

Structural Insight into Catalysis by the Flavin-Dependent NADH Oxidase (Pden_5119) of Paracoccus denitrificans

Abstract
:1. Introduction
2. Results
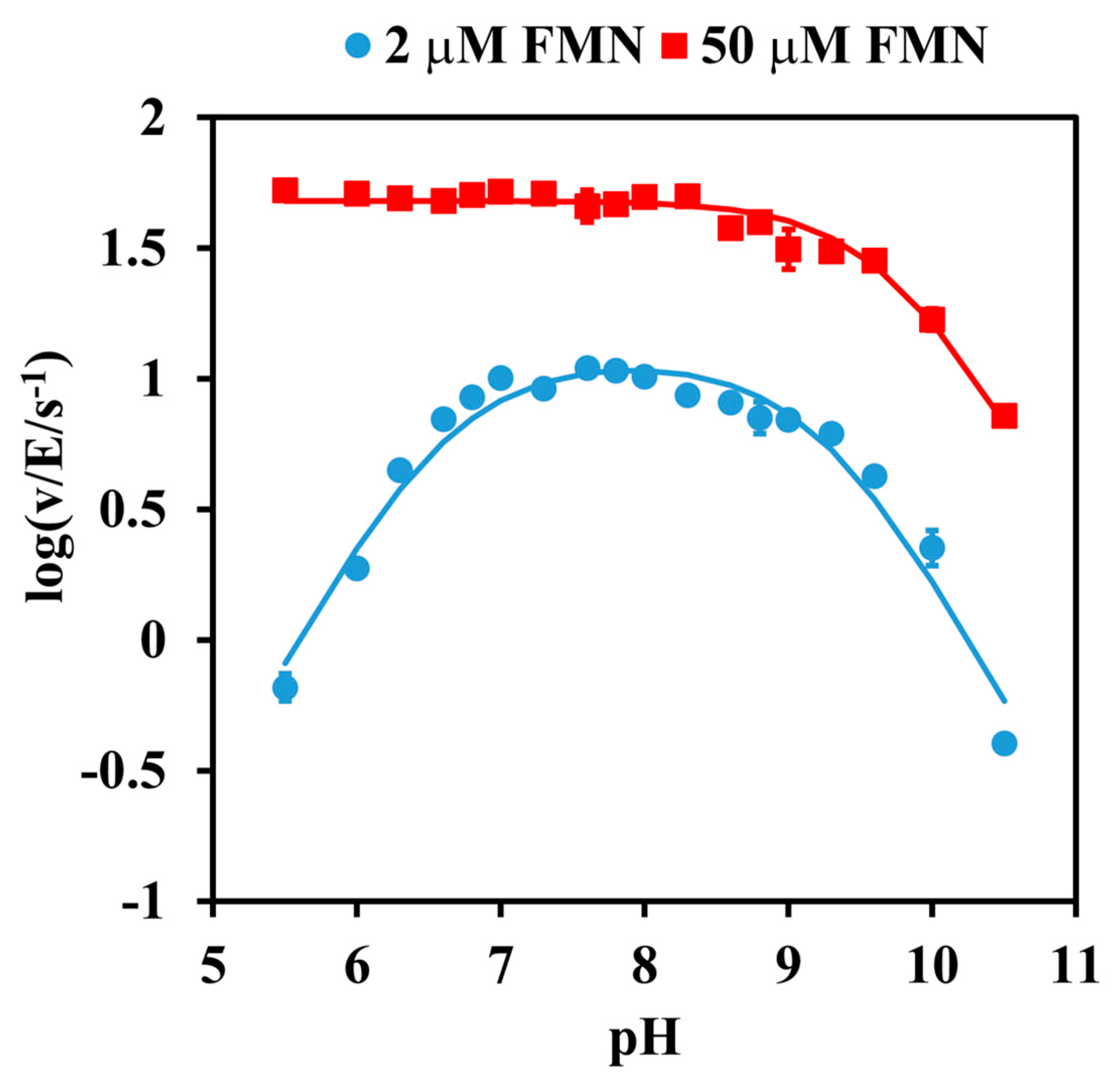
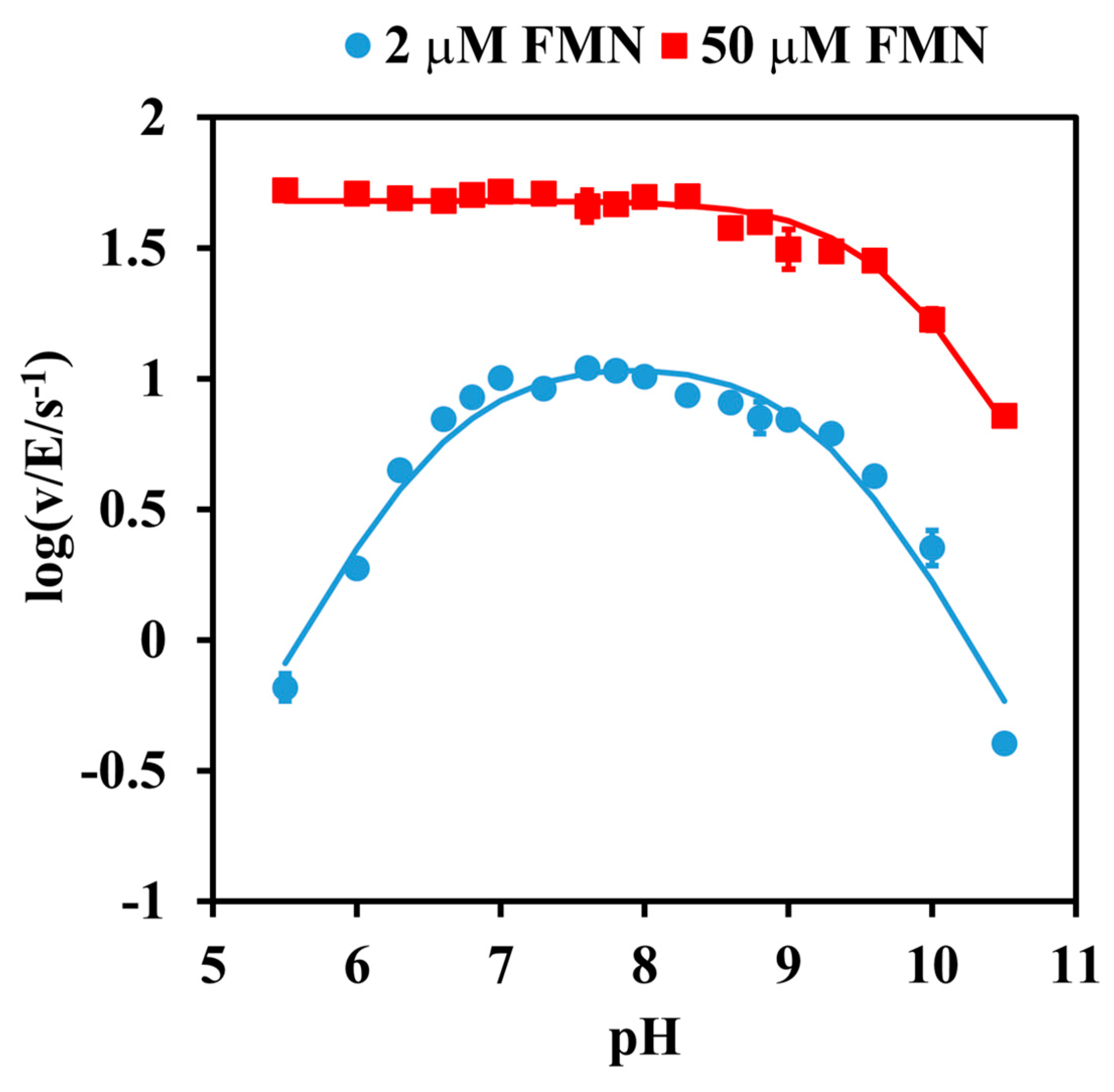
2.1. pH Dependence Analysis
