineage-specific diversity driven by recombination. PLoS Biol. 2022, 20, e3001890. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J.; Xiong, X.; Cheng, Z.M.; Chen, F. De novo assembly of plant complete genomes. Trop. Plants 2022, 1, 7. [Google Scholar] [CrossRef]
- Alhakami, H.; Mirebrahim, H.; Lonardi, S. A comparative evaluation of genome assembly reconciliation tools. Genome Biol. 2017, 18, 93. [Google Scholar] [CrossRef]
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 15 January 2023).
- Ayyangar, K.R. Taxonomy of the Cucurbitaceae. Bull. Nat. Inst. Sci. India 1967, 34, 380–396. [Google Scholar]
- Trivedi, R.N.R.R. Cytological studies in Cucumis and Citrus. Cytol. Tokyo 1970, 35, 561–569. [Google Scholar] [CrossRef]
- Singh, A.K.; Roy, R.P. Karyological Studies in Cucumis (L.). Caryologia Int. J. Cytol. Cytosyst. Cytogenet. 1974, 27, 153–160. [Google Scholar] [CrossRef]
- Ramachandran, C.S.V. Cytological analysis of the genome of cucumber (Cucumis sativus L.) and muskmclon (Cucumis melo L.). Z. Pflanzenzucht. 1986, 96, 25–38. [Google Scholar]
- Plader, W.; Hoshi, Y.; Malepszy, S. Sequential fluorescent staining with CMA and DAPI for somatic chromosome identification in cucumber (Cucumis sativus L.). J. Appl. Genet. 1998, 39, 249–258. [Google Scholar]
- Hoshi, Y.; Mori, M.; Matoba, H.; Tagashira, N.; Murata, T.; Plader, W.; Malepszy, S. Chromosomal Polymorphism of Two Pickling Cucumbers (Cucumis sativus L.) Revealed by Fluorescent Staining with CMA and DAPI. Cytologia 2008, 71, 41–48. [Google Scholar] [CrossRef][Green Version]
- Tagashira, N.; Plader, W.O.; Filipecki, M.; Yin, Z.; Wisniewska, A.; Gaj, P.; Szwacka, M.; Fiehn, O.; Hoshi, Y.; Kondo, K.; et al. The metabolic profiles of transgenic cucumber lines vary with different chromosomal locations of the transgene. Cell. Mol. Biol. Lett. 2005, 10, 697. [Google Scholar]
- Han, Y.H.; Zhang, Z.H.; Liu, J.H.; Lu, J.Y.; Huang, S.W.; Jin, W.W. Distribution of the tandem repeat sequences and karyotyping in cucumber (Cucumis sativus L.) by fluorescence in situ hybridization. Cytogenet. Genome Res. 2008, 122, 80–88. [Google Scholar] [CrossRef]
- Zhao, X.; Lu, J.; Zhang, Z.; Hu, J.; Huang, S.; Jin, W. Comparison of the distribution of the repetitive DNA sequences in three variants of Cucumis sativus reveals their phylogenetic relationships. J. Genet. Genom. 2011, 38, 39–45. [Google Scholar] [CrossRef]
- Yang, L.; Koo, D.H.; Li, D.; Zhang, T.; Jiang, J.; Luan, F.; Renner, S.S.; Henaff, E.; Sanseverino, W.; Garcia-Mas, J.; et al. Next-generation sequencing, FISH mapping and synteny-based modeling reveal mechanisms of decreasing dysploidy in Cucumis. Plant J. 2014, 77, 16–30. [Google Scholar] [CrossRef]
- Tagashira, N.; Hoshi, Y.; Yagi, K.; Pląder, W.; Malepszy, S. Cytogenetic comparison among three cultivars of cucumber (Cucumis sativus L.) by using post-heated DAPI band, 45S and 5S rDNA sites. Botany 2009, 4, 19–23. [Google Scholar] [CrossRef][Green Version]
- Zhang, Z.T.; Yang, S.Q.; Li, Z.A.; Zhang, Y.X.; Wang, Y.Z.; Cheng, C.Y.; Li, J.; Chen, J.F.; Lou, Q.F. Comparative chromosomal localization of 45S and 5S rDNAs and implications for genome evolution in Cucumis. Genome 2016, 59, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, S.; Sun, H.; Wang, X.; Tang, X.; Guo, S.; Zhang, Z.; Huang, S.; Xu, Y.; Weng, Y.; et al. CuGenDBv2: An updated database for cucurbit genomics. Nucleic Acids. Res. 2022, 51, D1457–D1464. [Google Scholar] [CrossRef] [PubMed]
- GitHub—Raymondkiu/Sequence-Stats: Generate Statistics from FASTQ and FASTA Files. Also Manipulate Sequences Such as Renaming Contigs and Converting FASTQ to FASTA. Written in Bash. All in One Place. n.d. Available online: https://github.com/raymondkiu/sequence-stats (accessed on 15 January 2023).
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Jenike, K.; Ou, S.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 2022, 23, 258. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.; Shen, H.T.; Lu, C.L. Multi-CSAR: A multiple reference-based contig scaffolder using algebraic rearrangements. BMC Syst. Biol. 2018, 12, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- A General Introduction to the E-Utilities—Entrez Programming Utilities Help—NCBI Bookshelf n.d. Available online: https://www.ncbi.nlm.nih.gov/books/NBK25497/ (accessed on 15 January 2023).
