1. Introduction
EphA2 is a receptor tyrosine kinase that has multiple roles in facilitating malignant progression. Although EphA2 was first studied in the context of neuronal migration during embryogenesis, it has since been shown to regulate cancer cell growth, migration, invasion, and angiogenesis [
1]. In addition, EphA2 is overexpressed in various cancers, including breast cancer [
2], esophageal cancer [
3], melanoma [
4], lung cancer [
5], prostate cancer [
6], ovarian cancer [
7,
8], and endometrial cancer [
9,
10]. Over the years, many therapeutic strategies have been developed to target EphA2, including tyrosine kinase inhibitors, monoclonal antibodies, immunoconjugates, aptamers, and short-interfering RNA (siRNA) [
11,
12]. We have previously demonstrated that delivery of EphA2 siRNA through 1,2-dioleoyl-
sn-glycero-3-phosphatidylcholine (DOPC) neutral liposome nanoparticles (EPHARNA) showed highly efficient in vivo delivery to the tumor, resulting in decreased tumor burden in mouse ovarian cancer models [
13]. In addition, EPHARNA combined well with paclitaxel and significantly reduced tumor growth in these preclinical models [
13]. A phase 1 trial of EPHARNA in patients with solid cancers is ongoing. However, rational combinations with EphA2-targeted therapy are not yet known; therefore, we aimed to identify novel therapeutic combinations through high-throughput chemical screens for use in endometrial cancer.
In the present study, we identified the Wee1 kinase inhibitor, MK1775, as a synergistic partner to EphA2-targeted therapy in endometrial cancer cells. To test our hypothesis that Wee1 inhibition sensitizes cells to EphA2-targeted therapy, we examined the anti-tumor effects of both agents in endometrial cancer mouse models and evaluated potential mechanisms of synergy. Our findings supported our hypothesis, justifying further investigation of this combination.
3. Discussion
In this study, we sought to identify novel agents for combination with EphA2-targeted therapy in endometrial cancer. One of the top hits identified in our high-throughput screen was the Wee1 kinase inhibitor, MK1775. We found EphA2- and Wee1-targeted therapies had synergistic effects in vitro, and the combination therapy led to enhanced anti-tumor efficacy in vivo.
Over two decades of preclinical research have identified EphA2 as a promising target for clinical translation. Previous studies have shown that EphA2 silencing was effective in reducing tumor burden in combination with chemotherapeutic agents such as paclitaxel and docetaxel [
13]. Here, we showed that silencing EphA2 expression with siRNA increases Wee1 activity, as evidenced by increased cdc2 phosphorylation, which regulates the G2/M cell cycle block and allows cells to evade cell death by inhibiting premature entry into mitosis. Therefore, increased Wee1 activity may serve as an adaptive cell survival mechanism to evade cell death upon EphA2 inhibition. Blocking such secondary cell survival loops shows promise in enhancing the therapeutic efficacy of EphA2 inhibition.
