

Mechanisms for the α-Adrenoceptor-Mediated Positive Inotropy in Mouse Ventricular Myocardium: Enhancing Effect of Action Potential Prolongation

Abstract
1. Introduction
2. Results
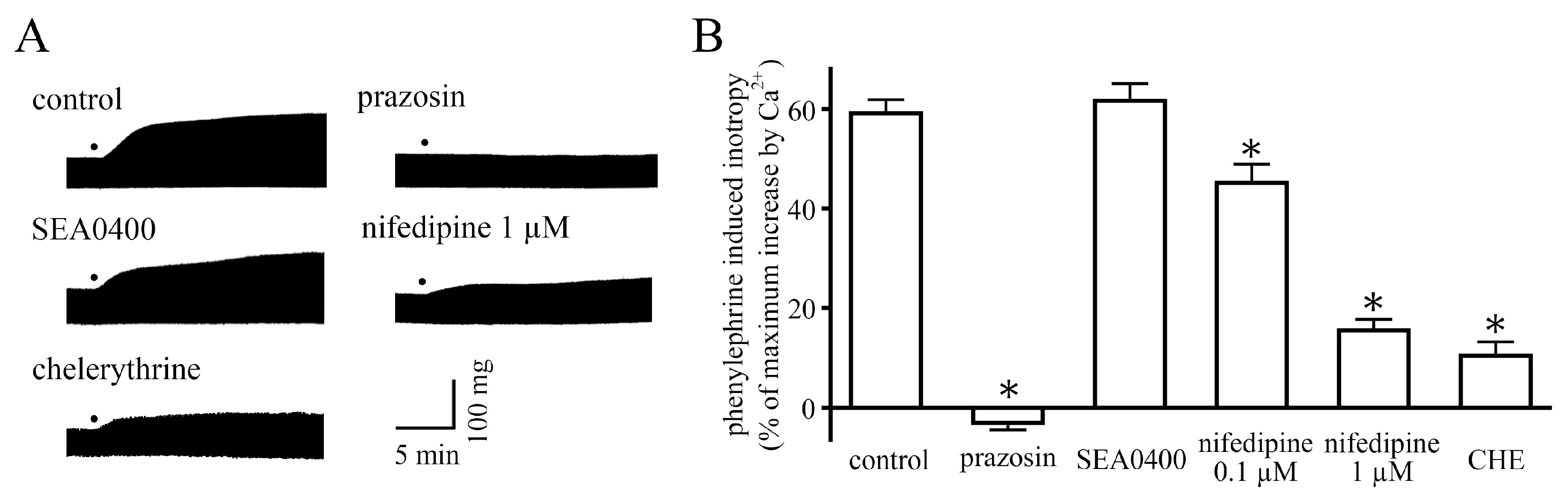
2.1. Measurement of Contractile Force
2.2. Measurement of Membrane Currents
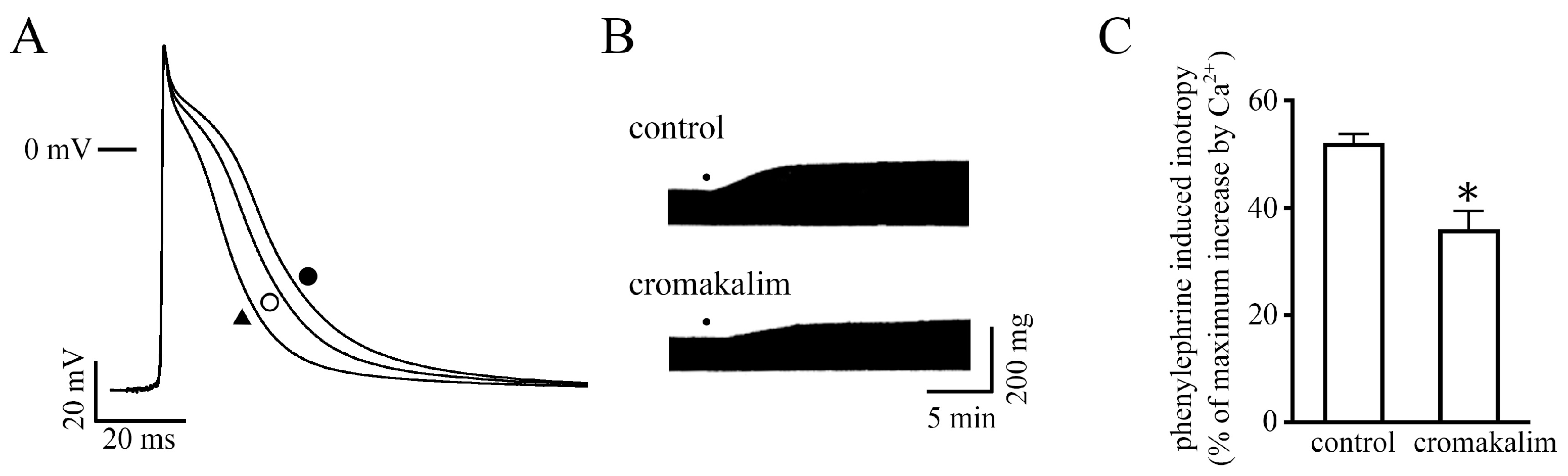
2.3. Measurement of Action Potentials
2.4. Effect of Changes in Action Potential Duration on the Positive Inotropy
3. Discussion
4. Materials and Methods
4.1. General
4.2. Measurement of Contractile Force
4.3. Measurement of Action Potentials
4.4. Isolation of Mouse Ventricular Cardiomyocytes
4.5. Measurement of Membrane Currents
4.6. Drugs and Chemicals
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woodcock, E.A.; Du, X.J.; Reichelt, M.E.; Graham, R.M. Cardiac α1-adrenergic drive in pathological remodeling. Cardiovas. Res. 2008, 77, 452–462. [Google Scholar] [CrossRef]
- Endoh, M. Signal transduction and Ca2+ signaling in intact myocardium. J. Pharmacol. Sci. 2006, 100, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M. Cardiac α1-adrenoceptors and inotropy. Myofilament Ca2+ sensitivity, intracellular Ca2+ mobilization, signaling pathway, and pathophysiological relevance. Circ. Res. 2016, 119, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Simpson, P.C.; Jensen, B.C. Cardiac α1A-adrenergic receptors: Emerging protective roles in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H725–H733. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Hiramoto, T.; Ishiwata, A.; Takanashi, M.; Inui, J. Myocardial α1-adrenoceptors mediate positive inotropic effect and changes in phosphatidyl inositol metabolism. Species differences in receptor distribution and the intracellular coupling process in mammalian ventricular myocardium. Circ. Res. 1991, 68, 1179–1190. [Google Scholar] [CrossRef]
- Tanaka, H.; Manita, S.; Matsuda, T.; Adachi, M.; Shigenobu, K. Sustained negative inotropism mediated by α-adrenoceptors in adult mouse myocardia: Developmental conversion from positive response in the neonate. Br. J. Pharmacol. 1995, 114, 673–677. [Google Scholar] [CrossRef]
- Tanaka, H.; Matsuda, T.; Adachi, M.; Shigenobu, K. Effect of sympathectomy on inotropic responsiveness to α-adrenoceptor stimulation in developing mouse myocardia. Can. J. Physiol. Pharmacol. 1995, 73, 1285–1288. [Google Scholar] [CrossRef]