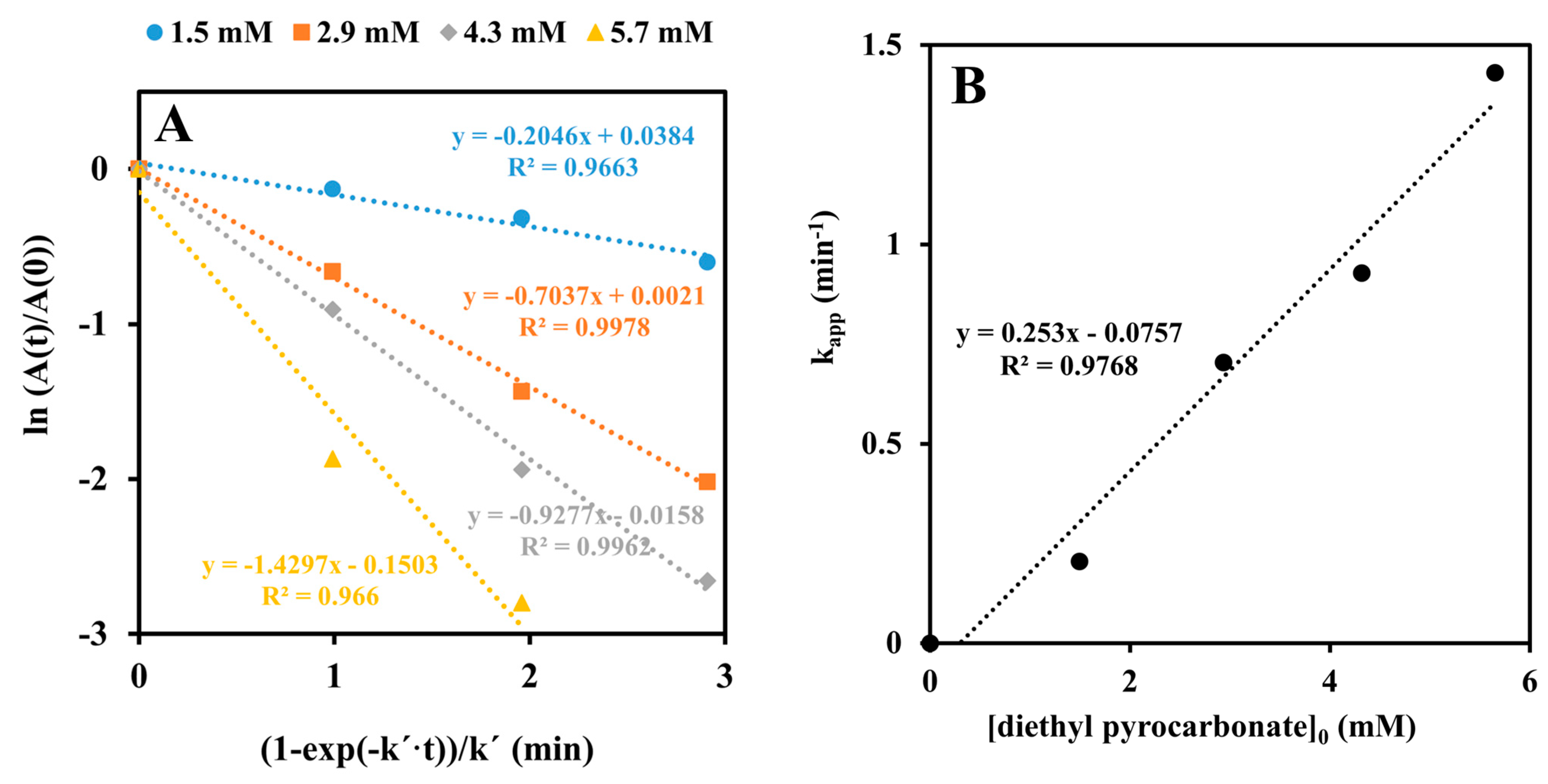
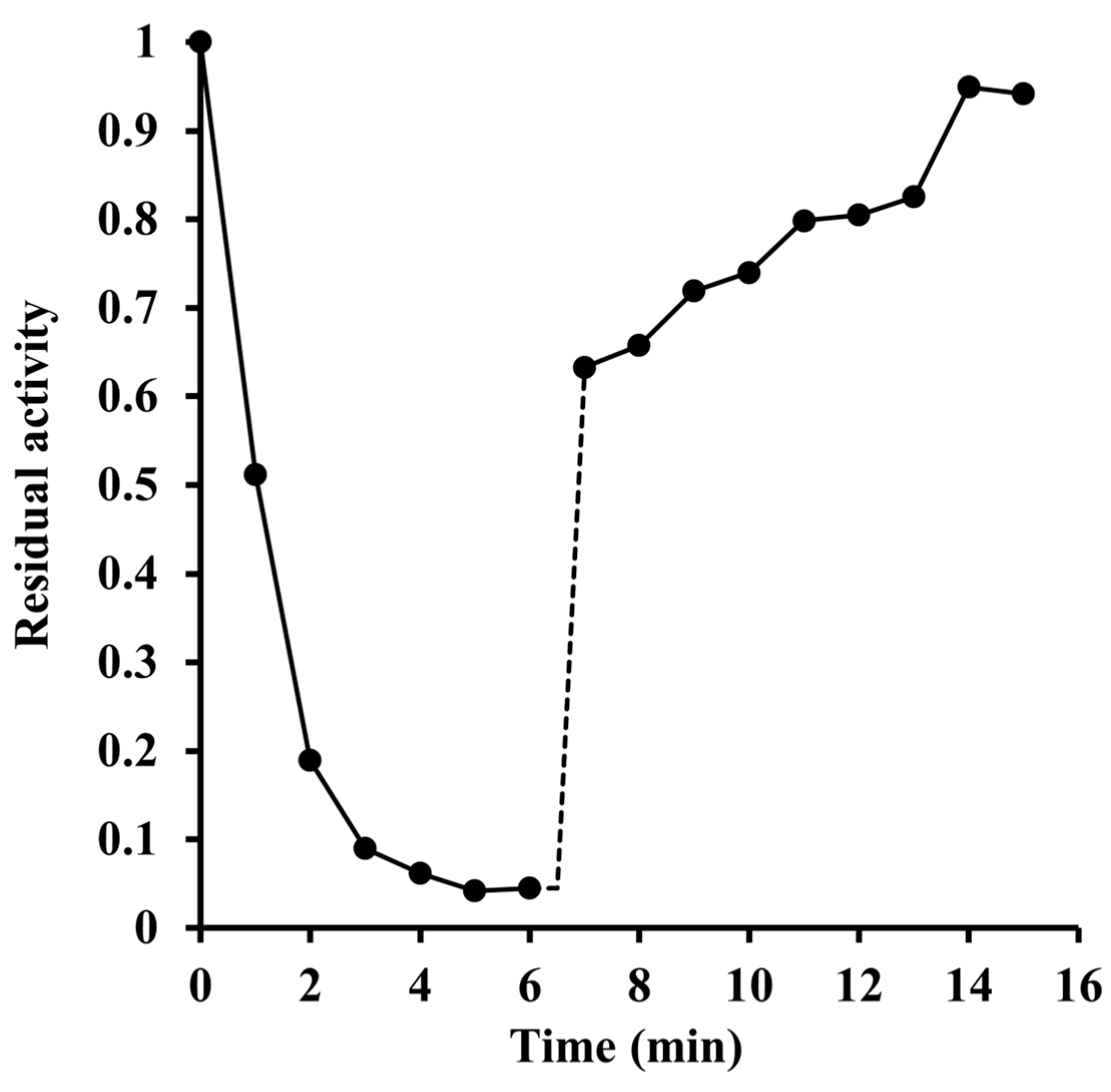
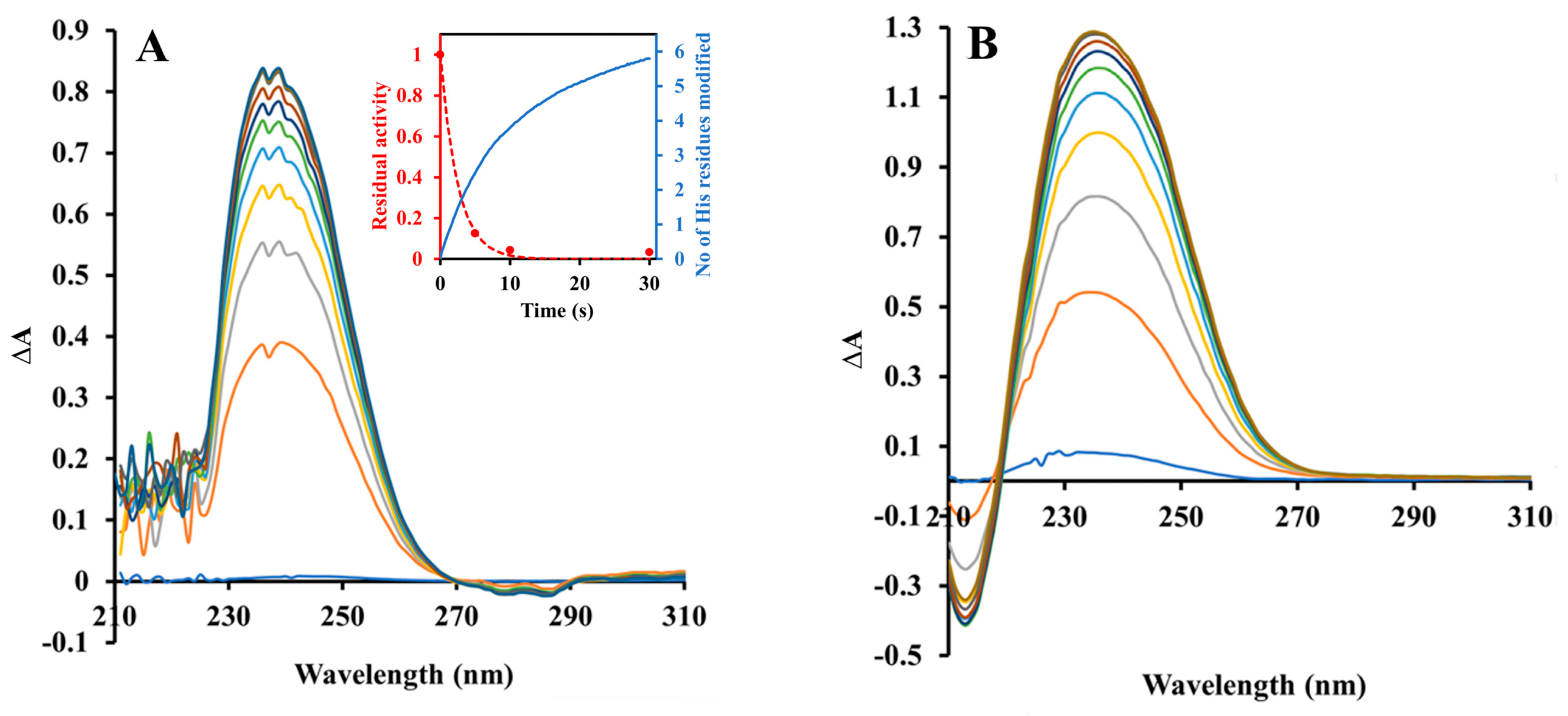