- Hoshi, Y.; Kido, M.; Yagi, K.; Tagashira, N.; Morikawa, A.; Nagano, K. Somatic chromosome differentiation in Cucumis melo L. and C. metuliferus E.Mey. ex Naudin. Chromosom. Bot. 2013, 8, 7–12. [Google Scholar] [CrossRef][Green Version]
- Sogin, M.L. Amplification of ribosomal RNA genes for molecular evolution studies. In PCR Protocols: A Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; pp. 307–314. [Google Scholar]
- Helm, M.A.; Hemleben, V. Characterization of a new prominent satellite DNA of Cucumis metuliferus and differential distribution of satellite DNA in cultivated and wild species of Cucumis and in related genera of Cucurbitaceae. Euphytica 1997, 94, 219–226. [Google Scholar] [CrossRef]
- Adobe Inc. Adobe Photoshop. Available online: https://www.adobe.com/products/photoshop.html (accessed on 15 January 2023).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Genome | Contigs | GC (%) | A (%) | T (%) | G (%) | C (%) | N50 | Max | Min | Median |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B10v3 | 3.4 × 108 | 8035 | 35.72 | 32.11 | 32.15 | 17.87 | 17.84 | 857,970 | 12,672,375 | 3684 | 15,301 |
| Gy14 | 2.6 × 108 | 8 | 30.01 | 29.86 | 29.78 | 15.00 | 15.00 | 33,288,019 | 41,698,299 | 23,759,298 | 32,825,499 |
| 9930 | 2.3 × 108 | 85 | 32.81 | 33.60 | 33.56 | 16.40 | 16.40 | 31,125,843 | 40,877,379 | 5476 | 160,015 |
| Number of Found Proteins in BLAST Databases | |||
|---|---|---|---|
| Identity Search Criteria | 9930_db | Gy14_db | Nr Database Narrowed to 3650 Taxon |
| =100 | 9519 | 7422 | 7311 |
| <100 AND ≥95 | 5008 | 4635 | 6002 |
| <95 AND ≥90 | 579 | 1335 | 1488 |
| <90 AND ≥80 | 315 | 906 | 718 |
| <80 | 671 | 1788 | 404 |
| Sum of found protein sequences | 16,092 | 16,086 | 15,923 |
| STC Sequence Identifier | Found Contig with BLASTN Program | Bioinformatics Consensus Result | Chromosome According to BAC Analysis Results |
|---|---|---|---|
| STC1_Bam_024_D19_M13 | ctg105 | Chr2 | Chr2 |
| STC1_Bam_007_M21_M13 | ctg184 | Chr4 | Chr4 |
| STC1_Bam_022_G02_M13 | ctg184 | Chr4 | Chr4 |
| STC1_Bam_001_J07_M13 | ctg184 | Chr4 | Chr3 |
| STC1_Bam_065_A04_M13 | ctg1673 | Chr5 | Chr5 |
| STC1_Bam_002_E06_M13 | ctg2607 | Chr3 | Chr1 |
| STC1_Bam_041_H23_M13 | ctg1047 | Chr7 | Chr7 |
| STC1_Bam_002_L09_M13 | ctg197 | Chr5 | Chr5 |
| STC1_Bam_062_O16_M13 | ctg775 | Chr5 | Chr5 |
| STC1_Bam_072_K05_M13 | ctg105 | Chr2 | Chr2 |
| STC1_Bam_038_P19_M13 | ctg1000 | Chr6 | Chr6 |
| STC1_Bam_065_L02_M13 | ctg775 | Chr5 | Chr5 |
| Contig Name | Chromosome B10v3 Markers | Chromosome 9930 RagTag | Chromosome Gy14 RagTag | Chromosome Blast 9930db | Chromosome Blast gy14db | Chromosome According to DArT-Seq Analysis | Chromosome According to BAC Analysis |
|---|---|---|---|---|---|---|---|
| ctg2607 | Chr3 | Chr3_9930 | Chr3_Gy14 | Chr3 | Chr3 | Chr3 | Chr1 |
| ctg105 | Chr2 | Chr2_9930 | Chr2_Gy14 | Chr2 | Chr2 | Chr2 | Chr2 |
| ctg184 | Chr4 | Chr4_9930 | Chr4_Gy14 | Chr4 | Chr4 | Chr4 | Chr4 |
| ctg197 | Chr5 | Chr5_9930 | Chr5_Gy14 | Chr5 | Chr5 | Chr5 | Chr5 |
| ctg1673 | Chr5 | Chr5_9930 | Chr5_Gy14 | Chr5 | Chr5 | Chr5 | Chr5 |
| ctg1047 | Chr7 | Chr7_9930 | Chr7_Gy14 | Chr7 | Chr7 | Chr7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turek, S.; Pląder, W.; Hoshi, Y.; Skarzyńska, A.; Pawełkowicz, M. Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data. Int. J. Mol. Sci. 2023, 24, 4011. https://doi.org/10.3390/ijms24044011
Turek S, Pląder W, Hoshi Y, Skarzyńska A, Pawełkowicz M. Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data. International Journal of Molecular Sciences. 2023; 24(4):4011. https://doi.org/10.3390/ijms24044011
Chicago/Turabian StyleTurek, Szymon, Wojciech Pląder, Yoshikazu Hoshi, Agnieszka Skarzyńska, and Magdalena Pawełkowicz. 2023. "Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data" International Journal of Molecular Sciences 24, no. 4: 4011. https://doi.org/10.3390/ijms24044011
APA StyleTurek, S., Pląder, W., Hoshi, Y., Skarzyńska, A., & Pawełkowicz, M. (2023). Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data. International Journal of Molecular Sciences, 24(4), 4011. https://doi.org/10.3390/ijms24044011