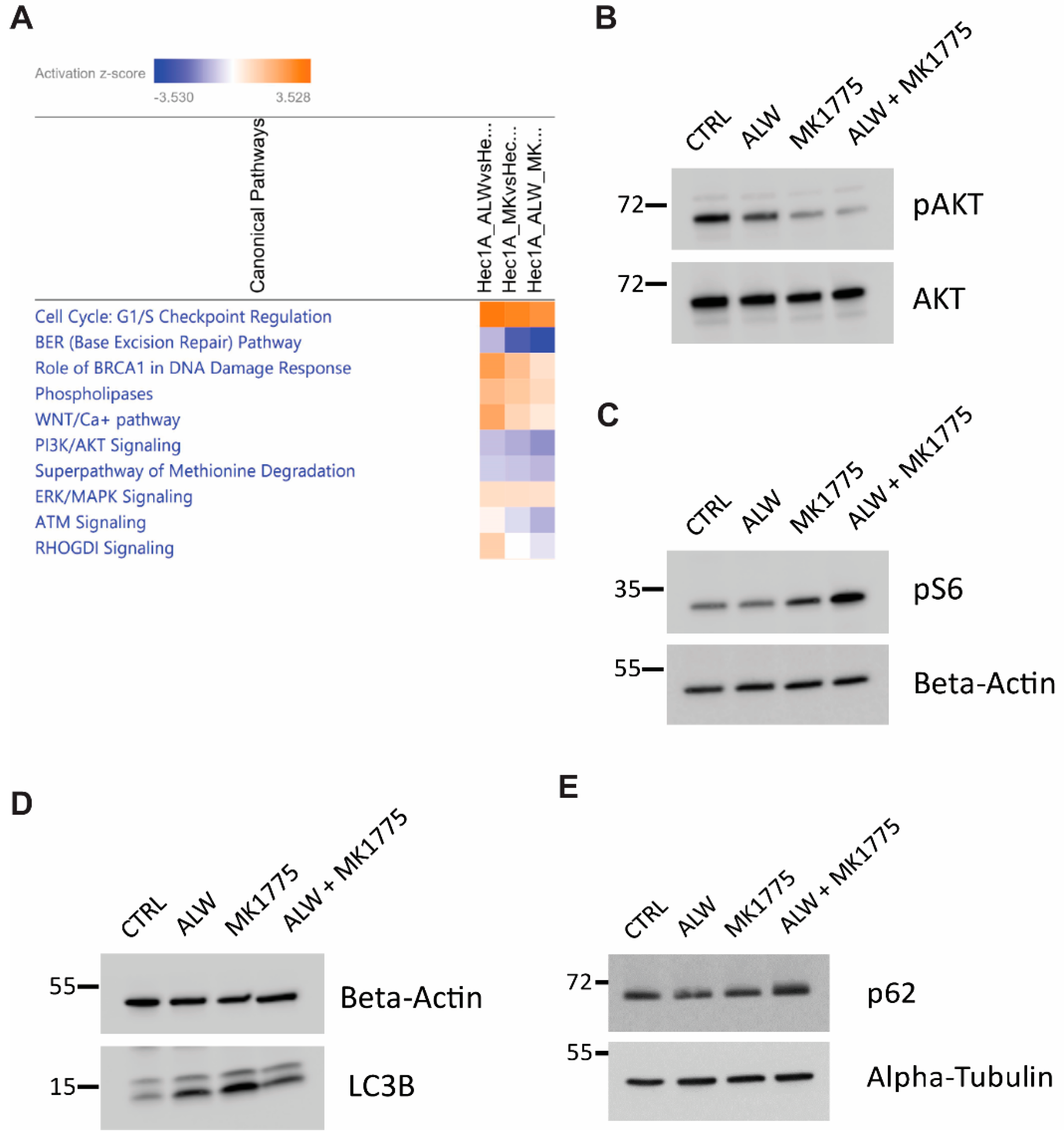
In vitro experiments confirmed the synergy between the EphA2- and Wee1-targeted drugs, as seen through an increase in apoptosis and the inhibition of colony-forming efficiency. Furthermore, RNA-Seq analysis confirmed the synergy was partly mediated by suppression of cell proliferation and of DNA damage response pathways. EphA2 has been shown to be highly expressed in many cancers and has been shown to regulate the PI3K-AKT signaling pathway. However, it has been shown to have opposing effects in different cancers. For example, in pancreatic cancer and hepatocellular carcinoma, ligand-induced EphA2 signaling activates AKT signaling by enhancing its phosphorylation [
14,
15]. In contrast, in glioblastoma, EphA2 signaling decreases AKT function by reduced phosphorylation [
16]. Here, we observed that EphA2 inhibition decreased AKT signaling modestly, with further reduction when EphA2 inhibition was combined with the Wee1 inhibitor MK1775. This suppression of AKT signaling with the combination treatment enhanced mTOR activity, leading to inhibition of cell survival autophagy and ultimately leading to cell death by apoptosis in the absence of efficient DNA damage response activation.
