
The Pre-Stroke Induction and Normalization of Insulin Resistance Respectively Worsens and Improves Functional Recovery

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
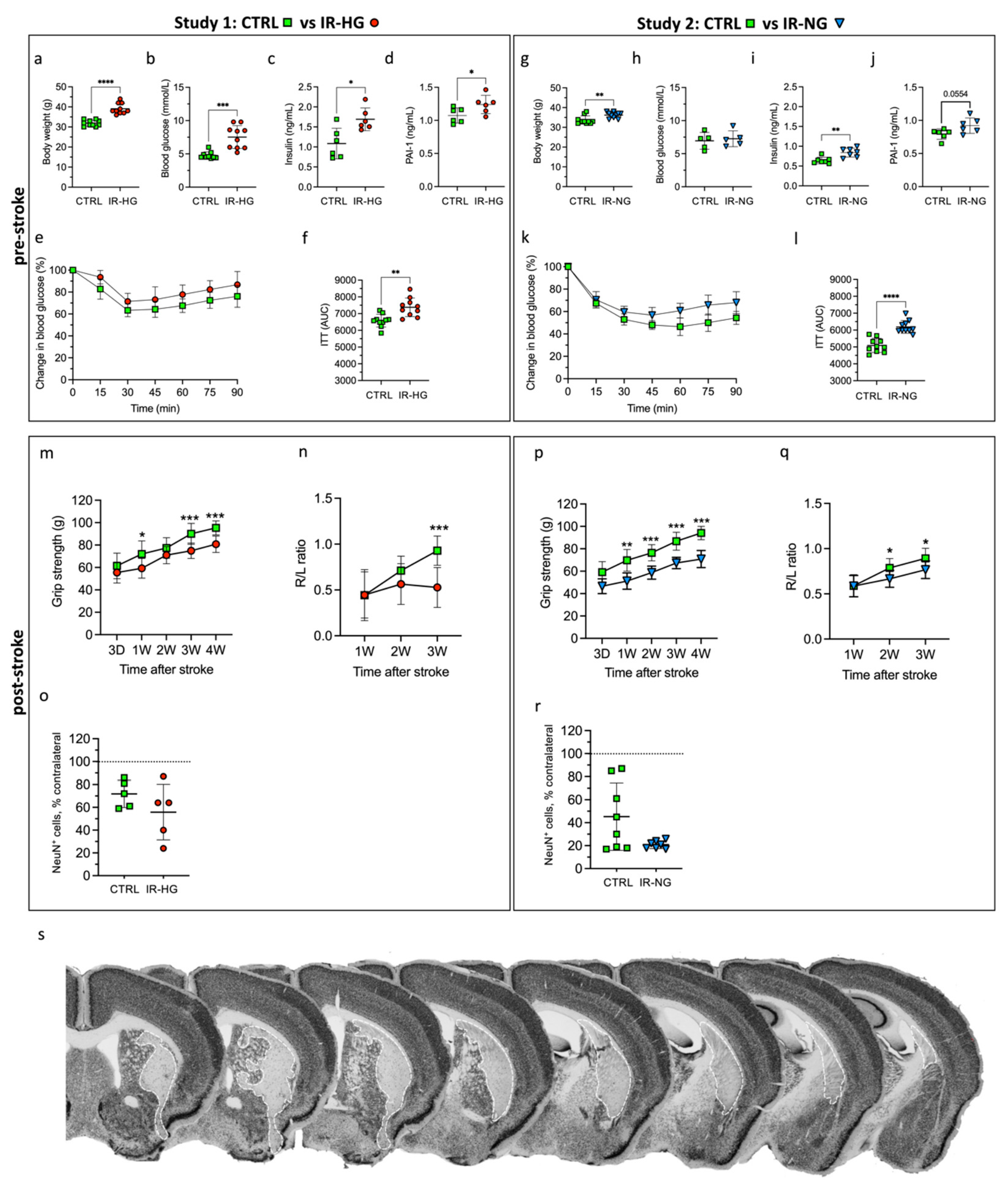
2.1. HFD Induces IR and Hyperglycemia while 30% Sucrose in Drinking Water Leads to Early IR without Hyperglycemia
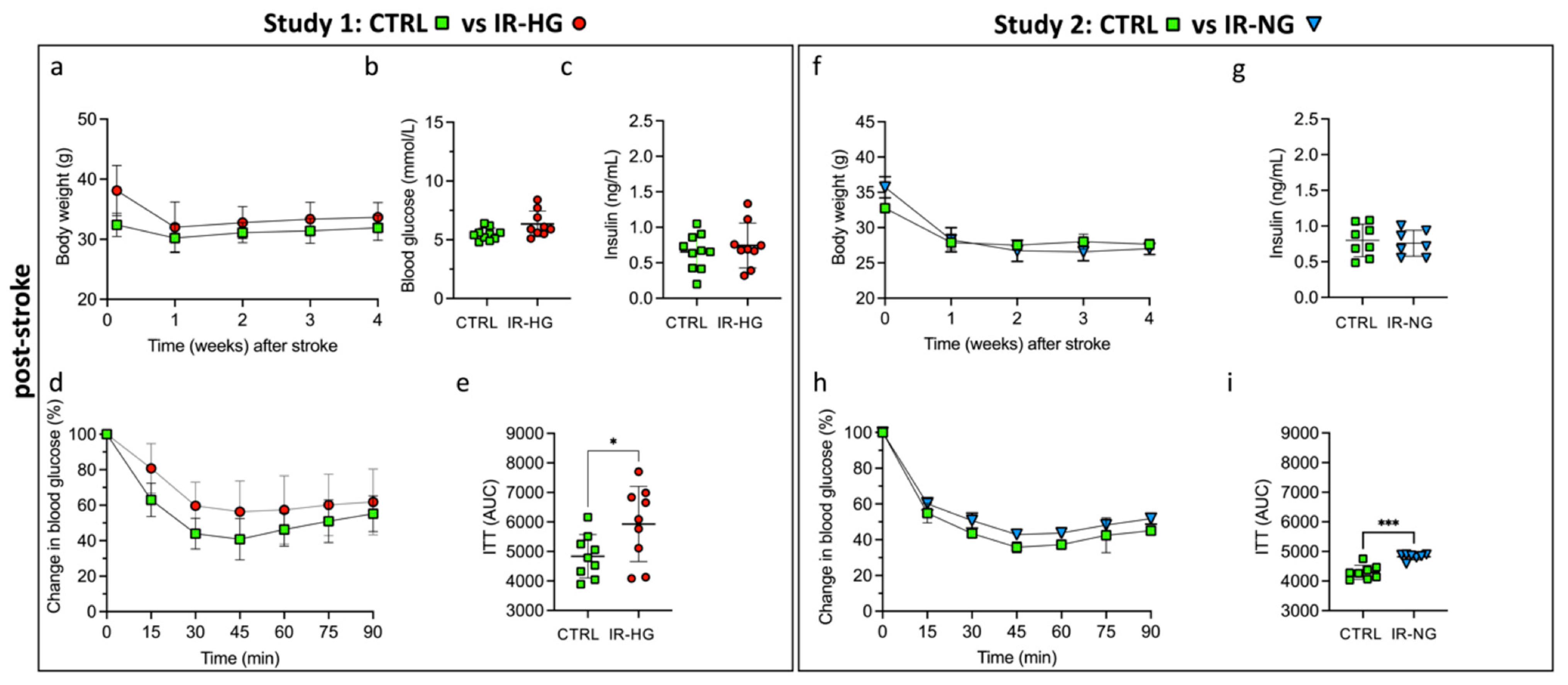
2.2. IR with or without Hyperglycemia Similarly Impairs Stroke Recovery
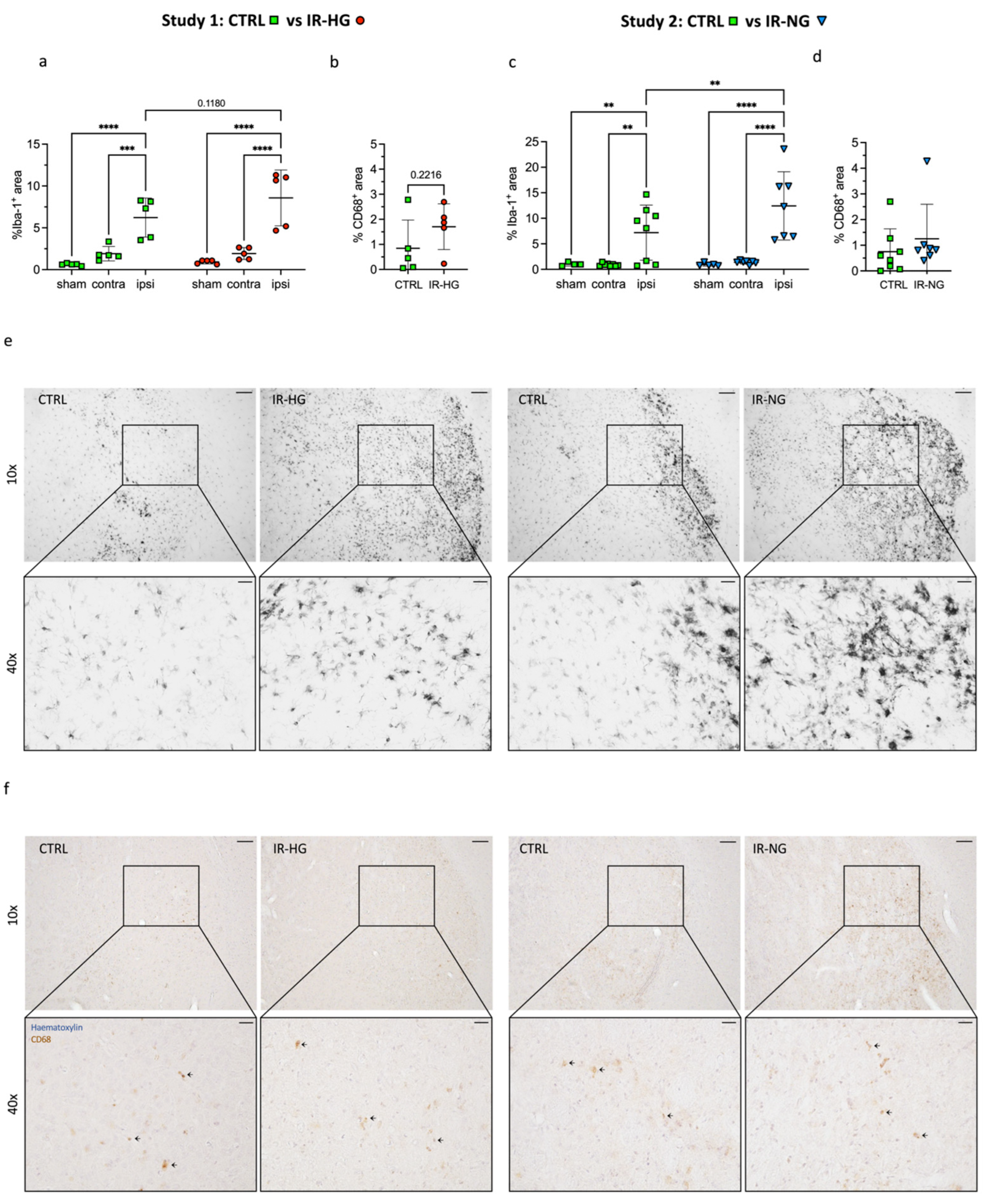
2.3. IR Significantly Increases Stroke-Induced Inflammation
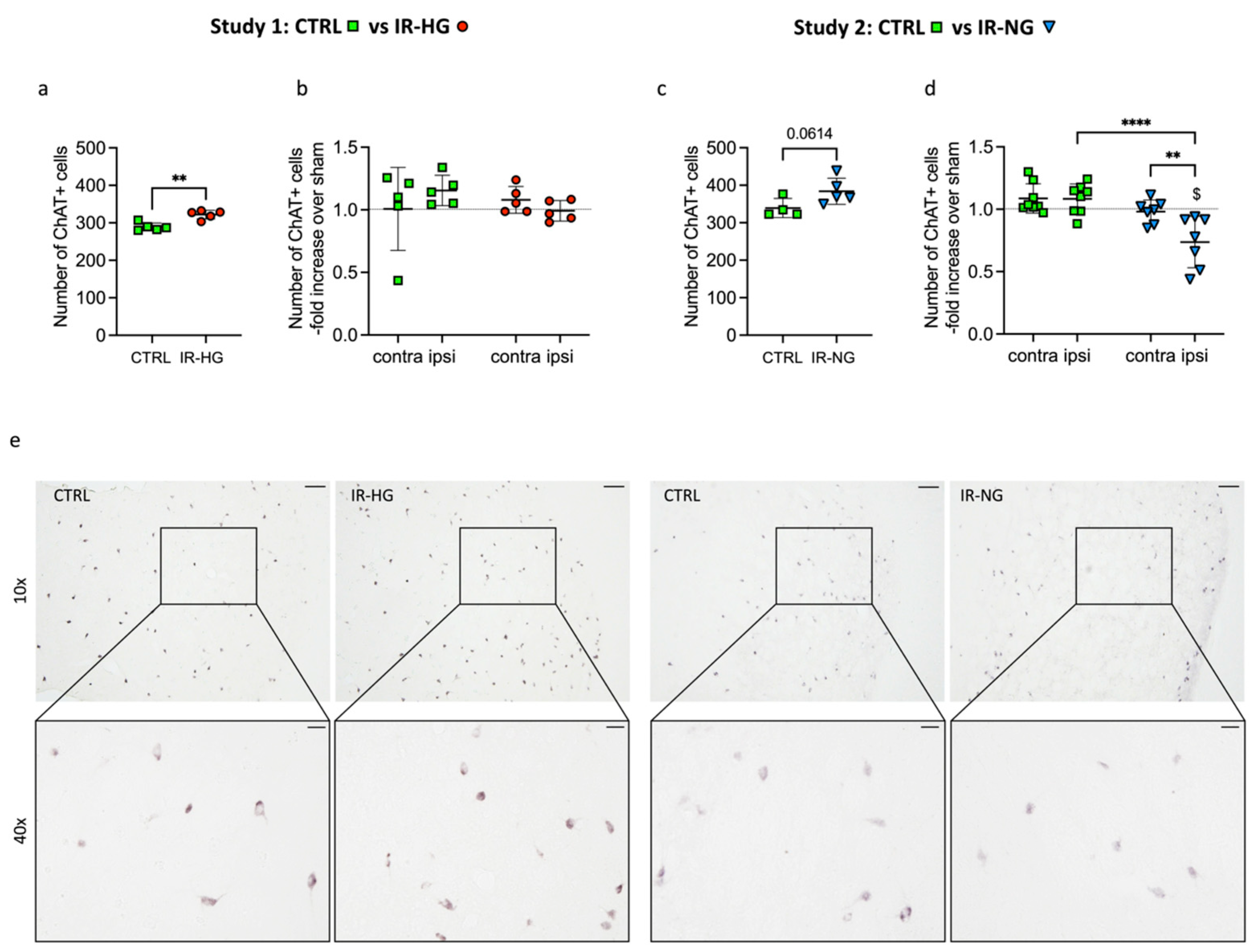
2.4. The Number of ChAT+ Interneurons Pre- and Post-Stroke Is Differently Regulated in IR-HG and IR-NG Mice
2.5. The Pre-Stroke Normalization of IR Improves Functional Recovery in Middle-Aged Mice
3. Discussion
4. Materials and Methods
4.1. Sample Size Calculation
4.2. Animals
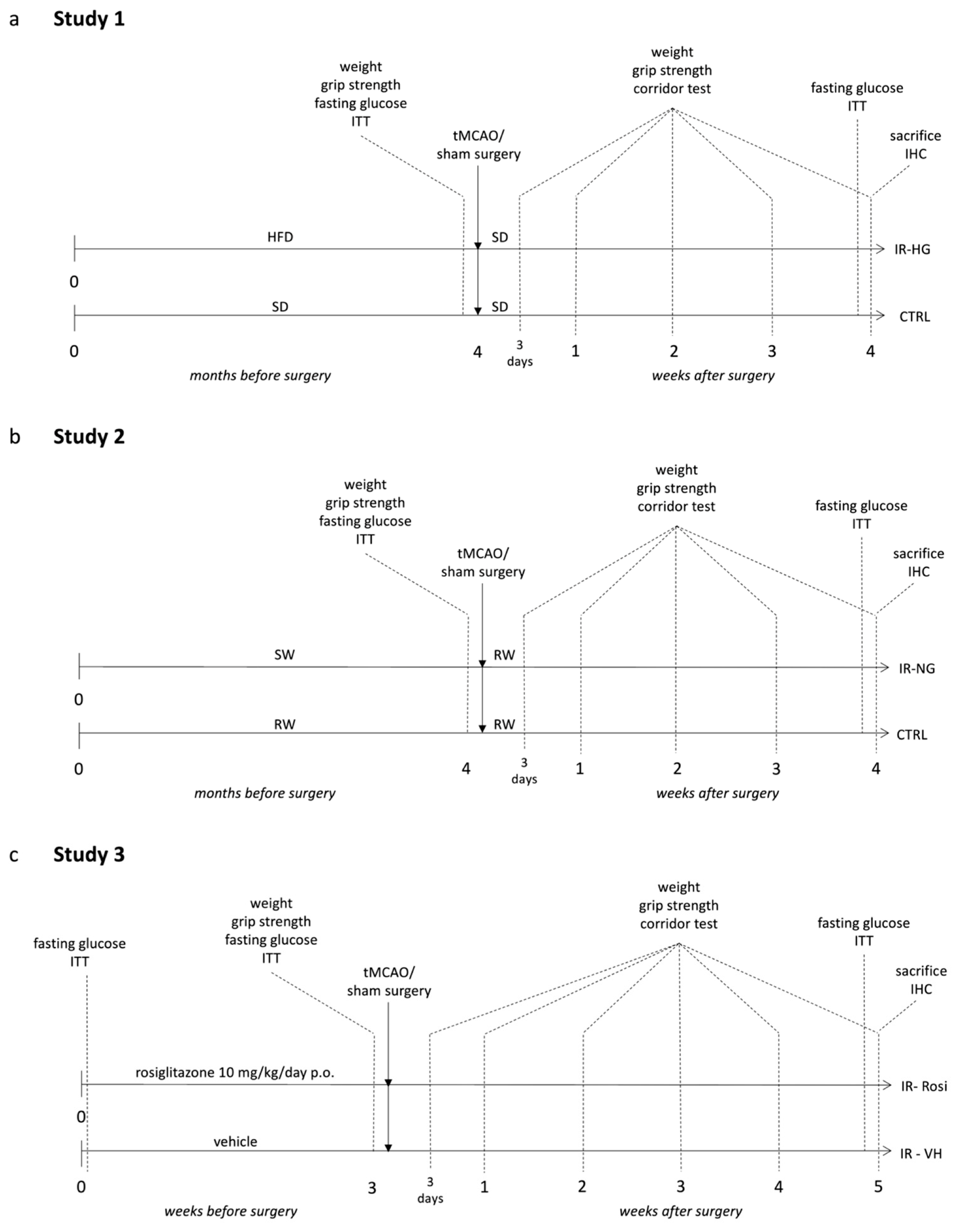
4.3. Experimental Design
4.3.1. Study 1 (Pre-Stroke IR and Fasting Hyperglycemia Were Induced by Chronic High-Fat Diet (HFD) Feeding)
4.3.2. Study 2 (Pre-Stroke IR, without Hyperglycemia, Was Induced by High Content of Sugar in the Drinking Water)
4.3.3. Study 3 (Pre-Stroke IR Was Induced by 10 Months of Aging and Normalized by Rosiglitazone Treatment)
4.4. Transient Middle Cerebral Artery Occlusion
4.5. Fasting Glycemia and ITT
4.6. Assessment of Sensorimotor Function
4.6.1. Forelimb Grip Strength
4.6.2. The Corridor Test
4.7. Serum and Tissue Collection
4.8. Immunohistochemistry (IHC)
4.9. Quantitative Microscopy and Image Analysis
4.9.1. Assessment of Stroke-Induced Brain Damage by Quantifying NeuN+ Surviving Neurons
4.9.2. Assessment of Neuroinflammation
4.9.3. Analysis of Cholinergic Interneurons
4.10. Insulin and Plasminogen Activator Inhibitor-1 (PAI-1) Enzyme-Linked Immunosorbent Assays (ELISA)
4.11. Data and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| BW | Body weight |
| CD68 | Cluster of differentiation 68 |
| ECA | External carotid artery |
| GABA | Gamma aminobutyric acid |
| HFD | High-fat diet |
| Iba-1 | Ionized calcium-binding adapter molecule 1 |
| ICA | Internal carotid artery |
| ITT | Insulin tolerance test |
| IR | Insulin resistance |
| MACE | Major adverse cardiovascular events |
| MCA | Middle cerebral artery |
| NeuN | Neuronal nuclei |
| PAI-1 | Plasminogen activator inhibitor 1 |
| PBS | Phosphate-buffered saline |
| SD | Standard laboratory diet |
| tMCAO | Transient middle cerebral artery occlusion |