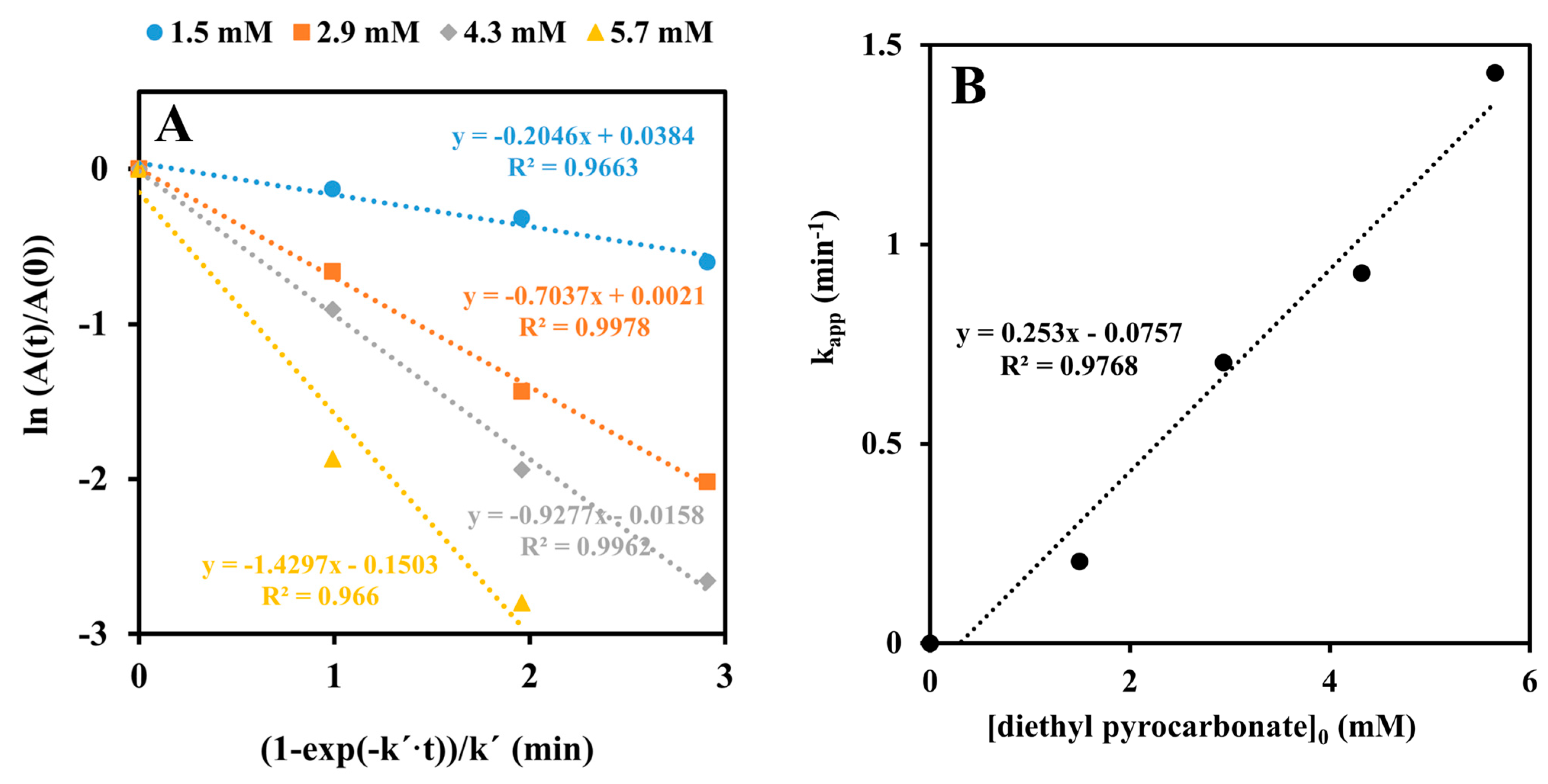
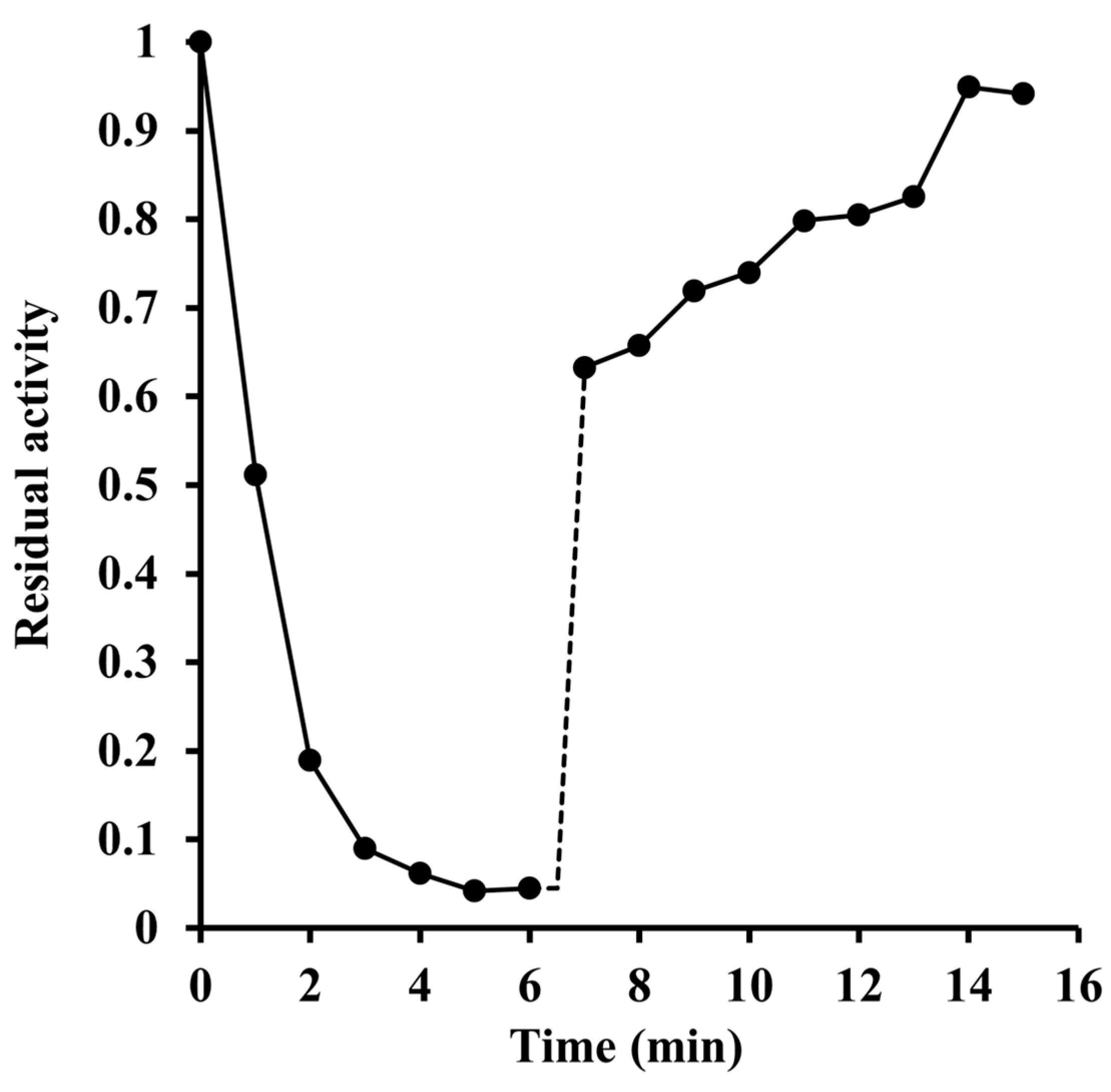
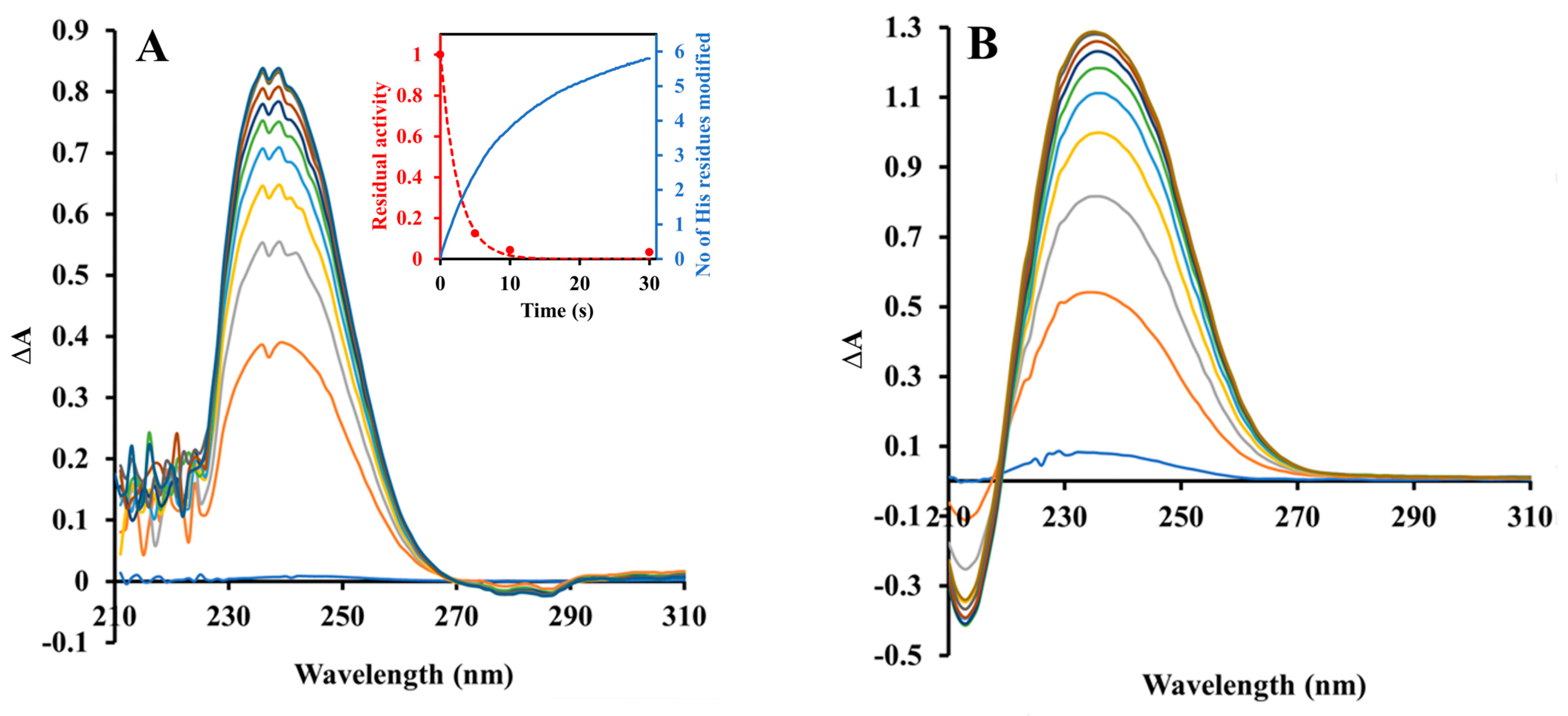
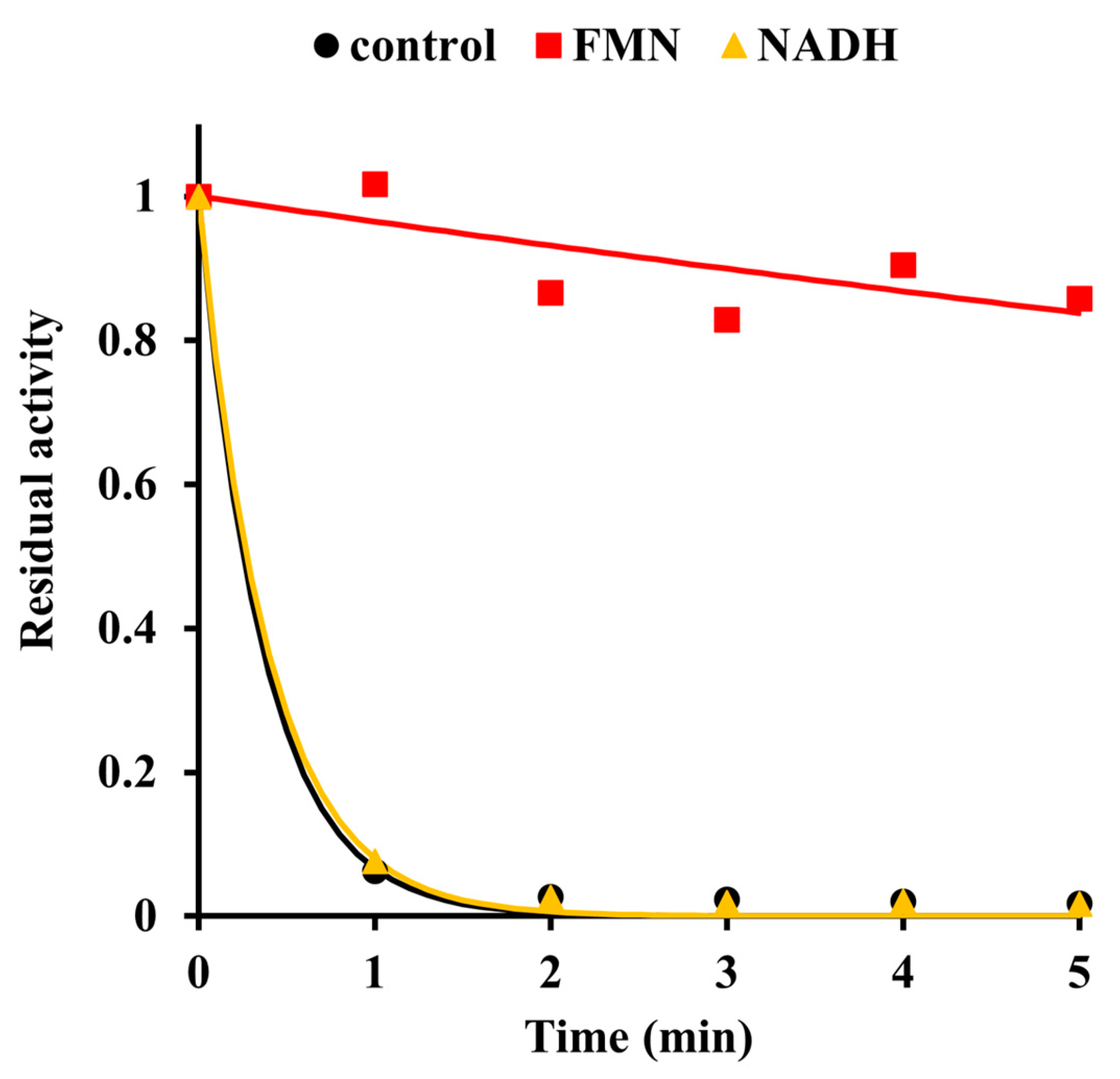
