
Bottom-Up Synthesis of De-Functionalized and Dispersible Carbon Spheres as Colloidal Adsorbent

 , , and
, , and
Abstract
:
1. Introduction
- How can the dispersibility of the individual particles be maintained during pyrolysis?
- How can the pore size distribution and surface chemistry of the aCS be tuned during the pyrolysis step?
- How does the surface of the synthesized aCS change over time, and how can it be stabilized?
2. Results and Discussion
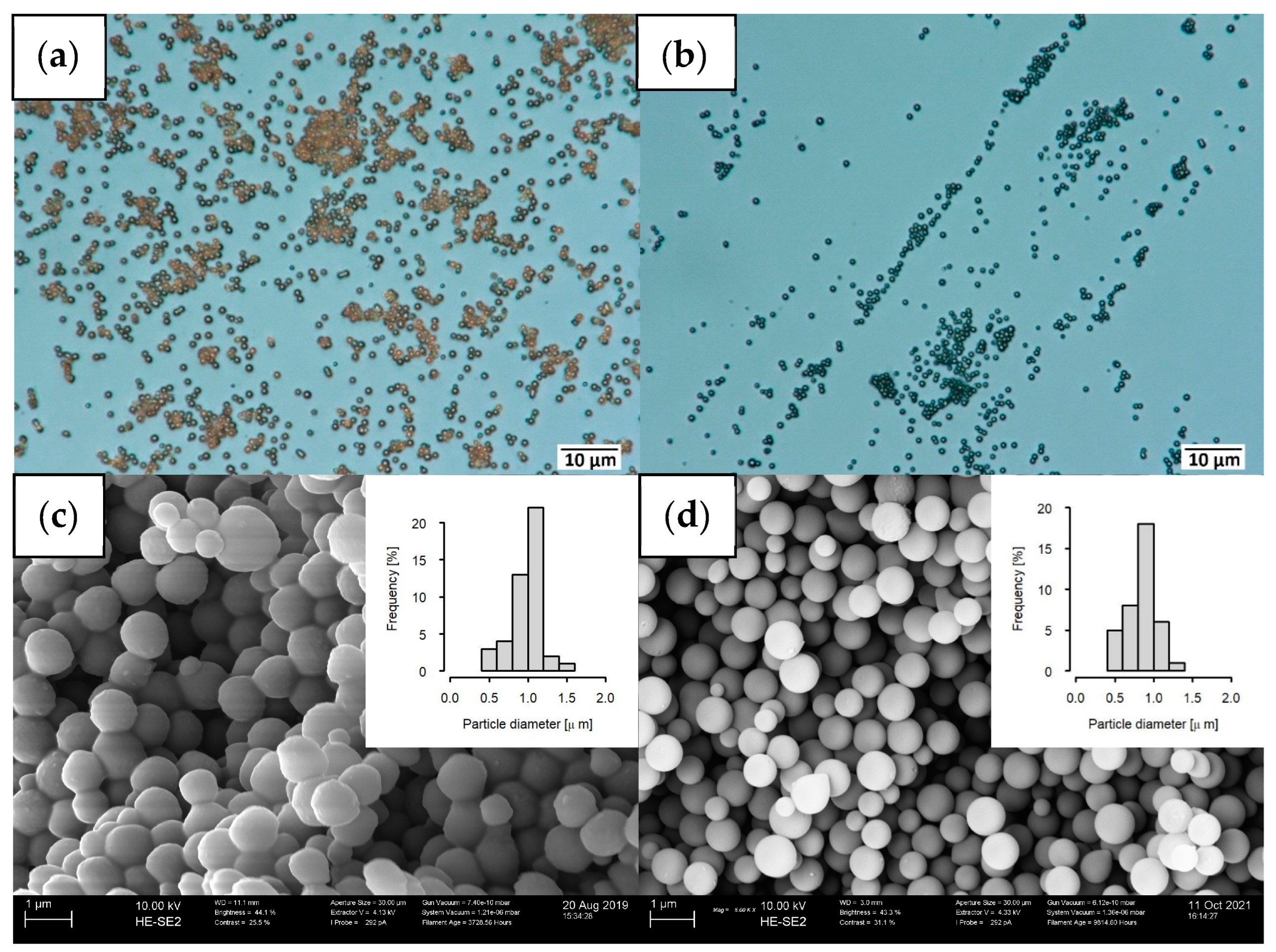
2.1. Maintaining the Excellent Dispersibility of Carbon Spheres after the Pyrolysis Step
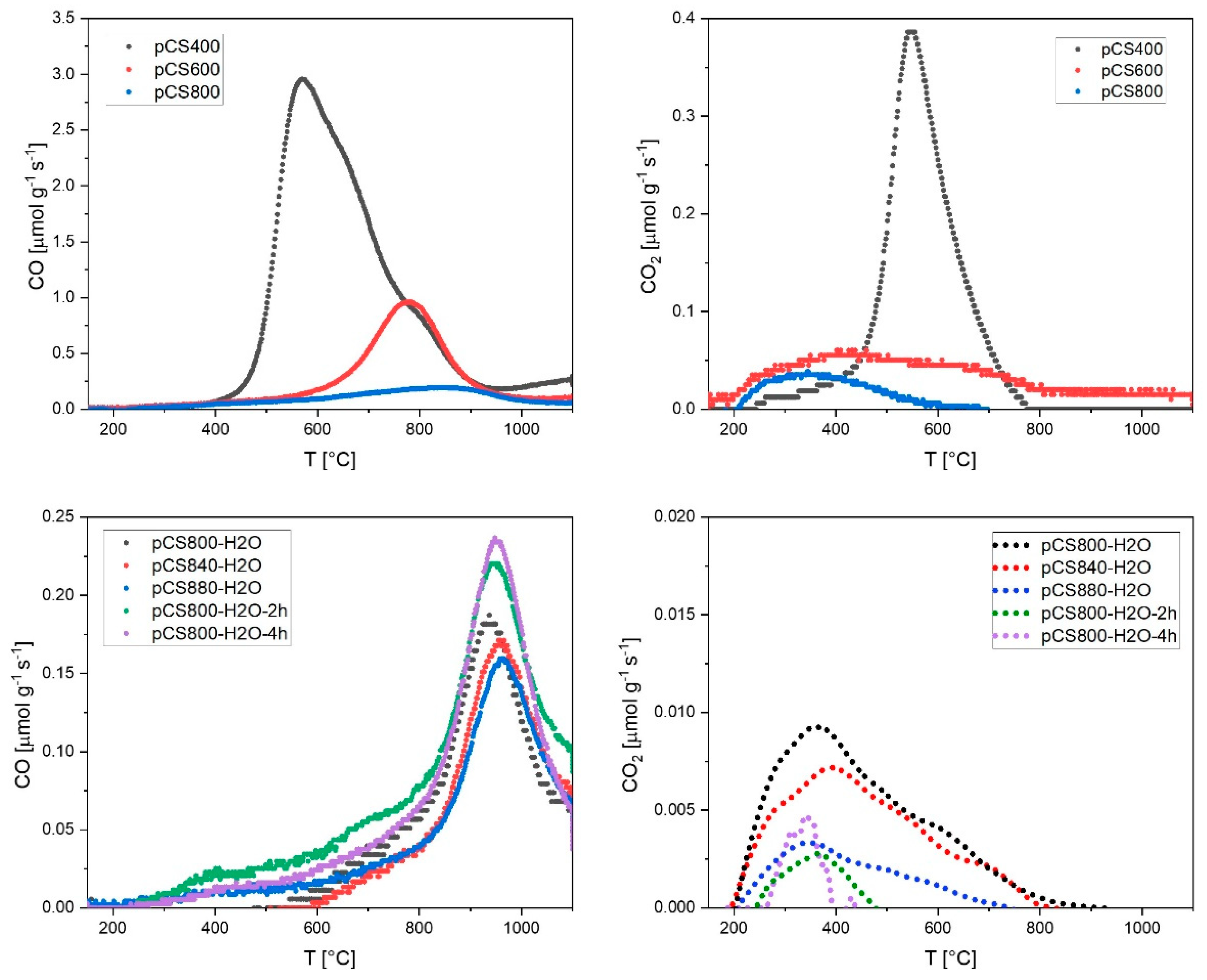
2.2. Tuning of Surface Properties and Mean Pore Diameters during the Pyrolysis Step
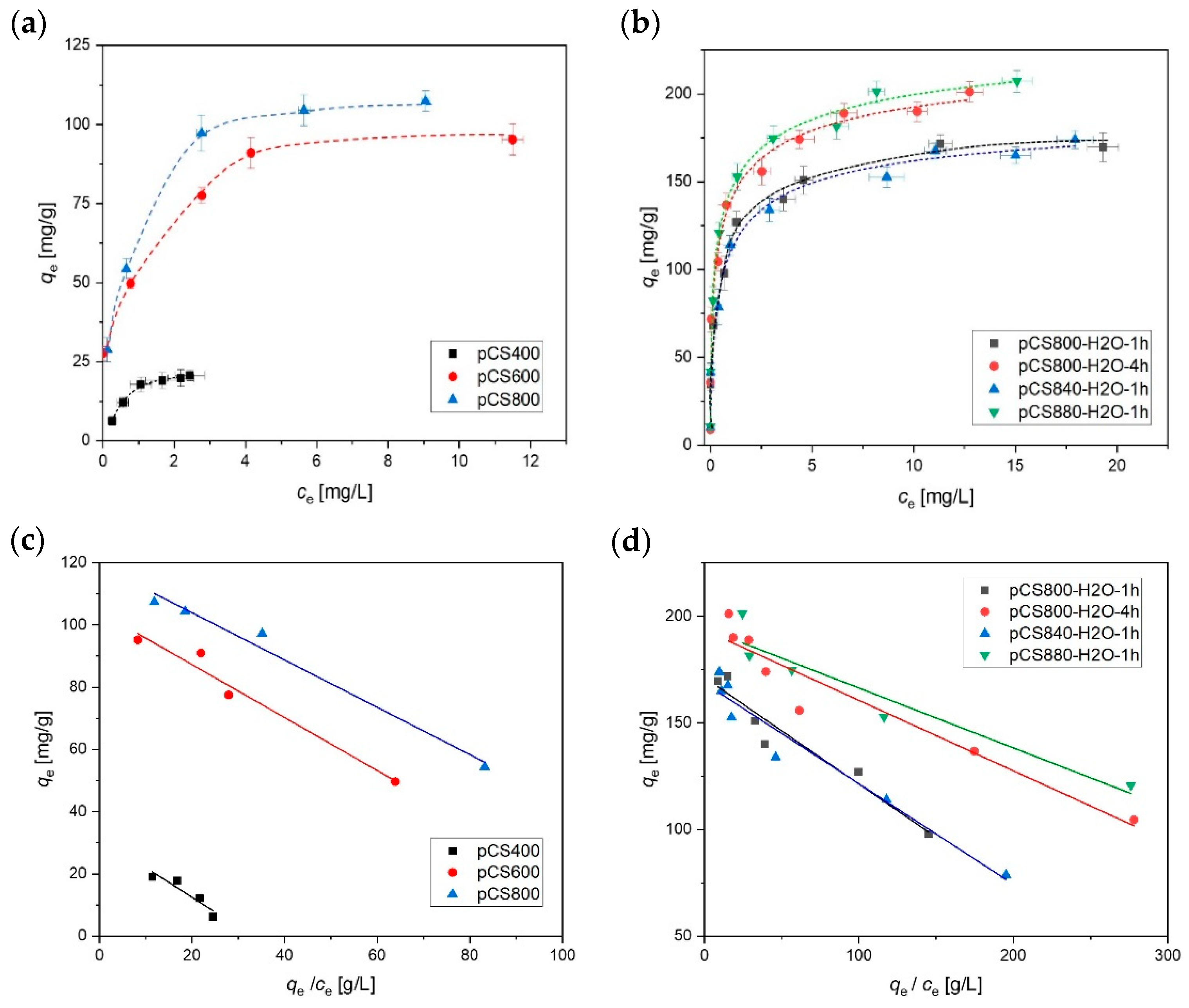
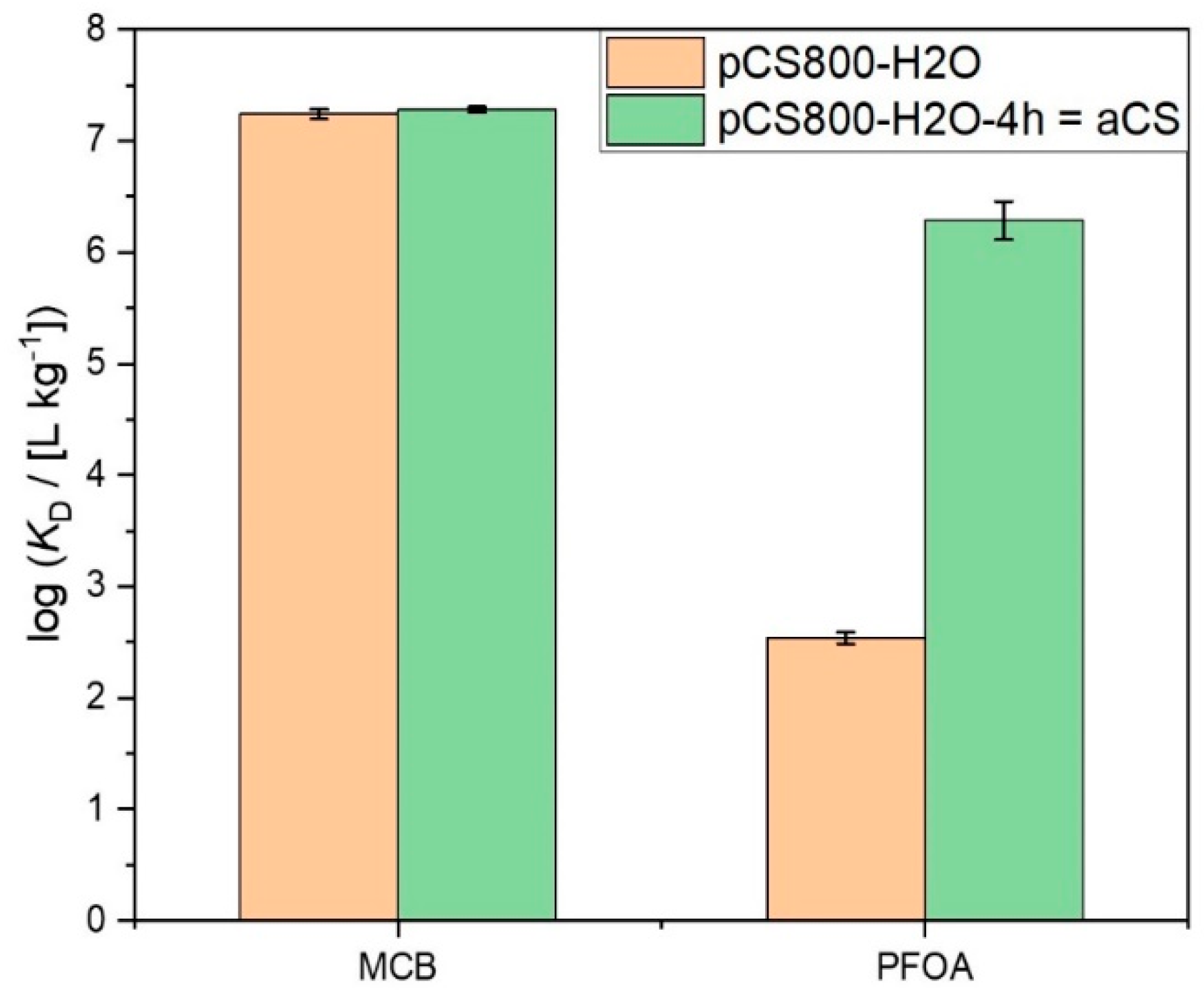