References
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef] [PubMed]
- The Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.J.; Deedwania, P.; Acharya, T.; Aguilar, D.; Bhatt, D.L.; Chyun, D.A.; Di Palo, K.E.; Golden, S.H.; Sperling, L.S.; American Heart Association Diabetes Committee of the Council; et al. Comprehensive Management of Cardiovascular Risk Factors for Adults With Type 2 Diabetes: A Scientific Statement From the American Heart Association. Circulation 2022, 145, e722–e759. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, H.; Nakayama, H.; Raaschou, H.O.; Olsen, T.S. Stroke in patients with diabetes. The Copenhagen Stroke Study. Stroke 1994, 25, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Megherbi, S.E.; Milan, C.; Minier, D.; Couvreur, G.; Osseby, G.V.; Tilling, K.; Di Carlo, A.; Inzitari, D.; Wolfe, C.D.; Moreau, T.; et al. Association between diabetes and stroke subtype on survival and functional outcome 3 months after stroke: Data from the European BIOMED Stroke Project. Stroke 2003, 34, 688–694. [Google Scholar] [CrossRef]
- Ullberg, T.; Zia, E.; Petersson, J.; Norrving, B. Changes in functional outcome over the first year after stroke: An observational study from the Swedish stroke register. Stroke 2015, 46, 389–394. [Google Scholar] [CrossRef]
- Luitse, M.J.; Biessels, G.J.; Rutten, G.E.; Kappelle, L.J. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012, 11, 261–271. [Google Scholar] [CrossRef]
- Yang, S.; Boudier-Reveret, M.; Kwon, S.; Lee, M.Y.; Chang, M.C. Effect of Diabetes on Post-stroke Recovery: A Systematic Narrative Review. Front. Neurol. 2021, 12, 747878. [Google Scholar] [CrossRef]
- Hou, D.; Zhong, P.; Ye, X.; Wu, D. Persistent hyperglycemia is a useful glycemic pattern to predict stroke mortality: A systematic review and meta-analysis. BMC Neurol. 2021, 21, 487. [Google Scholar] [CrossRef]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke 2001, 32, 2426–2432. [Google Scholar] [CrossRef]
- Baird, T.A.; Parsons, M.W.; Phan, T.; Butcher, K.S.; Desmond, P.M.; Tress, B.M.; Colman, P.G.; Chambers, B.R.; Davis, S.M. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke 2003, 34, 2208–2214. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.C.; Bruno, A.; Pauls, Q.; Hall, C.E.; Barrett, K.M.; Barsan, W.; Fansler, A.; Van de Bruinhorst, K.; Janis, S.; Durkalski-Mauldin, V.L.; et al. Intensive vs Standard Treatment of Hyperglycemia and Functional Outcome in Patients With Acute Ischemic Stroke: The SHINE Randomized Clinical Trial. JAMA 2019, 322, 326–335. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Krinock, M.J.; Singhal, N.S. Diabetes, stroke, and neuroresilience: Looking beyond hyperglycemia. Ann. N. Y. Acad. Sci. 2021, 1495, 78–98. [Google Scholar] [CrossRef]