At present, several EphA2-targeted therapeutics are being evaluated in clinical trials in various cancers in which EphA2 has an established oncogenic function [
10,
12]. Our findings provide further support to explore additional combination therapies that may synergize with EphA2-targeted therapeutics. In addition, recent ADAGIO phase 2 trial results of the Wee1 inhibitor adavosertib (AZD1775) demonstrating response rates of 30% in uterine serous carcinoma are promising and warrant further investigation [
17]. Furthermore, since EphA2 is overexpressed in ovarian serous carcinoma and is associated with poor clinical outcomes [
18], it is possible that the combination therapy with Epha2 inhibition may be a viable option for further improving clinical outcomes of patients with other histological subtypes such as serous carcinoma.
4. Materials and Methods
4.1. Cell Culture
Hec1A (RRID:CVCL_0293) and Ishikawa cells (RRID:CVCL_2529) were procured from ATCC and The University of Texas MD Anderson Cancer Center Characterized Cell Line Core, respectively. Cell lines were validated by short tandem repeat fingerprinting in the core facility. Cells were routinely screened for mycoplasma. Hec1A cells were grown in McCoy’s 5A medium (HyClone, Logan, UT, USA), and Ishikawa cells were grown in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA) and 0.1% gentamicin sulfate (Gemini Bioproducts, West Sacramento, CA, USA). Cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C. All experiments were conducted with cells at 70% to 80% confluence and cultured for fewer than 20 passages for in vitro work and for fewer than 10 passages for in vivo experiments.
4.2. siRNA Transfection
Cells were plated in six-well plates so that the cells could reach 60% to 70% confluence by the next day. For each well, 1.3 µg of siRNA was added to 150 µL of reduced serum medium (Opti-MEM, Thermo Fisher Scientific, Waltham, MA, USA), and in a separate tube, 8 µL of Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA) was incubated in 150 µL of Opti-MEM for 5 min. The siRNA/media mixture was added dropwise to the transfection reagent mixture, vortexed and then incubated for 15 to 20 min at room temperature. Wells to be transfected were washed once with PBS, and then 900 µL of Opti-MEM and 300 µL of siRNA mixture were added dropwise to each well. The plates were gently swirled and placed in the incubator for 4 to 6 h. The transfection medium was replaced with complete media. For the high-throughput screening, after 24 h of transfection, cells were trypsinized, counted, and seeded in clear-bottom 384-well plates. For Western blot analysis, cells were collected 48 to 72 h after transfection.
4.3. High-Throughput Screening
High-throughput chemical screens were performed by the Gulf Coast Consortia’s Combinatorial Drug Discovery Program at the Institute of Biosciences and Technology in Texas A&M Health Science Center, Houston, TX, USA. Hec1A endometrial cancer cells transfected with either siControl or siEphA2 were screened against two drug library collections: The Broad Collection-Informer Set (358 compounds) and Selleck Bioactives Collection (1150 compounds) libraries. A brief description of the contents of the libraries can be found at the following link (
https://ibt.tamu.edu/cores/high-throughput/core-libraries/approved-drugs.html, accessed on 12 July 2019). For screening assays, a total of 800 cells per well were suspended in 50 µL of media and seeded into black 384-well µClear plates (Greiner Bio-One International, Monroe, NC, USA) using a Multidrop Combi liquid dispenser (Thermo Fisher Scientific, Waltham, MA, USA). After cell seeding, the plates were kept at room temperature for 40 to 60 min before being moved into a cell culture incubator. The cells were grown overnight at 37 °C in a humidified chamber (>95% relative humidity) with 5% CO
2. The following day, 50 nl of drugs were transferred into each well using an Echo 550 acoustic dispensing platform (Labcyte, San Jose, CA, USA). A non-treated plate was immediately fixed with 4% paraformaldehyde and followed by nuclei staining with 4′,6-diamidino-2-phenylindole (DAPI) at the start of drug treatment (day 0) to estimate the number of cells present at the start of treatment.