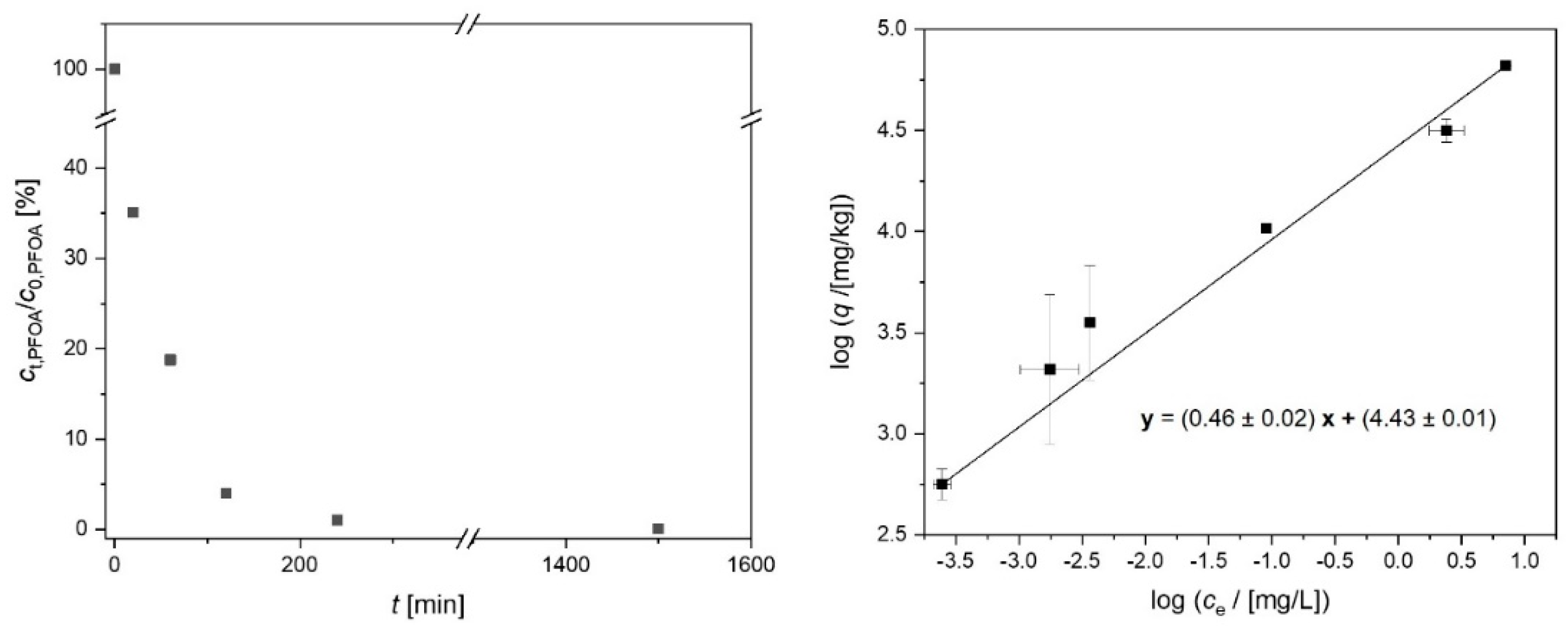
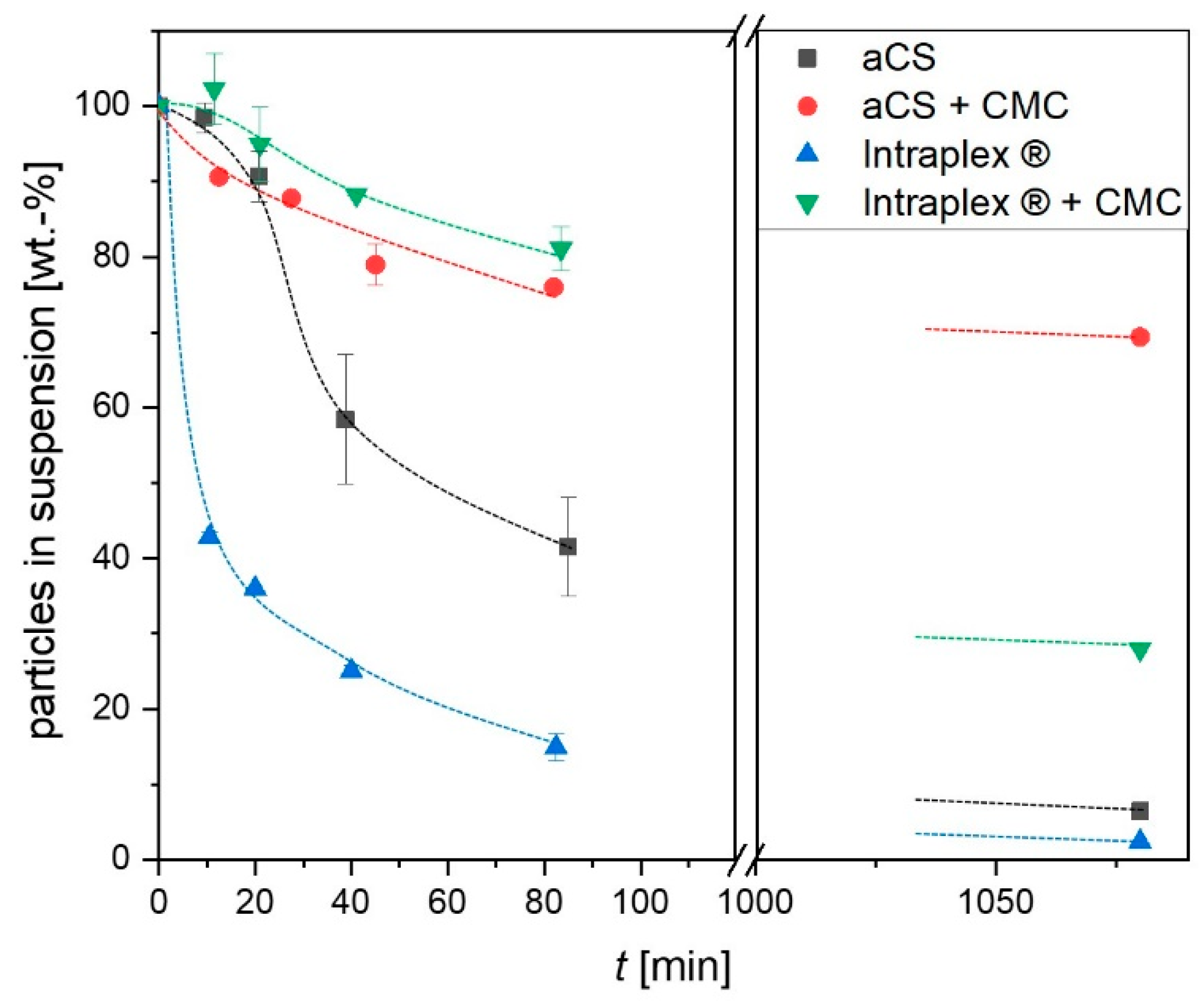
2.3. Suspension Stability of the Activated Carbon Spheres
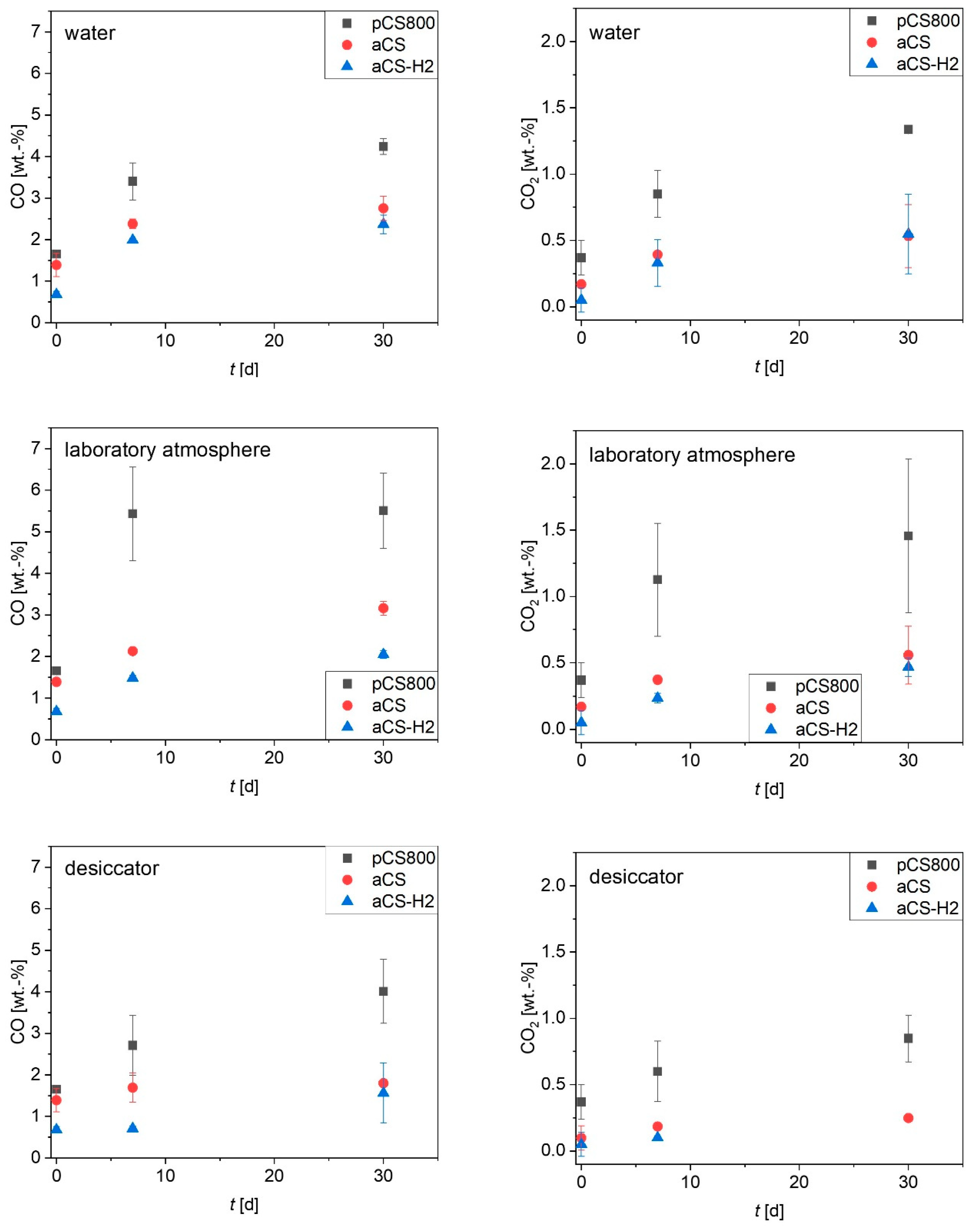
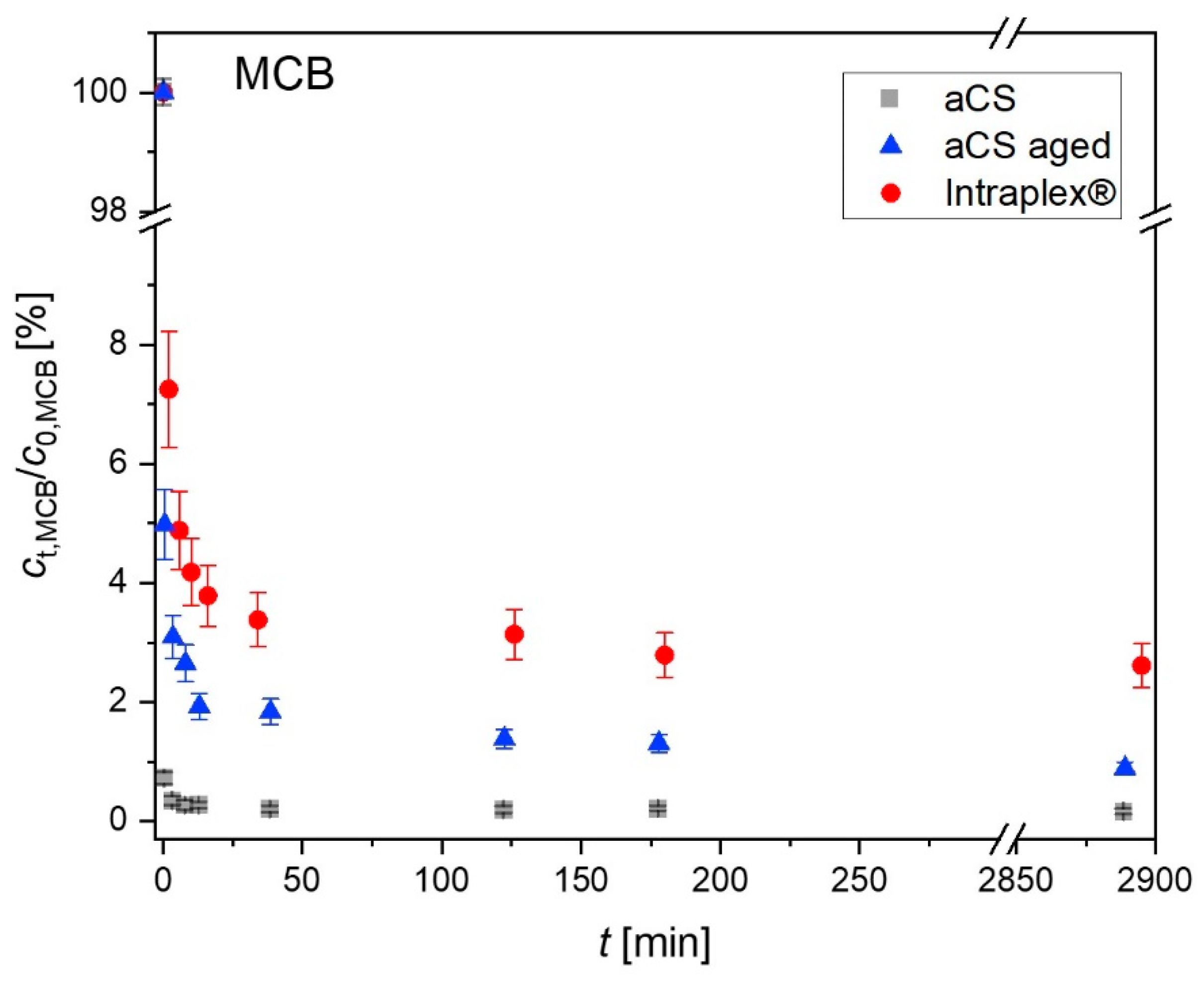
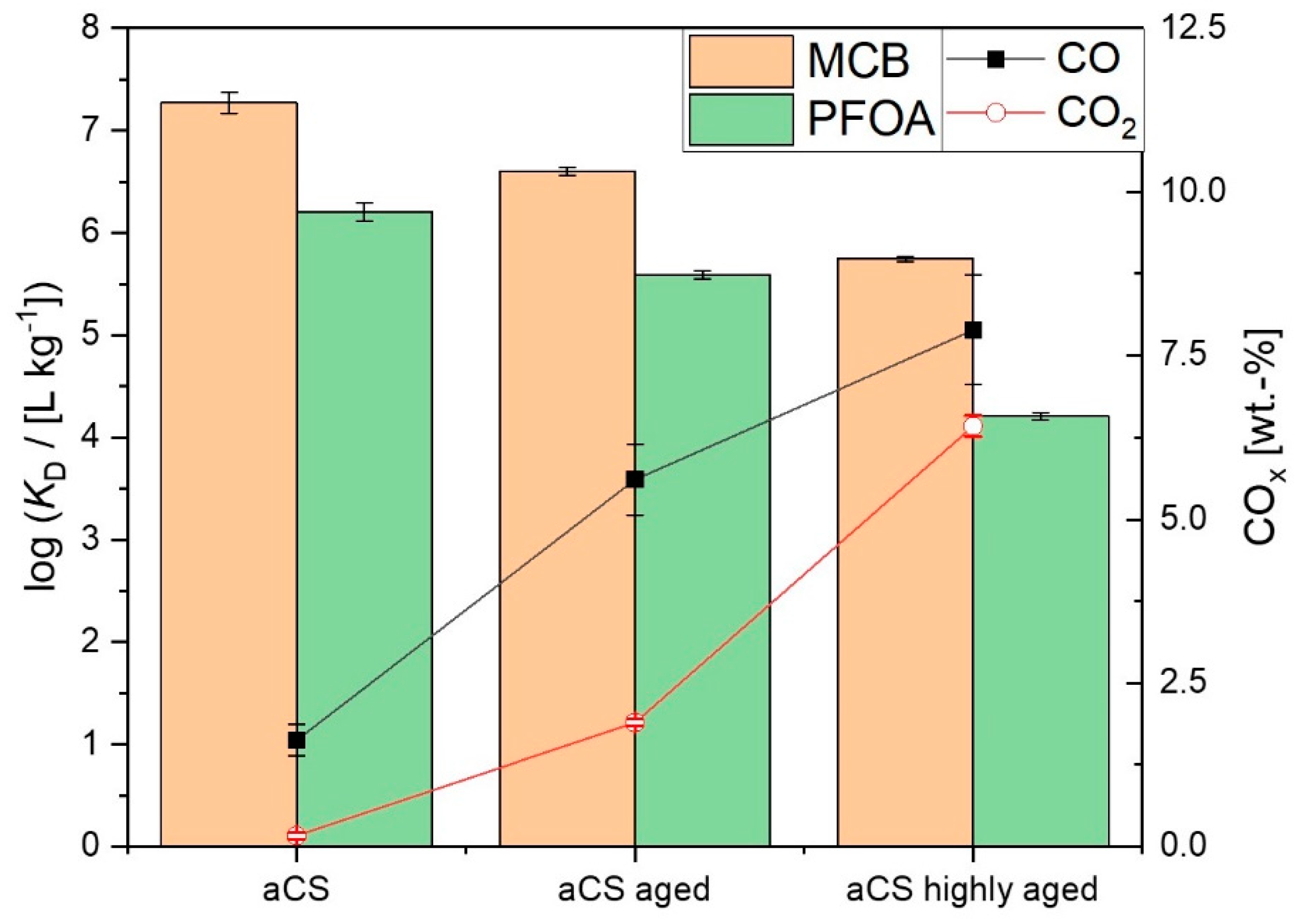
2.4. Ageing of the Activated Carbon Spheres
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Activated Carbon Spheres
3.3. Ageing of Activated Carbon Spheres
3.4. Adsorption Experiments
3.5. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Decrey, L.; Bonvin, F.; Bonvin, C.; Bonvin, E.; Kohn, T. Removal of trace organic contaminants from wastewater by superfine powdered activated carbon (SPAC) is neither affected by SPAC dispersal nor coagulation. Water Res. 2020, 185, 116302. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, F.; Jost, L.; Randin, L.; Bonvin, E.; Kohn, T. Super-fine powdered activated carbon (SPAC) for efficient removal of micropollutants from wastewater treatment plant effluent. Water Res. 2016, 90, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Regenesis, PlumeStop® Liquid Activated CarbonTM. 2022. Available online: https://regenesis.com/en/remediation-products/plumestop-liquid-activated-carbon/ (accessed on 21 May 2022).
- Bardos, P.; Merly, C.; Kvapil, P.; Koschitzky, H.-P. Status of nanoremediation and its potential for future deployment: Risk-benefit and benchmarking appraisals. Remediat. J. 2018, 28, 43–56. [Google Scholar] [CrossRef]
- Intrapore Particle-Based Ground-Water Remediation. Available online: https://intrapore.com/en/ (accessed on 21 May 2022).
