


Gain of Aggressive Histological and Molecular Patterns after Acquired Resistance to Novel Anti-EGFR Therapies in Non-Small Cell Lung Cancer
 , , ,
, , ,
Abstract
1. Introduction
1.1. Context
1.2. Objective
1.3. Method
2. Detailed Case Description
2.1. Patient Information and Molecular Profile of the Initial Tumor
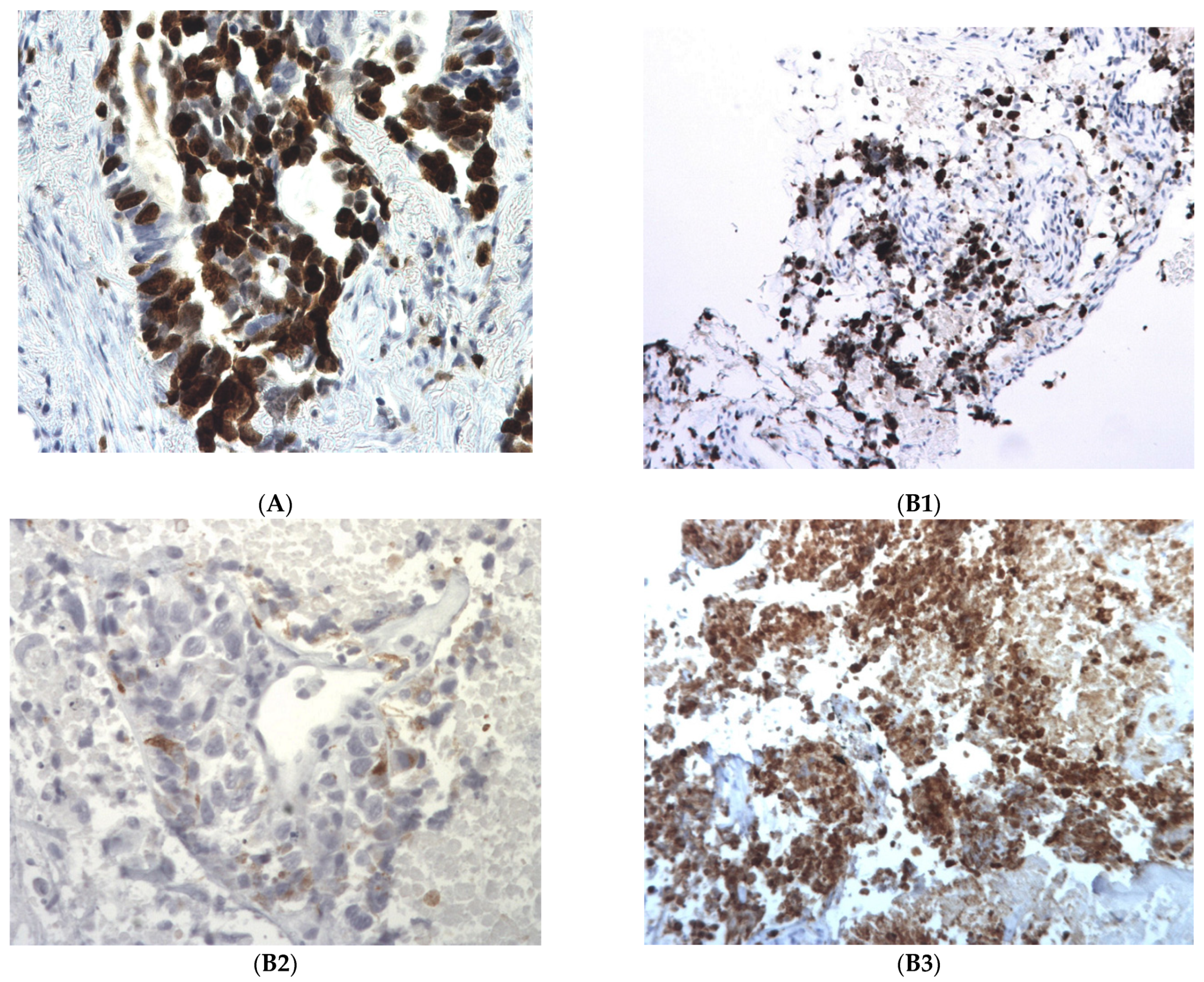
2.2. Pathological Findings: Differentiation in a More Aggressive Histology
2.3. Molecular Profile of the Tumor at Progression: Alteration in TP53 and RB1 Signaling Pathway
2.4. Transformation under Novel Anti-EGFR Therapies Patient by Patient
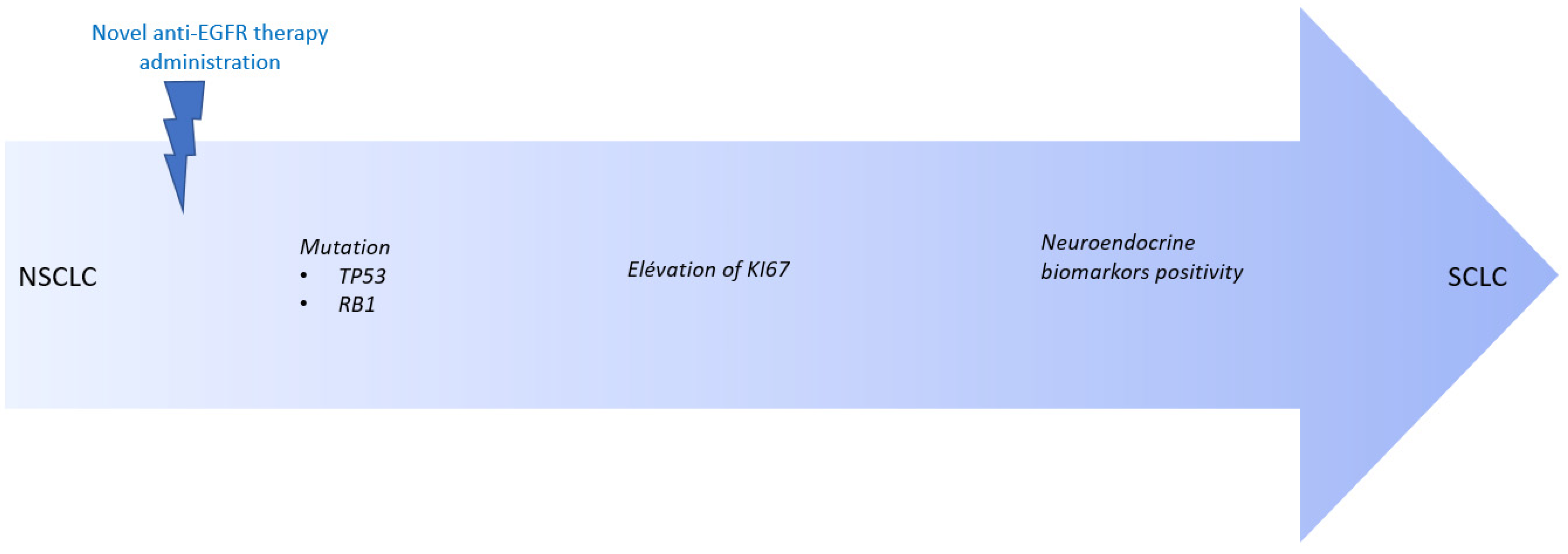
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Comprehensive Cancer Network. Non-Small Cell Lung Cancer (Version 7.2015). Available online: http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 30 June 2015).
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Wu, Y.L.; Planchard, D.; Lu, S.; Sun, H.; Yamamoto, N.; Kim, D.W.; Tan, D.S.W.; Yang, J.C.-H.; Azrif, M.; Mitsudomi, T.; et al. PanAsian adapted clinical practice guidelines for the management of patients with metastatic non-small-cell lung cancer: A CSCO ESMO initiative endorsed by JSMO, KSMO, MOS, SSO and TOS. Ann. Oncol. 2019, 30, 171–210. [Google Scholar] [PubMed]
- Minari, R.; Bordi, P.; Tiseo, M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: Review on emerged mechanisms of resistance. Transl. Lung Cancer Res. 2016, 5, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Riess, J.W.; Gandara, D.R.; Frampton, G.M.; Madison, R.; Peled, N.; Bufill, J.A.; Dy, G.K.; Ou, S.-H.I.; Stephens, P.J.; McPherson, J.D.; et al. Diverse EGFR exon 20 insertions and co-occurring molecular alterations identified by comprehensive genomic profiling of NSCLC. J. Thorac. Oncol. 2018, 13, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Goerge, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.-Y.; Kim, S.-W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Baik, C.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.; Beraud, C.; Cabel, L.; Fontugne, J.; Lassalle, M.; Krucker, C.; Dufour, F.; Groeneveld, C.S.; Dixon, V.; Meng, X.; et al. Integrated molecular and pharmacological characterization of patient derived xenografts from bladder and ureteral cancers identifies new potential therapies. Front. Oncol. 2022, 12, 930731. [Google Scholar] [CrossRef] [PubMed]