In the primary screen, the drug libraries were tested at three concentrations (1 µM, 0.1 µM, and 0.01 µM) with a fixed volume of dimethyl sulfoxide (DMSO) (0.1% v/v) and two biological replicates. Each assay plate contained a fixed concentration of the drugs in addition to a negative control (0.1% DMSO) and two positive controls (etoposide and dasatinib). After 72 h of incubation, plates were fixed with 0.4% paraformaldehyde and nuclei stained with DAPI using an integrated HydroSpeed plate washer (Tecan Life Sciences, Männedorf, Switzerland) and Multidrop Combi dispenser. Plates were imaged on an IN Cell Analyzer 6000 laser-based confocal imaging platform (GE Healthcare Bio-Sciences, Marlborough, MA, USA), and nuclei were counted using IN Cell Developer Toolbox software (version 1.6). To evaluate the cells’ response to the drug screen, we performed curve fitting followed by a calculation of area under the curve values.
4.4. Orthogonal Cell Viability Assay
To evaluate the cytotoxicity of ALW-II-41-27 (ALW; ApexBio Technology, Houston, TX, USA) and MK1775 (ApexBio Technology, Houston, TX, USA) both alone and in combination, cells were plated in a 96-well plate at a starting density of 2000 cells per well for Hec1A cells and 1000 cells per well for Ishikawa cells. After 24 h, the medium was aspirated, and 100 µL of fresh medium containing serial dilutions of individual drugs was placed over the cells. After 72 h of incubation, the medium was aspirated, and cells were incubated with 0.05% MTT solution for 1 h. The supernatant was removed, and the formazan crystals were dissolved in 100 µL DMSO. The plates were read at 570 nm by a uQuant microplate spectrophotometer (BioTek, Winooski, VT, USA). Triplicate biological experiments were performed. Dose–response curves were plotted using Prism 8.0.0 (GraphPad Software, San Diego, CA, USA), the combination index was determined by CompuSyn software [
19] (ComboSyn, combosyn.com, accessed on 12 July 2019), and synergy assessment was performed using the Bliss model in the SynergyFinder web application [
20] (
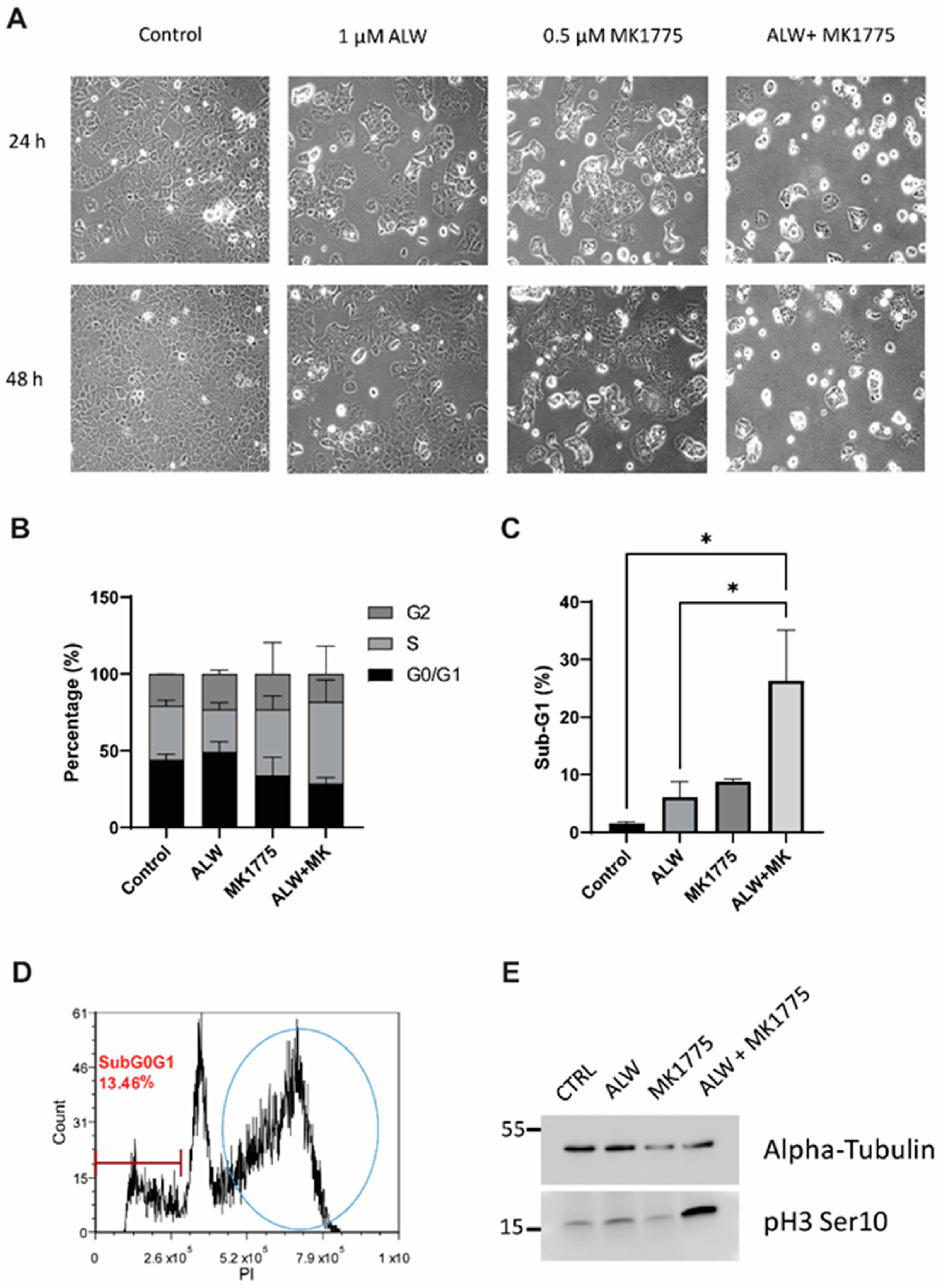
https://synergyfinder.fimm.fi, accessed on 29 July 2019). The Bliss synergy scores in this platform indicate synergy if they are greater than 10, additivity if they are between −10 and 10, and antagonism if they are less than −10. Based on the cell viability results for all future experiments, Hec1A cells were treated with DMSO, 1 µM ALW, 0.5 µM MK1775, and the combination of 1 µM ALW and 0.5 µM MK1775. Ishikawa cells were treated with DMSO, 0.5 µM ALW, 0.25 µM MK1775, and the combination of 0.5 µM ALW and 0.25 µM MK1775.