- Zabala, A.; Darsalia, V.; Lind, M.; Svensson, A.M.; Franzen, S.; Eliasson, B.; Patrone, C.; Jonsson, M.; Nystrom, T. Estimated glucose disposal rate and risk of stroke and mortality in type 2 diabetes: A nationwide cohort study. Cardiovasc. Diabetol. 2021, 20, 202. [Google Scholar] [CrossRef]
- Schmidt, K.; Power, M.C.; Ciarleglio, A.; Nadareishvili, Z.; Group, I.S. Post-stroke cognitive impairment and the risk of stroke recurrence and death in patients with insulin resistance. J. Stroke Cerebrovasc. Dis. 2022, 31, 106744. [Google Scholar] [CrossRef]
- Karampatsi, D.; Zabala, A.; Wilhelmsson, U.; Dekens, D.; Vercalsteren, E.; Larsson, M.; Nystrom, T.; Pekny, M.; Patrone, C.; Darsalia, V. Diet-induced weight loss in obese/diabetic mice normalizes glucose metabolism and promotes functional recovery after stroke. Cardiovasc. Diabetol. 2021, 20, 240. [Google Scholar] [CrossRef]
- Kernan, W.N.; Inzucchi, S.E.; Viscoli, C.M.; Brass, L.M.; Bravata, D.M.; Shulman, G.I.; McVeety, J.C.; Horwitz, R.I. Impaired insulin sensitivity among nondiabetic patients with a recent TIA or ischemic stroke. Neurology 2003, 60, 1447–1451. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Glucose intolerance and aging. Diabetes Care 1981, 4, 493–501. [Google Scholar] [CrossRef]
- Cowie, C.C.; Rust, K.F.; Ford, E.S.; Eberhardt, M.S.; Byrd-Holt, D.D.; Li, C.; Williams, D.E.; Gregg, E.W.; Bainbridge, K.E.; Saydah, S.H.; et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care 2009, 32, 287–294. [Google Scholar] [CrossRef]
- Ago, T.; Matsuo, R.; Hata, J.; Wakisaka, Y.; Kuroda, J.; Kitazono, T.; Kamouchi, M.; Fukuoka Stroke Registry, I. Insulin resistance and clinical outcomes after acute ischemic stroke. Neurology 2018, 90, e1470–e1477. [Google Scholar] [CrossRef] [PubMed]