2.2. Inactivation by Amino Acid Modifying Reagents
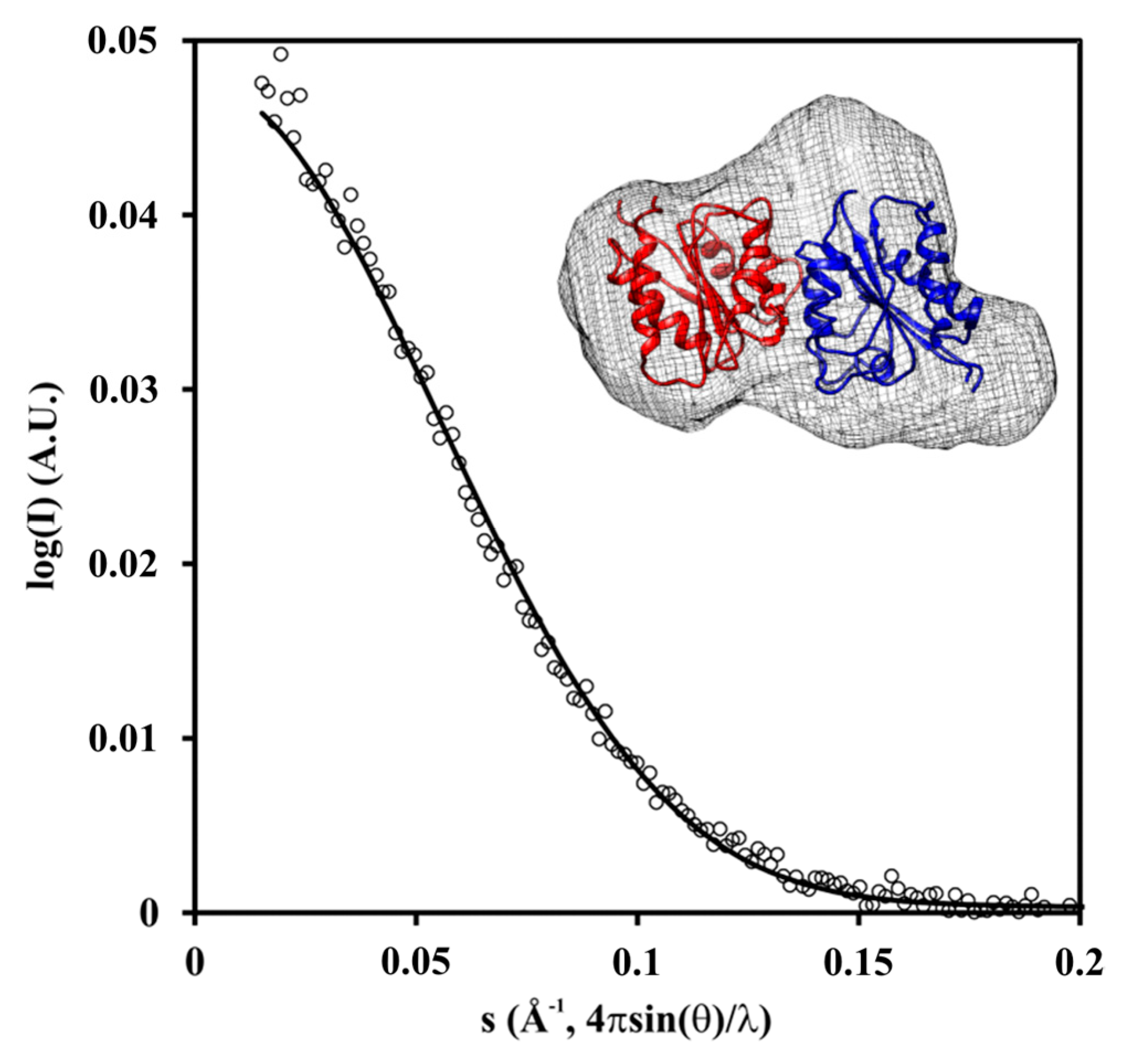