4.5. Cell Cycle and Apoptosis Analysis
For the cell cycle assay, control and drug-treated cells were trypsinized, washed with PBS twice, fixed in ice-cold 70% ethanol, and stored at –20 °C. On the day of analysis, cells were washed twice with PBS and then incubated in 50 µg/mL of propidium iodide (PI) solution containing 0.5 µg/mL RNase A for 4 h in the dark and analyzed by flow cytometry. For the apoptosis assay, the cell supernatant as well as trypsinized cells were mixed and pelleted and then washed with PBS. The apoptosis assay was performed using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, Franklin Lakes, NJ, USA). After Annexin V–fluorescein isothiocyanate (FITC) and PI staining, cells were analyzed by flow cytometry.
4.6. Colony Formation Assay
Cells were seeded at a density of 500 to 1000 cells per well in a 12-well plate, and the cells were left to grow in the incubator for 10 to 14 days with the respective drug combinations for both Hec1A and Ishikawa cells. After visible colonies containing more than 50 cells appeared, the plates were fixed with a solution containing glutaraldehyde (6.0%, v/v) and crystal violet (0.5%, w/v) for 15 to 20 min at room temperature. After that, the crystal-violet-fixing solution was decanted, and the plates were washed in water three to five times and then left to dry at room temperature. The plates were imaged, and the number of colonies was counted.
4.7. Western Blotting
Harvested cells were spun down at 2000 rpm for 5 min, washed with ice-cold PBS, and then pelleted at 3000 rpm for 3 min. Cell pellets were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors and quantified using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein (20 µg) were boiled at 95 °C for 10 min, run on an SDS-PAGE gel (8–12%), transferred onto a nitrocellulose membrane, incubated in 5% milk (in Tris buffered saline-Tween 20 [TBS-T]) for 1 h, and then incubated overnight in the appropriate primary antibodies (listed below). Blots were washed with TBS-T thrice for 5 min each and then incubated with corresponding secondary antibodies (1:2500 dilution, GE Healthcare, Chicago, IL, USA) for 1 h. Enhanced chemiluminescence substrate (ECL; Thermo Fisher Scientific, Waltham, MA, USA) was then added to the blots for 1 min, and immunoblot images were captured using an Azure Biosystems imaging machine (Azure Biosystems, Dublin, CA, USA).