- Fonarow, G.C.; Reeves, M.J.; Zhao, X.; Olson, D.M.; Smith, E.E.; Saver, J.L.; Schwamm, L.H.; for the Get With the Guidelines–Stroke Steering Committee and Investigators. Age-related differences in characteristics, performance measures, treatment trends, and outcomes in patients with ischemic stroke. Circulation 2010, 121, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Aberg, D.; Aberg, N.D.; Jood, K.; Holmegaard, L.; Redfors, P.; Blomstrand, C.; Isgaard, J.; Jern, C.; Svensson, J. Homeostasis model assessment of insulin resistance and outcome of ischemic stroke in non-diabetic patients—A prospective observational study. BMC Neurol. 2019, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.F.; Zhang, H.S.; Wang, J.; Gao, Y.Y.; Mao, J.N.; Hang, C.H.; Li, W. Insulin resistance in ischemic stroke: Mechanisms and therapeutic approaches. Front. Endocrinol. 2022, 13, 1092431. [Google Scholar] [CrossRef] [PubMed]
- Pintana, H.; Lietzau, G.; Augestad, I.L.; Chiazza, F.; Nystrom, T.; Patrone, C.; Darsalia, V. Obesity-induced type 2 diabetes impairs neurological recovery after stroke in correlation with decreased neurogenesis and persistent atrophy of parvalbumin-positive interneurons. Clin. Sci. 2019, 133, 1367–1386. [Google Scholar] [CrossRef] [PubMed]
- Chiazza, F.; Pintana, H.; Lietzau, G.; Nystrom, T.; Patrone, C.; Darsalia, V. The Stroke-Induced Increase of Somatostatin-Expressing Neurons is Inhibited by Diabetes: A Potential Mechanism at the Basis of Impaired Stroke Recovery. Cell Mol. Neurobiol. 2020, 41, 591–603. [Google Scholar] [CrossRef]
- Zhang, L.; Chopp, M.; Zhang, Y.; Xiong, Y.; Li, C.; Sadry, N.; Rhaleb, I.; Lu, M.; Zhang, Z.G. Diabetes Mellitus Impairs Cognitive Function in Middle-Aged Rats and Neurological Recovery in Middle-Aged Rats After Stroke. Stroke 2016, 47, 2112–2118. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, J.; Li, Y.; Wu, Y.; Liu, N.; Shi, S.X.; Lin, L.; Yuan, J.; Wang, S.; Ning, M.M.; et al. Delayed rFGF21 Administration Improves Cerebrovascular Remodeling and White Matter Repair After Focal Stroke in Diabetic Mice. Transl. Stroke Res. 2021, 13, 311–325. [Google Scholar] [CrossRef]
- Augestad, I.L.; Dekens, D.; Karampatsi, D.; Elabi, O.; Zabala, A.; Pintana, H.; Larsson, M.; Nystrom, T.; Paul, G.; Darsalia, V.; et al. Normalisation of glucose metabolism by exendin-4 in the chronic phase after stroke promotes functional recovery in male diabetic mice. Br. J. Pharmacol. 2021, 179, 677–694. [Google Scholar] [CrossRef]
- Prakash, R.; Li, W.; Qu, Z.; Johnson, M.A.; Fagan, S.C.; Ergul, A. Vascularization pattern after ischemic stroke is different in control versus diabetic rats: Relevance to stroke recovery. Stroke 2013, 44, 2875–2882. [Google Scholar] [CrossRef]