- Chu, C.; Thai, K.; Park, K.W.; Wang, P.; Makwana, O.; Lovett, D.H.; Simpson, P.C.; Baker, A.J. Intraventricular and interventricular cellular heterogeneity inotropic responses to α1-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H946–H953. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Canan, B.D.; Kilic, A.; Whitson, B.A.; Baker, A.J. Human myocardium has a robust α1A-subtype adrenergic receptor inotropic response. J. Cardiovas. Pharmacol. 2018, 72, 136–142. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Karpinski, E.; Pang, P.K. The L-type calcium channel current is increased by alpha-1 adrenoceptor activation in neonatal rat ventricular cells. J. Pharmacol. Exp. Ther. 1994, 271, 935–943. [Google Scholar]
- Liu, S.J.; Kennedy, R.H. α1-Adrenergic activation of L-type Ca current in rat ventricular myocytes: Perforated patch-clamp recordings. Am. J. Physiol. 1998, 274, H2203–H2207. [Google Scholar] [CrossRef]
- Zhang, S.; Hiraoka, M.; Hirano, Y. Effects of alpha1-adrenergic Stimulation on L-type Ca2+ Current in Rat Ventricular Myocytes. J. Mol. Cell. Cardiol. 1998, 30, 1955–1965. [Google Scholar] [CrossRef]
- Fedida, D.; Shimoni, Y.; Giles, W.R. α-Adrenergic modulation of the transient outward current in rabbit atrial myocytes. J. Physiol. 1990, 423, 257–277. [Google Scholar] [CrossRef]
- Tohse, N.; Nakaya, H.; Hattori, Y.; Endou, M.; Kanno, M. Inhibitory effect mediated by α1-adrenoceptors on transient outward current in isolated rat ventricular cells. Pflugers Arch. 1990, 415, 575–581. [Google Scholar] [CrossRef]
- Nishimaru, K.; Kobayashi, M.; Matsuda, T.; Tanaka, Y.; Tanaka, H.; Shigenobu, K. α-Adrenoceptor stimulation-mediated negative inotropism and enhanced Na+-Ca2+ exchange in mouse ventricle. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H132–H141. [Google Scholar] [CrossRef]
- Tanaka, H.; Namekata, I.; Takeda, K.; Kazama, A.; Shimizu, Y.; Moriwaki, R.; Hirayama, W.; Sato, A.; Kawanishi, T.; Shigenobu, K. Unique excitation-contraction characteristics of mouse myocardium as revealed by SEA0400, a specific inhibitor of Na+-Ca2+ exchanger. Naunyn Schmiedebergs Arch. Pharmacol. 2005, 371, 526–534. [Google Scholar] [CrossRef]
- Kim, J.S.; Kang, H.S.; Kim, J.S. α1-adrenoceptor-mediated negative inotropic effect caused by intracellular ionic activities in guinea-pig papillary muscle. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2005, 52, 498–505. [Google Scholar] [CrossRef]
- Hirano, S.; Kusakari, Y.; O-Uchi, J.; Morimoto, S.; Kawai, M.; Hongo, K.; Kurihara, S. Intracellular mechanism of the negative inotropic effect induced by α1-adrenoceptor stimulation in mouse myocardium. J. Physiol. Sci. 2006, 56, 297–304. [Google Scholar] [CrossRef]
- Kamata, K.; Satoh, T.; Tanaka, H.; Shigenobu, K. Changes in electrophysiological and mechanical responses of the rat papillary muscle to α- and β-agonist in streptozotocin-induced diabetes. Can. J. Physiol. Pharmacol. 1997, 75, 781–788. [Google Scholar] [CrossRef]