- Cuella-Martin, R.; Oliveira, C.; Lockstone, H.E.; Snellenberg, S.; Grolmusova, N.; Chapman, J.R. 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol. Cell. 2016, 64, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Levine, A. Spontaneous and inherited TP53 genetic alterations. Oncogene 2021, 40, 5975–5983. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, B.; Shim, J.H.; Lee, S.H.; Park, W.Y.; Choi, Y.L.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K. Concurrent Genetic Alterations Predict the Progression to Target Therapy in EGFR-Mutated Advanced NSCLC. J. Thorac. Oncol. 2019, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Petracc, E.; Delmonte, A.; Bronte, G.; Chiadini, E.; Ludovini, V.; Dubini, A.; Papi, M.; Baglivo, S.; De Luigi, N.; et al. Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J. Clin. Med. 2020, 9, 1047. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Minari, R.; Mazzaschi, G.; Gnetti, L.; La Monico, S.; Alfieri, R.; Campanini, N.; Verze, M.; Olivani, A.; Ventura, L.; et al. Small Cell Lung Cancer Transformation as a Resistance Mechanism to Osimertinib in Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: Case Report and Literature Review. Front. Oncol. 2021, 11, 642190. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| # | Age | Molecular Characteristics at Baseline | Pathological Characteristics at Baseline before Treatment | Treatment Administered | Delay under Treatment before Progression | Molecular Characteristics at Progression | Pathological Characteristics at Progression after Treatment |
|---|---|---|---|---|---|---|---|
| 1 | 56 | Exon 21 EGFR L858R (c.2573T>G), AF of 52% | Differenciated LUAD Ki67 30% Chromogranin A − CD56 − Synaptophysin − | patritumab deruxtecan | 15 months | EGFR c.2573T>G/p.(Leu858Arg), AF of 35% RB1 c.2106+1G>A/p.?, AF of 41% with LOH TP53 c.920−1G>A/p.?, AF of 52% with LOH | Differenciated LUAD Ki67 60% Chromogranin A − CD56 − Synaptophysin − |
| 2 | 66 | EGFR exon 20 Insertion, c.2313_2314insGTC/p.(Asn771_Pro772insVal), AF of 10% | Differenciated LUAD Ki67 20% Chromogranin A − CD56 − Synaptophysin − | amivantamab | 17 months | EGFR exon 20 Insertion, c.2313_2314insGTC/p.(Asn771_Pro772insVal), AF of 51% RB1 mutation c.1389+1_1389+2del/p.? (AF of 35%), heterozygocy with LOH TP53 mutation c.301A>T/p. (Lys101*), AF of 37% with LOH RBM10 mutation c.2398G>T/p.(Glu800*), AF of 24% | Differenciated LUAD Ki67 > 90% Chromogranin A − CD56 − Synaptophysin − |