- Ergul, A.; Abdelsaid, M.; Fouda, A.Y.; Fagan, S.C. Cerebral neovascularization in diabetes: Implications for stroke recovery and beyond. J. Cereb. Blood Flow Metab. 2014, 34, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wang, J.; Wang, Y.; Dai, X.; Xu, F.; Gao, X.; Johnson, J.; Xu, N.; Leak, R.K.; Hu, X.; et al. Diabetes Mellitus Impairs White Matter Repair and Long-Term Functional Deficits After Cerebral Ischemia. Stroke 2018, 49, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liu, N.; Wang, Q.; Yu, Z.; Lin, L.; Yuan, J.; Guo, S.; Ahn, B.J.; Wang, X.J.; Li, X.; et al. Endocrine Regulator rFGF21 (Recombinant Human Fibroblast Growth Factor 21) Improves Neurological Outcomes Following Focal Ischemic Stroke of Type 2 Diabetes Mellitus Male Mice. Stroke 2018, 49, 3039–3049. [Google Scholar] [CrossRef] [PubMed]
- Augestad, I.L.; Pintana, H.; Larsson, M.; Krizhanovskii, C.; Nystrom, T.; Klein, T.; Darsalia, V.; Patrone, C. The Regulation of Glycemia in the Recovery Phase After Stroke Counteracts the Detrimental Effect of Obesity-Induced Type 2 Diabetes on Neurological Recovery. Diabetes 2020, 69, 1961–1973. [Google Scholar] [CrossRef]
- Shukla, V.; Shakya, A.K.; Perez-Pinzon, M.A.; Dave, K.R. Cerebral ischemic damage in diabetes: An inflammatory perspective. J. Neuroinflamm. 2017, 14, 21. [Google Scholar] [CrossRef]
- Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170. [Google Scholar] [CrossRef]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: A systematic review and meta-analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef]
- Gritton, H.J.; Howe, W.M.; Romano, M.F.; DiFeliceantonio, A.G.; Kramer, M.A.; Saligrama, V.; Bucklin, M.E.; Zemel, D.; Han, X. Unique contributions of parvalbumin and cholinergic interneurons in organizing striatal networks during movement. Nat. Neurosci. 2019, 22, 586–597. [Google Scholar] [CrossRef]
- Goncalves, D.F.; Guzman, M.S.; Gros, R.; Massensini, A.R.; Bartha, R.; Prado, V.F.; Prado, M.A.M. Striatal Acetylcholine Helps to Preserve Functional Outcomes in a Mouse Model of Stroke. ASN Neuro. 2020, 12, 1759091420961612. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef]
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Haring, H.U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Kim, C.K.; Kim, M.K.; Seo, W.K.; Oh, K. Insulin resistance is associated with poor functional outcome after acute ischemic stroke in non-diabetic patients. Sci. Rep. 2021, 11, 1229. [Google Scholar] [CrossRef] [PubMed]