- Kamata, K.; Satoh, T.; Matsumoto, T.; Noguchi, E.; Taguchi, K.; Kobayashi, T.; Tanaka, H.; Shigenobu, K. Enhancement of methoxamine-induced contractile responses of rat ventricular muscle in streptozotocin-induced diabetes is associated with α1A adrenoceptor upregulation. Acta. Physiol. 2006, 188, 173–183. [Google Scholar] [CrossRef]
- Kanae, H.; Hamaguchi, S.; Wakasugi, Y.; Kusakabe, T.; Kato, K.; Namekata, I.; Tanaka, H. Pathological prolongation of action potential duration as a cause of the reduced alpha-adrenoceptor-mediated negative inotropy in streptozotocin-induced diabetic mice myocardium. J. Pharmacol. Sci. 2017, 135, 131–133. [Google Scholar] [CrossRef]
- Sekine, T.; Kusano, H.; Nishimaru, K.; Tanaka, Y.; Tanaka, H.; Shigenobu, K. Developmental conversion of inotropism by endothelin I and angiotensin II from positive to negative in mice. Eur. J. Pharmacol. 1999, 374, 411–415. [Google Scholar] [CrossRef]
- Hamaguchi, S.; Kawakami, Y.; Honda, Y.; Nemoto, K.; Sano, A.; Namekata, I.; Tanaka, H. Developmental changes in excitation-contraction mechanisms of the mouse ventricular myocardium as revealed by functional and confocal imaging analyses. J. Pharmacol. Sci. 2013, 123, 167–175. [Google Scholar] [CrossRef]
- Woo, S.H.; Lee, C.O. Role of PKC in the effects of alpha1-adrenergic stimulation on Ca2+ transients, contraction and Ca2+ current in guinea-pig ventricular myocytes. Pflugers. Arch. 1999, 437, 335–344. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishimaru, K.; Aikawa, T.; Hirayama, W.; Tanaka, Y.; Shigenobu, K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br. J. Pharmacol. 2002, 135, 1096–1100. [Google Scholar] [CrossRef]
- Shen, J.B.; Jiang, B.; Pappano, A.J. Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. J. Pharmacol. Exp. Ther. 2000, 294, 562–570. [Google Scholar]
- Herbert, J.M.; Augereau, J.M.; Gleye, J.; Maffrand, J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac Na/Ca exchange function in rabbit, mouse and man: What’s the difference? J. Mol. Cell. Cardiol. 2002, 34, 369–373. [Google Scholar] [CrossRef]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef]
- Nishimaru, K.; Tanaka, Y.; Tanaka, H.; Shigenobu, K. Pharmacological evidence for involvement of phospholipase D, protein kinase C, and sodium-calcium exchanger in α-adrenoceptor-mediated negative inotropy in adult mouse ventricle. J. Pharmacol. Sci. 2003, 92, 196–202. [Google Scholar] [CrossRef]
- Fedida, D.; Bouchard, R.A. Mechanisms for the positive inotropic effect of α1-adrenoceptor stimulation in rat cardiac myocytes. Circ. Res. 1992, 71, 673–688. [Google Scholar] [CrossRef]