- Coperchini, F.; Awwad, O.; Rotondi, M.; Santini, F.; Imbriani, M.; Chiovato, L. Thyroid disruption by perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA). J. Endocrinol. Investig. 2017, 40, 105–121. [Google Scholar] [CrossRef]
- Holbech, H.; Matthiessen, P.; Hansen, M.; Schüürmann, G.; Knapen, D.; Reuver, M.; Flamant, F.; Sachs, L.; Kloas, W.; Hilscherova, K.; et al. ERGO: Breaking down the wall between human healtz and environmental testing of endocrine disrupters. Int. J. Mol. Sci. 2020, 21, 2954. [Google Scholar] [CrossRef]
- Cordner, A.; Goldenman, G.; Birnbaum, L.; Brown, M.; Miller, M.F.; Mueller, R.; Patton, S.; Salvatore, D.H.; Trasande, L. The true cost of PFAS and the benefits of acting now. Environ. Sci. Technol. 2021, 55, 9630–9633. [Google Scholar] [CrossRef]
- McGregor, R. In situ treatment of PFAS-impacted groundwater using colloidal activated carbon. Remediat. J. 2018, 28, 33–41. [Google Scholar] [CrossRef]
- Carey, G.R.; McGregor, R.; Pham, A.L.; Sleep, B.; Hakimabadi, S. Evaluating the logevity of a PFAS in situ colloidal activated carbon remedy. Remediation 2019, 29, 17–31. [Google Scholar] [CrossRef]
- Georgi, A.; Schierz, A.; Mackenzie, K.; Kopinke, F.-D. Colloidal activated carbon for in-situ groundwater remediation—Transport characteristics and adsorption of organic compounds in water-saturated sediment columns. J. Contam. Hydrol. 2015, 179, 76–88. [Google Scholar] [CrossRef]
- Balda, M.; Mackenzie, K.; Kopinke, F.-D.; Georgi, A. Uniform and dispersible carbonaceous microspheres as quasi-liquid sorbent. Chemosphere 2022, 307, 136079. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y. Colloidal carbon spheres and their core/shell structures with noble-metal nanoparticles. Angew. Chem. 2004, 116, 607–611. [Google Scholar] [CrossRef]
- Yang, J.; Li, J.-Y.; Qian, J.-Q.; Lian, H.-Z.; Chen, H.-Y. Solid phase extraction of magnetic carbon doped Fe3O4 nanoparticles. J. Chromatogr. A 2014, 1325, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Takaesu, H.; Matsui, H.; Nishimura, Y.; Matsushita, T.; Shirasaki, N. Micro-milling super-fine powdered activated carbon decreases adsorption capacity by introducing oxygen/hydrogen-containing functional groups on carbon surface from water. Water Res. 2019, 155, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, N.; Kopinke, F.-D.; Georgi, A. What is specific in adsorption of perfluoroalkyl acids on carbon materials? Chemosphere 2021, 273, 128520. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.C.; Bahaddi, Y. New insight on the chemical role of water vapour in the ageing of activated carbon. Carbon 1993, 31, 821–825. [Google Scholar] [CrossRef]
- Menéndez, J.A.; Philips, J.; Xia, B.; Radovic, L.R. On the modification and characterization of chemical surface properties of activated carbon: In the search of carbons with stable basic properties. Langmuir 1996, 12, 4404–4410. [Google Scholar] [CrossRef]
- Osmond, N.M. Activated carbon fibre adsorbent materials. Adsorp. Sci. Technol. 2000, 18, 529–539. [Google Scholar] [CrossRef]
- Saha, N.; Xin, D.; Chiu, P.C.; Toufiq Reza, M. Effect of pyrolysis temperature on acidic oxygen-containing functional groups and electron storage capacities of pyrolyzed hydrochars. ACS Sustain. Chem. Eng. 2019, 7, 8387–8396. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfao, J.J.M. Characterization of active sites on carbon catalysts. Ind. Eng. Chem. Res. 2007, 46, 4110–4115. [Google Scholar] [CrossRef]
- Azzaz, A.A.; Khiari, B.; Jellali, S.; Ghimbeu, C.M.; Jeguirim, M. Hydrochars production, characterization and application for wastewater treatment: A review. Renew. Sustain. Energy Rev. 2020, 127, 109882. [Google Scholar] [CrossRef]
- Román, S.; Nabais Valente, J.M.; Ledesma, B.; González, M.T.; Laginhas, C.; Titirici, M.M. Production of low-cost adsorbents with tunable surface chemistry by conjunction of hydrothermal carbonization and activation processes. Microporous Mesoporous Mater. 2013, 165, 127–133. [Google Scholar] [CrossRef]
- Chegini, G.; Briens, C.; Pjontek, D. Production and characterization of adsorbents from a hydrothermal char by pyrolysis, carbon dioxide and steam activation. Biomass Conv. Bioref. 2022. [Google Scholar] [CrossRef]
- Mestre, A.S.; Tyszko, E.; Andrade, M.A.; Galhetas, M.; Freire, C.; Carvalho, A.P. Sustainable activated carbons prepared from a sucrose-derived hydrochar: Remarkable adsorbents for pharmaceutical compounds. RSC Adv. 2015, 5, 19696–19707. [Google Scholar] [CrossRef]
- Schüürmann, G. QSAR analysis of the acute fish toxicity of organic phosphorothionates using theoretically derived molecular descriptors. Environ. Toxicol. Chem. 1990, 9, 417–428. [Google Scholar] [CrossRef]
- Krahn, K.M.; Cornelissen, G.; Castro, G.; Arp, H.P.H.; Asimakopoulos, A.G.; Wolf, R.; Holmstad, R.; Zimmemann, A.R.; Sørmo, E. Sewage sludge biochars as effective PFAS-sorbents. J. Hazard. Mater. 2023, 445, 130449. [Google Scholar] [CrossRef]