- Calleja, A.I.; Garcia-Bermejo, P.; Cortijo, E.; Bustamante, R.; Rojo Martinez, E.; Gonzalez Sarmiento, E.; Fernandez-Herranz, R.; Arenillas, J.F. Insulin resistance is associated with a poor response to intravenous thrombolysis in acute ischemic stroke. Diabetes Care 2011, 34, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Bas, D.F.; Ozdemir, A.O.; Colak, E.; Kebapci, N. Higher Insulin Resistance Level is Associated with Worse Clinical Response in Acute Ischemic Stroke Patients Treated with Intravenous Thrombolysis. Transl. Stroke Res. 2016, 7, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, W.; Jing, J.; Zheng, H.; Jia, Q.; Li, H.; Zhao, X.; Liu, L.; Wang, Y.; He, Y.; et al. Pancreatic beta-Cell Function and Prognosis of Nondiabetic Patients With Ischemic Stroke. Stroke 2017, 48, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, T.; Matsuo, R.; Hata, J.; Nakamura, K.; Wakisaka, Y.; Kamouchi, M.; Kitazono, T.; Ago, T.; Investigators, F.S.R. beta-Cell Function and Clinical Outcome in Nondiabetic Patients With Acute Ischemic Stroke. Stroke 2021, 52, 2621–2628. [Google Scholar] [CrossRef]
- Jing, J.; Pan, Y.; Zhao, X.; Zheng, H.; Jia, Q.; Mi, D.; Chen, W.; Li, H.; Liu, L.; Wang, C.; et al. Insulin Resistance and Prognosis of Nondiabetic Patients With Ischemic Stroke: The ACROSS-China Study (Abnormal Glucose Regulation in Patients With Acute Stroke Across China). Stroke 2017, 48, 887–893. [Google Scholar] [CrossRef]
- Kawano, Y.; Edwards, M.; Huang, Y.; Bilate, A.M.; Araujo, L.P.; Tanoue, T.; Atarashi, K.; Ladinsky, M.S.; Reiner, S.L.; Wang, H.H.; et al. Microbiota imbalance induced by dietary sugar disrupts immune-mediated protection from metabolic syndrome. Cell 2022, 185, 3501–3519.e20. [Google Scholar] [CrossRef]
- Herman, M.A.; Birnbaum, M.J. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef]
- Bejot, Y.; Bailly, H.; Durier, J.; Giroud, M. Epidemiology of stroke in Europe and trends for the 21st century. Presse Med. 2016, 45, e391–e398. [Google Scholar] [CrossRef]
- Spence, J.D.; Viscoli, C.; Kernan, W.N.; Young, L.H.; Furie, K.; DeFronzo, R.; Abdul-Ghani, M.; Dandona, P.; Inzucchi, S.E. Efficacy of lower doses of pioglitazone after stroke or transient ischaemic attack in patients with insulin resistance. Diabetes Obes Metab. 2022, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Saver, J.L.; Liao, H.W.; Lin, C.H.; Ovbiagele, B. Pioglitazone for Secondary Stroke Prevention: A Systematic Review and Meta-Analysis. Stroke 2017, 48, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zuo, W.; Yin, L.; Gu, T.; Wang, S.; Fang, Z.; Wang, B.; Dong, H.; Hou, W.; Zuo, Z.; et al. Pioglitazone attenuates ischaemic stroke aggravation by blocking PPARgamma reduction and inhibiting chronic inflammation in diabetic mice. Eur. J. Neurosci. 2022, 56, 4948–4961. [Google Scholar] [CrossRef]