The following antibodies were used: anti-EphA2 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-phosphorylated (phospho) cdc2 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-cdc2 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), Cell Cycle and Apoptosis WB Cocktail (pCdk/pHH3/Actin/cleaved PARP) (1:250 dilution; Abcam, Cambridge, UK); Apoptosis and DNA damage WB Cocktail (pH2A.X/GAPDH/cleaved PARP) (1:250 dilution; Abcam, Cambridge, UK); anti-phospho S6 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-S6 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-AKT (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA); anti-pAKT (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA); anti-cleaved caspase 3 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA); anti-P62 (1:3000; BD Biosciences, Franklin Lakes, NJ, USA); anti-GAPDH (1:5000; Thermo Fisher Scientific, Waltham, MA, USA), anti-LC3B (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-alpha-tubulin (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), and antibeta-actin (1:3000; Sigma-Aldrich, St. Louis, MO, USA).
4.8. Liposomal Nanoparticle Preparation
For in vivo delivery, siRNAs were incorporated into DOPC liposomes as described earlier [
13]. In brief, DOPC and siRNA were mixed in a ratio of 1:10 (
w/
w) siRNA:DOPC in the presence of excess tertiary butanol. Tween 20 was added to the siRNA/DOPC mixture in a ratio of 1:19 (Tween-20:siRNA/DOPC). The mixture was vortexed, frozen in an acetone/dry-ice bath, and lyophilized. Before in vivo administration, this preparation was hydrated with magnesium- and calcium-free PBS to achieve a desired concentration of 5 µg of siRNA in 200 µL volume per dose per mouse.
4.9. In Vivo Model of Endometrial Cancer
Female nude mice aged 4–8 weeks were purchased from Taconic Biosciences, USA. All mice were housed at The University of Texas MD Anderson Cancer Center animal facility under specific pathogen-free conditions. All animal-related experiments were approved by the Institutional Animal Care and Use Committee of MD Anderson Cancer Center. The right uterine horns of 8-week-old female athymic nude mice were injected with five million Hec1A cells in 100 µL of Hank’s Balanced Salt Solution (HyClone, Logan, UT, USA) to the uterine horn. For the second model, one million Ishikawa-Luc cells were injected into the peritoneal cavity of 6–8-week-old female mice. After eight days, mice were randomized to four groups (10 mice per group): siControl-DOPC nanoparticles (NPs), siEphA2-DOPC NPs, siControl-DOPC NPs with MK1775, and siEphA2-DOPC NPs with MK1775. The siRNA-DOPC NPs were given to mice twice a week intraperitoneally, and MK1775 (30 mg/kg) was administered daily by oral gavage. Once mice from any group became moribund, all mice were euthanized; mouse weight, tumor weight, ascites volume, and number of nodules were recorded.
4.10. RNA-Seq Analysis
Hec1A cells were plated in six-well plates at a density of 100,000 cells per well, in triplicate, and incubated overnight. After 24 h, cells were treated with DMSO, 1 µM ALW, 0.5 µM MK1775, or the combination of 1 µM ALW and 0.5 µM MK1775 for 8 h, after which the RNA was extracted using Direct-zol RNA Miniprep Plus kit (ZYMO Research, Irvine, CA, USA). RNA quality was determined by RNA integrity number using a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and the samples were shipped to Novogene (Sacramento, CA, USA) for RNA-Seq analysis on the Illumina NovaSeq 6000 platform. Downstream analysis was performed using a combination of programs, including hisat2, DEseq2, and ClusterProfiler software. Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) (Qiagen, Hilden, Germany).
4.11. Statistical Analysis
Statistics were performed using unpaired t-tests for comparisons between two groups and one-way ANOVAs with the Tukey post hoc test for multiple comparisons between more than two groups (Prism). Statistical significance was defined as a p value of <0.05.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