- Tokuno, T.; Muraki, K.; Watanabe, M.; Imaizumi, Y. Effects of K+ channel modulators on the relationship between action potential duration and Ca2+ transients in single ventricular myocytes of the guinea pig. Jpn. J. Pharmacol. 1999, 80, 243–553. [Google Scholar] [CrossRef][Green Version]
- Wang, G.Y.; Yeh, C.C.; Jensen, B.C.; Mann, M.J.; Simpson, P.C.; Baker, A.J. Heart failure switches the RV α1-adrenergic inotropic response from negative to positive. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H913–H920. [Google Scholar] [CrossRef]
- Cowley, P.M.; Wang, G.; Ghang, A.N.; Makwana, O.; Swigart, P.M.; Lovett, D.H.; Stull, J.T.; Simpson, P.C.; Baker, A.J. The α1A-adrenergic subtype mediates increased contraction of failing right ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H888–H896. [Google Scholar] [CrossRef]
- Odagiri, F.; Inoue, H.; Sugihara, M.; Suzuki, T.; Murayama, T.; Shioya, T.; Konishi, M.; Nakazato, Y.; Daida, H.; Sakurai, T.; et al. Effects of candesartan on electrical remodeling in the hearts of inherited dilated cardiomyopathy model mice. PLoS ONE 2014, 9, e101838. [Google Scholar] [CrossRef]
- MacCarthy, P.A.; Grocott-Mason, R.; Prendergast, B.D.; Shar, A.M. Contrasting inotropic effects of endogenous endothelin in the normal and failing human heart: Studies with an intracoronary ET(A) receptor antagonist. Circulation 2000, 101, 142–147. [Google Scholar] [CrossRef]
- Beuckelmann, D.J.; Näbauer, M.; Erdmann, E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ. Res. 1993, 73, 379–385. [Google Scholar] [CrossRef]
- Kääb, S.; Nuss, H.B.; Chiamvimonvat, N.; O’Rourke, B.; Pak, P.H.; Kass, D.A.; Marban, E.; Tomaselli, G.F. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ. Res. 1996, 78, 262–273. [Google Scholar] [CrossRef]
- Zang, Y.L.; Xia, L. Cellular mechanism of cardiac alternans: An unresolved chicken or egg problem. J. Zhejiang Univ. Sci. B. 2014, 15, 201–211. [Google Scholar] [CrossRef]
- Hamaguchi, S.; Kariya, M.; Fukuma-Ozaki, A.; Namekata, I.; Tanaka, H. Contribution of ATP-mediated positive feedback to sympathetic nerve-induced positive inotropy in guinea pig ventricular myocardium. Biol. Pharm. Bull. 2021, 44, 458–460. [Google Scholar] [CrossRef]
- Hamaguchi, S.; Abe, K.; Komatsu, M.; Kainuma, J.; Namekata, I.; Tanaka, H. Positive lusitropic effect of quercetin on isolated ventricular myocardia from normal and streptozotocin-induced diabetic mice. Biol. Pharm. Bull. 2021, 44, 1894–1897. [Google Scholar] [CrossRef] [PubMed]