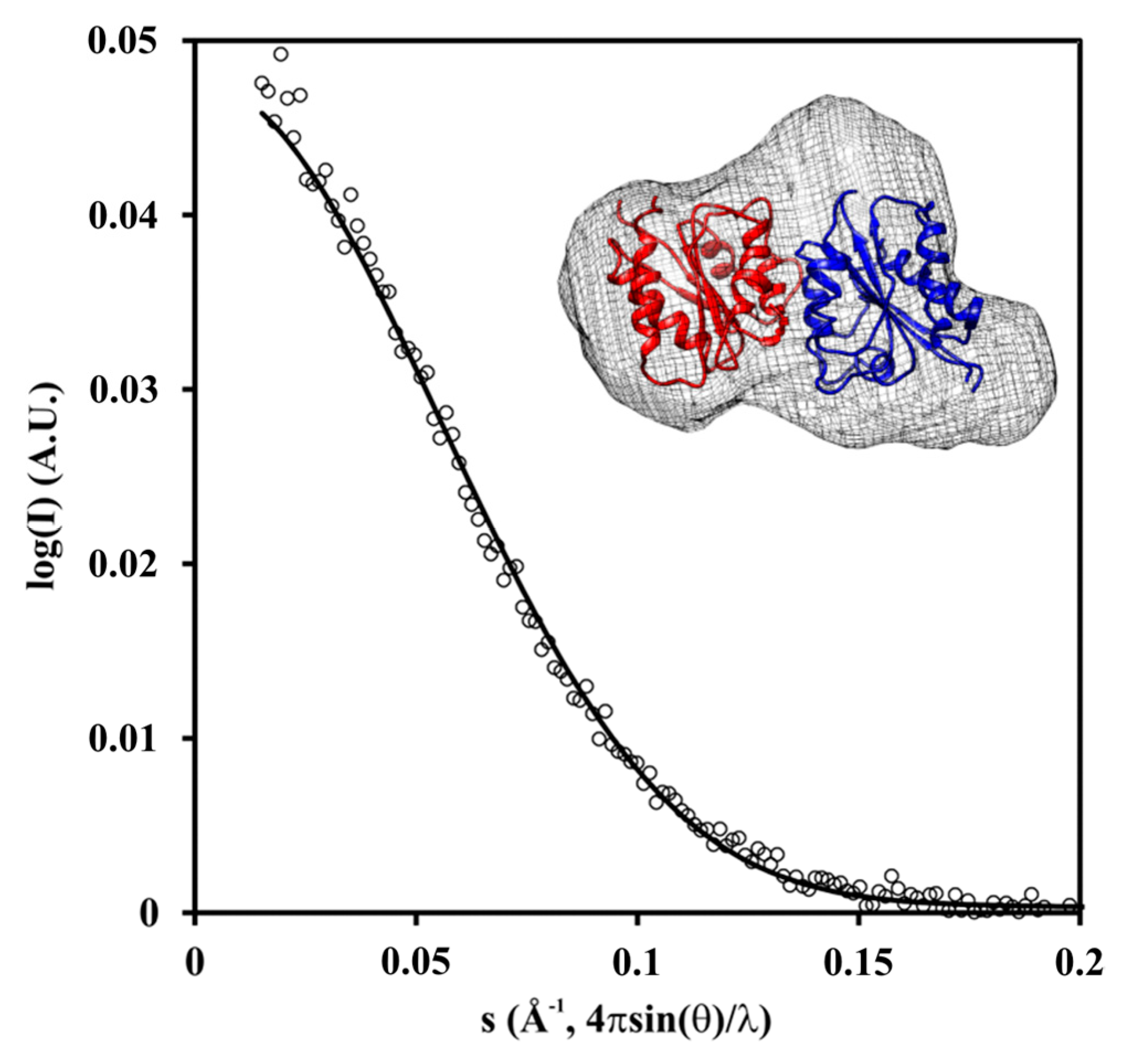
2.3. Crystal and Solution Structure
2.4. Search for Homology Proteins
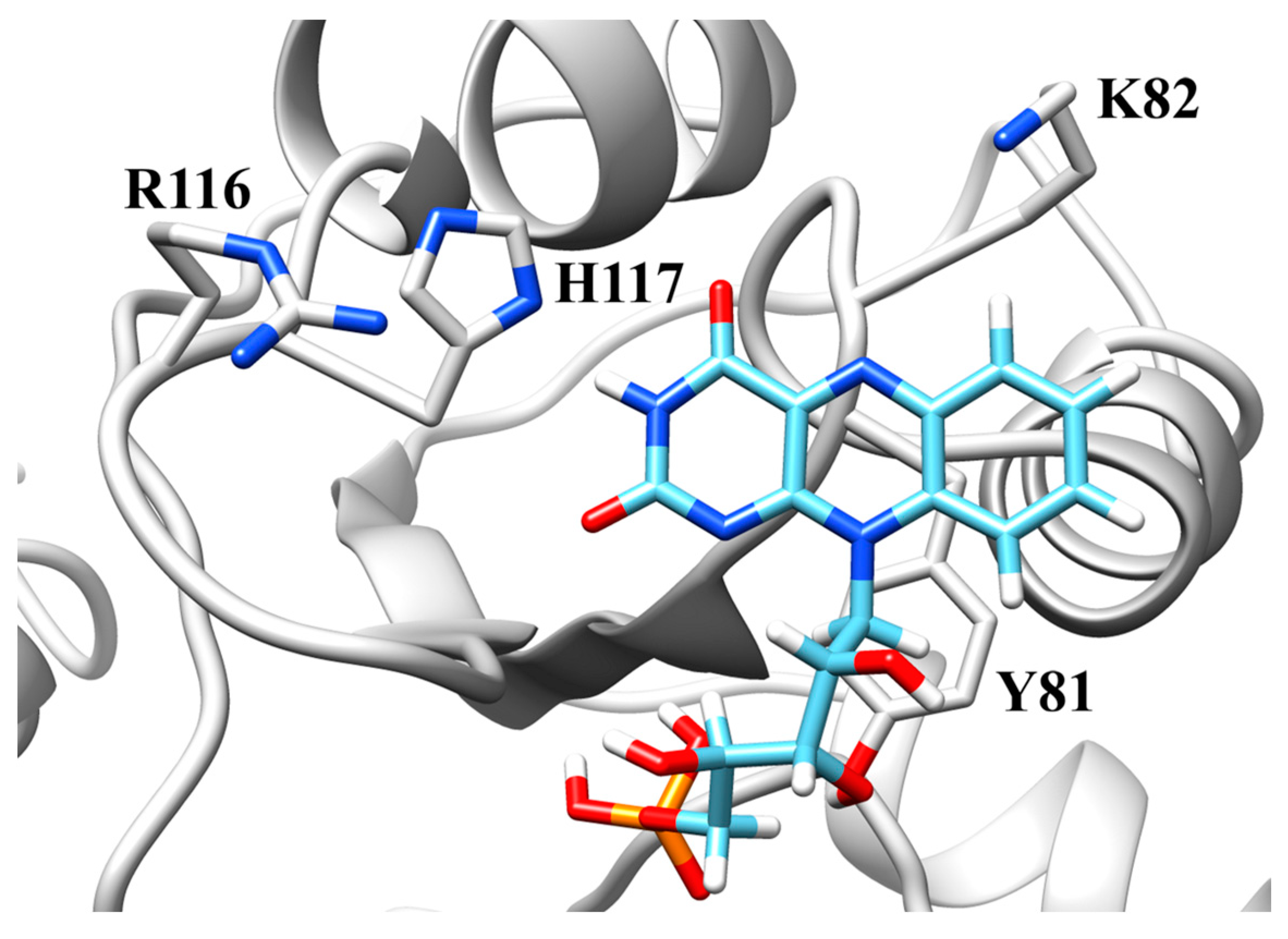