- White, A.T.; Murphy, A.N. Administration of thiazolidinediones for neuroprotection in ischemic stroke: A pre-clinical systematic review. J. Neurochem. 2010, 115, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Komleva, Y.; Chernykh, A.; Lopatina, O.; Gorina, Y.; Lokteva, I.; Salmina, A.; Gollasch, M. Inflamm-Aging and Brain Insulin Resistance: New Insights and Role of Life-style Strategies on Cognitive and Social Determinants in Aging and Neurodegeneration. Front. Neurosci. 2020, 14, 618395. [Google Scholar] [CrossRef]
- Shibahara, T.; Ago, T.; Tachibana, M.; Nakamura, K.; Yamanaka, K.; Kuroda, J.; Wakisaka, Y.; Kitazono, T. Reciprocal Interaction Between Pericytes and Macrophage in Poststroke Tissue Repair and Functional Recovery. Stroke 2020, 51, 3095–3106. [Google Scholar] [CrossRef]
- Shibahara, T.; Ago, T.; Nakamura, K.; Tachibana, M.; Yoshikawa, Y.; Komori, M.; Yamanaka, K.; Wakisaka, Y.; Kitazono, T. Pericyte-Mediated Tissue Repair through PDGFRbeta Promotes Peri-Infarct Astrogliosis, Oligodendrogenesis, and Functional Recovery after Acute Ischemic Stroke. Eneuro 2020, 7, 0474-19.2020. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Chen, W.; Cai, W.; Hoover, B.; Kahn, C.R. Insulin action in the brain: Cell types, circuits, and diseases. Trends Neurosci. 2022, 45, 384–400. [Google Scholar] [CrossRef]
- Fu, J.; Yu, M.G.; Li, Q.; Park, K.; King, G.L. Insulin’s actions on vascular tissues: Physiological effects and pathophysiological contributions to vascular complications of diabetes. Mol. Metab. 2021, 52, 101236. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lin, Y.W.; Clemont, A.; Feener, E.P.; Hein, K.D.; Igarashi, M.; Yamauchi, T.; White, M.F.; King, G.L. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J. Clin. Investig. 1999, 104, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis-A new target in stroke? Neurosci. Lett. 2019, 689, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.E.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Martinez-Rachadell, L.; Navarrete, M.; Pose-Utrilla, J.; Davila, J.C.; Pignatelli, J.; Diaz-Pacheco, S.; Guerra-Cantera, S.; Viedma-Moreno, E.; Palenzuela, R.; et al. Insulin regulates neurovascular coupling through astrocytes. Proc. Natl. Acad. Sci. USA 2022, 119, e2204527119. [Google Scholar] [CrossRef]