- Namekata, I.; Hiiro, H.; Odaka, R.; Saito, T.; Hamaguchi, S.; Tsukamoto, T.; Ishikawa, R.; Katayama, Y.; Kondo, Y.; Tanaka, H. Inhibitory effect of a late sodium current blocker, NCC-3902, on the automaticity of the guinea pig pulmonary vein myocardium. Biol. Pharm. Bull. 2022, 45, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Hiiro, H.; Hashimoto, T.; Mizoguchi, M.; Kaneko, M.; Deguchi, N.; Takahashi, Y.; Hamaguchi, S.; Namekata, I.; Tanaka, H. Negative inotropic effects of class I antiarrhythmics on guinea pig ventricular myocardium: Correlation with L-type Ca2+ channel blockade. Biol. Pharm. Bull. 2023, 46, 133–137. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Cromakalim | ||||
|---|---|---|---|---|---|
| before | Phenylephrine | before | Cromakalim | +Phenylephrine | |
| RP (mV) | −77.3 ± 0.8 | −77.6 ± 1.0 | −78.6 ± 0.8 | −78.1 ± 0.3 | −78.2 ± 0.4 |
| OS (mV) | 28.0 ± 2.2 | 28.3 ± 2.4 | 25.2 ± 3.4 | 25.9 ± 3.3 | 27.0 ± 2.5 |
| AMP (mV) | 105.3 ± 2.5 | 105.9 ± 2.6 | 103.8 ± 3.6 | 104.1 ± 3.5 | 105.2 ± 2.8 |
| APD20 (ms) | 7.0 ± 0.7 | 12.1 ± 1.0 * | 6.6 ± 0.7 | 4.8 ± 0.7 * | 8.9 ± 1.1 * |
| APD50 (ms) | 14.9 ± 1.4 | 23.0 ± 2.0 * | 18.2 ± 0.8 | 11.9 ± 0.7 * | 20.0 ± 1.1 |
| APD90 (ms) | 40.5 ± 4.8 | 67.9 ± 5.0 * | 47.7 ± 5.0 | 30.6 ± 1.6 * | 49.7 ± 2.8 |
| max (V/s) | 248.3 ± 21.5 | 244.9 ± 20.0 | 193.8 ± 13.0 | 202.5 ± 18.9 | 199.6 ± 19.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamaguchi, S.; Morinou, I.; Shiseki, Y.; Mikami, A.; Seki, M.; Namekata, I.; Tanaka, H. Mechanisms for the α-Adrenoceptor-Mediated Positive Inotropy in Mouse Ventricular Myocardium: Enhancing Effect of Action Potential Prolongation. Int. J. Mol. Sci. 2023, 24, 3926. https://doi.org/10.3390/ijms24043926
Hamaguchi S, Morinou I, Shiseki Y, Mikami A, Seki M, Namekata I, Tanaka H. Mechanisms for the α-Adrenoceptor-Mediated Positive Inotropy in Mouse Ventricular Myocardium: Enhancing Effect of Action Potential Prolongation. International Journal of Molecular Sciences. 2023; 24(4):3926. https://doi.org/10.3390/ijms24043926
Chicago/Turabian StyleHamaguchi, Shogo, Ikue Morinou, Yuko Shiseki, Ayako Mikami, Maika Seki, Iyuki Namekata, and Hikaru Tanaka. 2023. "Mechanisms for the α-Adrenoceptor-Mediated Positive Inotropy in Mouse Ventricular Myocardium: Enhancing Effect of Action Potential Prolongation" International Journal of Molecular Sciences 24, no. 4: 3926. https://doi.org/10.3390/ijms24043926
APA StyleHamaguchi, S., Morinou, I., Shiseki, Y., Mikami, A., Seki, M., Namekata, I., & Tanaka, H. (2023). Mechanisms for the α-Adrenoceptor-Mediated Positive Inotropy in Mouse Ventricular Myocardium: Enhancing Effect of Action Potential Prolongation. International Journal of Molecular Sciences, 24(4), 3926. https://doi.org/10.3390/ijms24043926