| 3 | 58 | Del 19 EGFR c.2240_2257del/p., AF of 49% EGFR c.2369C>T/p.(Thr790Met), AF of 12% TP53 mutation c.395A>G (p.Lys132Arg) (AF of 90%) with LOH | Differenciated LUAD Ki67 10% Chromogranin A − CD56 − Synaptophysin − | patritumab deruxtecan | 4 months | Del 19 EGFR c.2240_2257del/p., AF of 34.2% EGFR c.2369C>T/p.(Thr790Met), AF of 9% TP53 c.395A>G/p.(Lys132Arg), AF of 75% with LOH MYC amplification PIK3CB c.3151G>A/p.(Glu1051Lys), AF of 59%) | Undifferenciated Large cell carcinoma Ki67 90% Chromogranin A + CD56 – Synaptophysin + |
| 4 | 66 | EGFR exon 20 Insertion c.2284-5_2290dup/p.(Ala763_Tyr764insPheGlnGluAla), AF of 18% | Differenciated LUAD Ki67 10% Chromogranin A − CD56 − Synaptophysin − | amivantamab | 24 months | EGFR exon 20 Insertion c.2284-5_2290dup/p. p.(Ala763_Tyr764insPheGlnGluAla), AF of 16% FGFR1 c.1731C>A/p.(Asn577Lys), AF of 2% TP53BP1 mutation c.2702-4del, p.?, AF of 12% | Differenciated LUAD Ki67 50% Chromogranin A − CD56 − Synaptophysin − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basse, C.; Trabelsi-Grati, O.; Masliah, J.; Callens, C.; Kamal, M.; Freneaux, P.; Klijanienko, J.; Bieche, I.; Girard, N. Gain of Aggressive Histological and Molecular Patterns after Acquired Resistance to Novel Anti-EGFR Therapies in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2023, 24, 3802. https://doi.org/10.3390/ijms24043802
Basse C, Trabelsi-Grati O, Masliah J, Callens C, Kamal M, Freneaux P, Klijanienko J, Bieche I, Girard N. Gain of Aggressive Histological and Molecular Patterns after Acquired Resistance to Novel Anti-EGFR Therapies in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2023; 24(4):3802. https://doi.org/10.3390/ijms24043802
Chicago/Turabian StyleBasse, Clémence, Olfa Trabelsi-Grati, Julien Masliah, Céline Callens, Maud Kamal, Paul Freneaux, Jerzy Klijanienko, Ivan Bieche, and Nicolas Girard. 2023. "Gain of Aggressive Histological and Molecular Patterns after Acquired Resistance to Novel Anti-EGFR Therapies in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 24, no. 4: 3802. https://doi.org/10.3390/ijms24043802
APA StyleBasse, C., Trabelsi-Grati, O., Masliah, J., Callens, C., Kamal, M., Freneaux, P., Klijanienko, J., Bieche, I., & Girard, N. (2023). Gain of Aggressive Histological and Molecular Patterns after Acquired Resistance to Novel Anti-EGFR Therapies in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 24(4), 3802. https://doi.org/10.3390/ijms24043802