- Mackenzie, K.; Georgi, A. NZVI synthesis and characterization. In Nanoscale Zerovalent Iron Particles for Environmental Restoration; Phenrat, T., Lowry, G.V., Eds.; Springer International Publishing AG: Basel, Switzerland, 2019; pp. 45–95. [Google Scholar]
- Zhou, J.; Saeidi, N.; Wick, L.Y.; Xie, Y.; Kopinke, F.-D.; Georgi, A. Efficient removal of trifluoroacetic acid from water using surface-modified activated carbon and electro-assisted desorption. J. Hazard. Mater. 2022, 436, 129051. [Google Scholar] [CrossRef]
- Pignatello, J.J.; Mitch, W.A.; Xu, W. Activity and reactivity of pyrogenic carbonaceous matter toward organic compounds. Environ. Sci. Technol. 2017, 51, 8893–8908. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.; Balda, M.; Presser, V.; Kopinke, F.-D.; Georgi, A. Electro-assisted removal of polar and ionic organic compounds from water using activated carbon felts. Chem. Eng. J. 2022, 433, 133544. [Google Scholar] [CrossRef]
- Yao, C.; Chen, T. A new simplified method for estimating film mass transfer and surface diffusion coefficients from batch adsorption kinetic data. Chem. Eng. J. 2015, 265, 93–99. [Google Scholar] [CrossRef]
- Ocampo-Pérez, R.; Leyva-Ramos, R.; Sanchez-Polo, M.; Utrilla-Rivera, J. Role of pore volume and surface diffusion in the adsorption of aromatic compounds on activated carbon. Adsorption 2013, 19, 945–957. [Google Scholar] [CrossRef]
- Du, Z.; Deng, S.; Bei, Y.; Huang, Q.; Wang, B.; Huang, J.; Yu, G. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents—A review. J. Hazard. Mater. 2014, 274, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Fabregat-Palau, J.; Vidal, M.; Rigol, A. Examining sorption of perfluoroalkyl substances (PFAS) in biochars and other carbon-rich materials. Chemosphere 2022, 302, 134733. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 30 January 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Conditions (Gas Atmosphere, Final Temperature, Heating Rate and Holding Time) | |||||
|---|---|---|---|---|---|
| Fixed Parameters | Varied Parameter | Sample Name | Surface Area by Applying DFT Method [m2 g−1] | Mean Pore Diameter [nm] | Carbon Content [wt.-%] |
| 1 K min−1, N2, 1 h | 400 °C | pCS400 | 386 ± 5 | 0.60 ± 0.01 | 78 ± 1 |
| 600 °C | pCS600 | 653 ± 8 | 0.58 ± 0.01 | 88 ± 1 | |
| 800 °C | pCS800 | 877 ± 10 | 0.56 ± 0.01 | 92 ± 2 | |
| 1 K min−1, H2O/N2, 1 h | 800 °C | pCS800-H2O | 954 ± 11 | 0.58 ± 0.01 | 96 ± 2 |
| 840 °C | pCS840-H2O | 1178 ± 14 | 0.63 ± 0.01 | 98 ± 2 | |
| 880 °C | pCS880-H2O | 1386 ± 17 | 0.68 ± 0.01 | 96 ± 2 | |
| 1 K min−1, H2O/N2, 800 °C | 2 h | pCS800-H2O-2h | 1031 ± 12 | 0.60 ± 0.01 | 87 ± 2 |
| 4 h | pCS800-H2O-4h = aCS | 1015 ± 12 | 0.60 ± 0.01 | 91 ± 2 | |
| 1 K min−1, H2O/N2, 800 °C, 4 h | post-treatment at 900 °C, 2 h, 10 vol.-% H2/N2 | aCS-H2 | 1030 ± 12 | 0.61 ± 0.01 | 92 ± 2 |
| Sample Name | qmax (exp) | qmax (fit) | KL | R2 |
|---|---|---|---|---|
| [mg g−1] | [mg g−1] | [L mg−1] | ||
| pCS400 | 20 ± 1 | 32 ± 5 | 1.0 ± 0.3 | 0.81 |
| pCS600 | 104 ± 3 | 104 ± 4 | 1.2 ± 0.2 | 0.95 |
| pCS800 | 106 ± 2 | 119 ± 3 | 1.3 ± 0.1 | 0.97 |
| pCS800-H2O | 171 ± 1 | 171 ± 5 | 2.0 ± 0.3 | 0.94 |
| pCS840-H2O | 169 ± 4 | 168 ± 4 | 2.1 ± 0.2 | 0.95 |
| pCS880-H2O | 204 ± 3 | 194 ± 7 | 3.6 ± 0.6 | 0.89 |
| aCS | 196 ± 6 | 193 ± 5 | 3.0 ± 0.4 | 0.92 |
| AC Sample | r0 [mg g−1 s−1] | ke∙104 [s−1] | kf∙104 [m s−1] | Ds∙1020 [m2 s−1] |
|---|---|---|---|---|
| aCS | 1.2 | 7.9 | 1.3 | 2.3 |
| Aged aCS | 1.3 | 5.4 | 1.4 | 1.0 |
| Intraplex® | 1.2 | 1.1 | 1.3 | 25.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balda, M.; Mackenzie, K.; Woszidlo, S.; Uhlig, H.; Möllmer, J.; Kopinke, F.-D.; Schüürmann, G.; Georgi, A. Bottom-Up Synthesis of De-Functionalized and Dispersible Carbon Spheres as Colloidal Adsorbent. Int. J. Mol. Sci. 2023, 24, 3831. https://doi.org/10.3390/ijms24043831
Balda M, Mackenzie K, Woszidlo S, Uhlig H, Möllmer J, Kopinke F-D, Schüürmann G, Georgi A. Bottom-Up Synthesis of De-Functionalized and Dispersible Carbon Spheres as Colloidal Adsorbent. International Journal of Molecular Sciences. 2023; 24(4):3831. https://doi.org/10.3390/ijms24043831
Chicago/Turabian StyleBalda, Maria, Katrin Mackenzie, Silke Woszidlo, Hans Uhlig, Jens Möllmer, Frank-Dieter Kopinke, Gerrit Schüürmann, and Anett Georgi. 2023. "Bottom-Up Synthesis of De-Functionalized and Dispersible Carbon Spheres as Colloidal Adsorbent" International Journal of Molecular Sciences 24, no. 4: 3831. https://doi.org/10.3390/ijms24043831
APA StyleBalda, M., Mackenzie, K., Woszidlo, S., Uhlig, H., Möllmer, J., Kopinke, F.-D., Schüürmann, G., & Georgi, A. (2023). Bottom-Up Synthesis of De-Functionalized and Dispersible Carbon Spheres as Colloidal Adsorbent. International Journal of Molecular Sciences, 24(4), 3831. https://doi.org/10.3390/ijms24043831