- O’Grady, J.P.; Dean, D.C., 3rd; Yang, K.L.; Canda, C.M.; Hoscheidt, S.M.; Starks, E.J.; Merluzzi, A.; Hurley, S.; Davenport, N.J.; Okonkwo, O.C.; et al. Elevated Insulin and Insulin Resistance are Associated with Altered Myelin in Cognitively Unimpaired Middle-Aged Adults. Obesity 2019, 27, 1464–1471. [Google Scholar] [CrossRef]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef]
- West, M.J.; Slomianka, L.; Gundersen, H.J. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991, 231, 482–497. [Google Scholar] [CrossRef]
- West, M.J. Stereological methods for estimating the total number of neurons and synapses: Issues of precision and bias. Trends Neurosci. 1999, 22, 51–61. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Physiol. 2020, 598, 3793–3801. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vercalsteren, E.; Karampatsi, D.; Dekens, D.; Letsiou, A.; Zabala, A.; Romanitan, M.; Klein, T.; Nyström, T.; Darsalia, V.; Patrone, C. The Pre-Stroke Induction and Normalization of Insulin Resistance Respectively Worsens and Improves Functional Recovery. Int. J. Mol. Sci. 2023, 24, 3989. https://doi.org/10.3390/ijms24043989
Vercalsteren E, Karampatsi D, Dekens D, Letsiou A, Zabala A, Romanitan M, Klein T, Nyström T, Darsalia V, Patrone C. The Pre-Stroke Induction and Normalization of Insulin Resistance Respectively Worsens and Improves Functional Recovery. International Journal of Molecular Sciences. 2023; 24(4):3989. https://doi.org/10.3390/ijms24043989
Chicago/Turabian StyleVercalsteren, Ellen, Dimitra Karampatsi, Doortje Dekens, Aikaterini Letsiou, Alexander Zabala, Mihaela Romanitan, Thomas Klein, Thomas Nyström, Vladimer Darsalia, and Cesare Patrone. 2023. "The Pre-Stroke Induction and Normalization of Insulin Resistance Respectively Worsens and Improves Functional Recovery" International Journal of Molecular Sciences 24, no. 4: 3989. https://doi.org/10.3390/ijms24043989
APA StyleVercalsteren, E., Karampatsi, D., Dekens, D., Letsiou, A., Zabala, A., Romanitan, M., Klein, T., Nyström, T., Darsalia, V., & Patrone, C. (2023). The Pre-Stroke Induction and Normalization of Insulin Resistance Respectively Worsens and Improves Functional Recovery. International Journal of Molecular Sciences, 24(4), 3989. https://doi.org/10.3390/ijms24043989