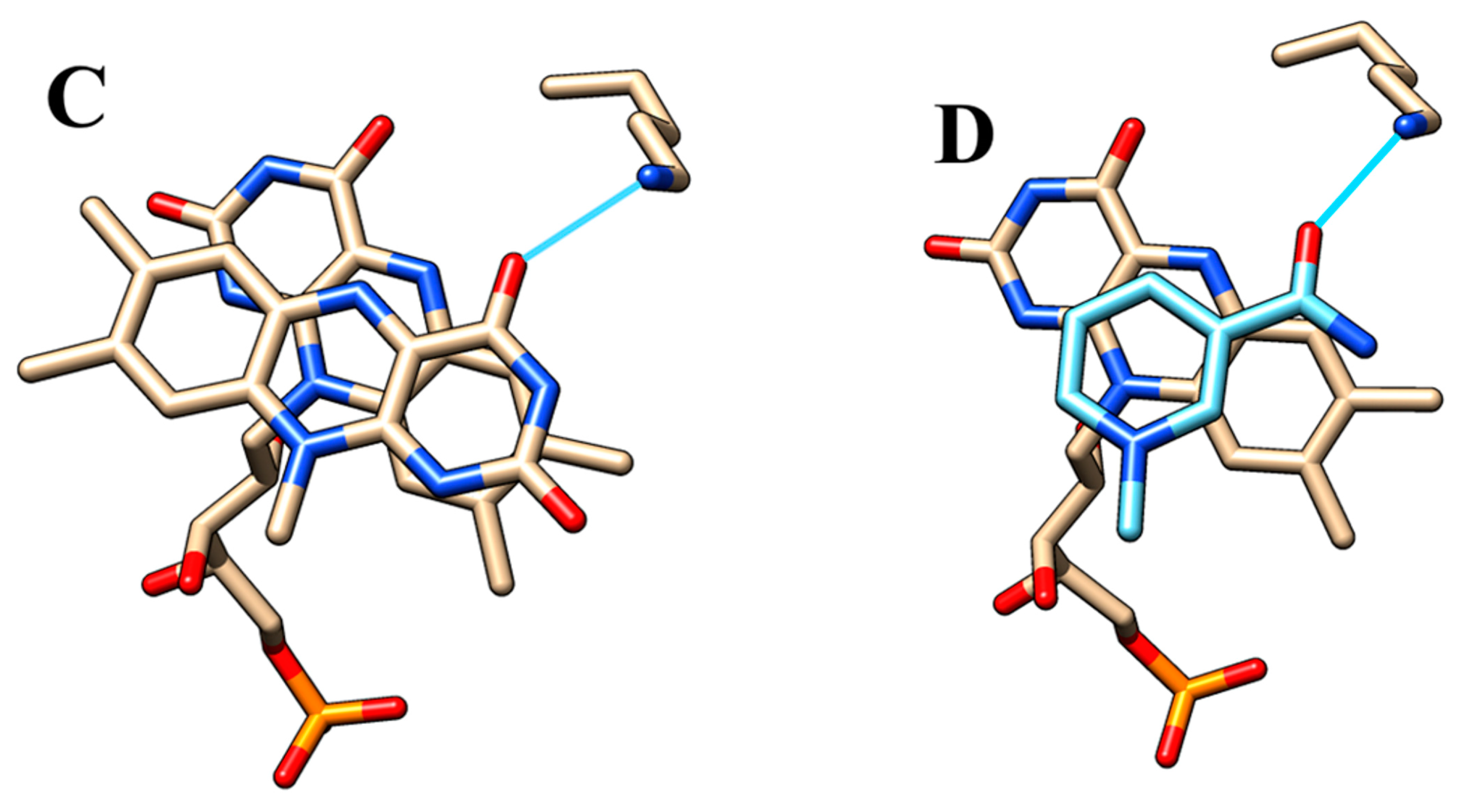
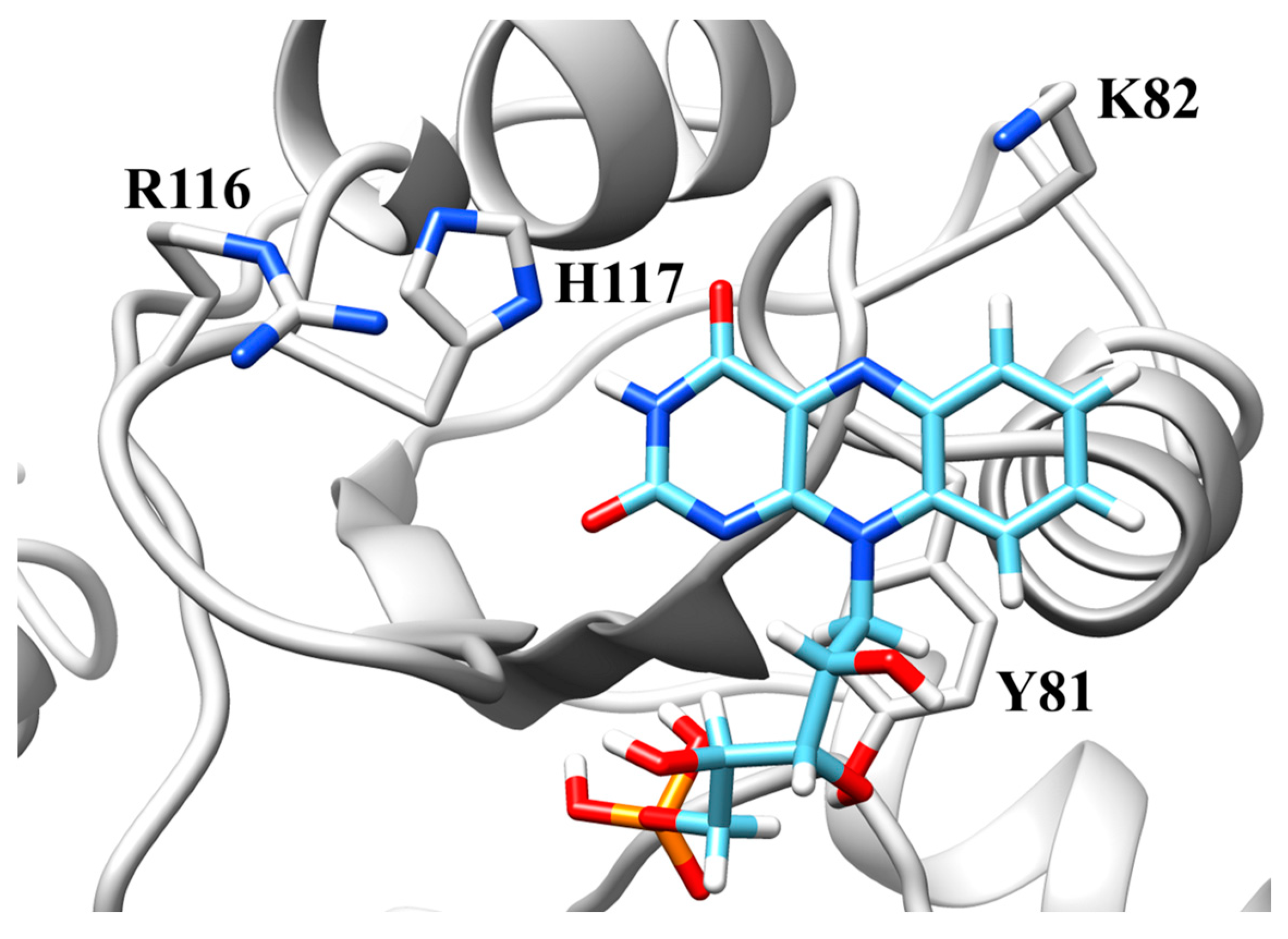
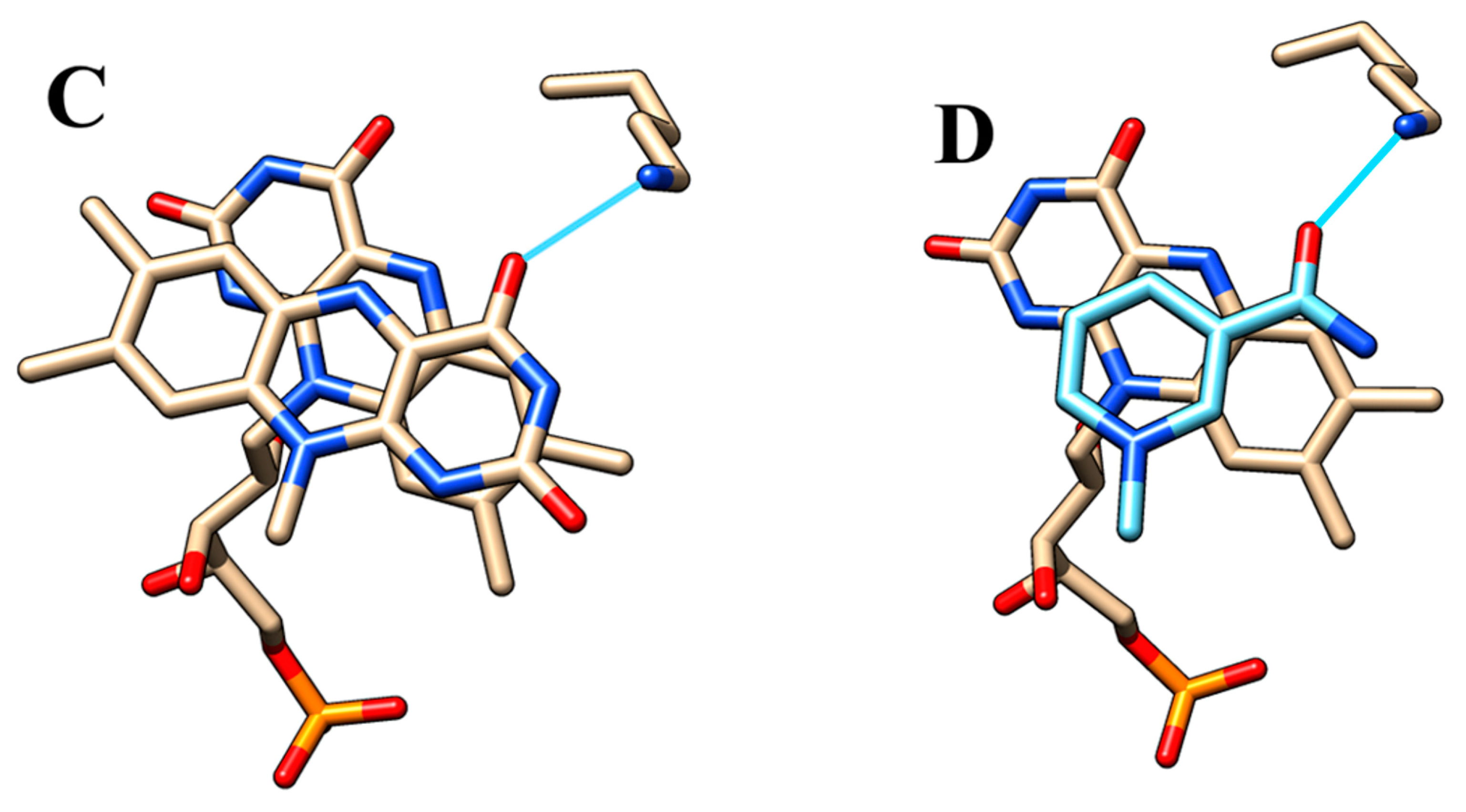
2.5. Active Site Model
2.6. Site-Directed Mutagenesis and Mutant Characterization
2.7. Kinetic Isotope Effect in Hydride Transfer from NADH
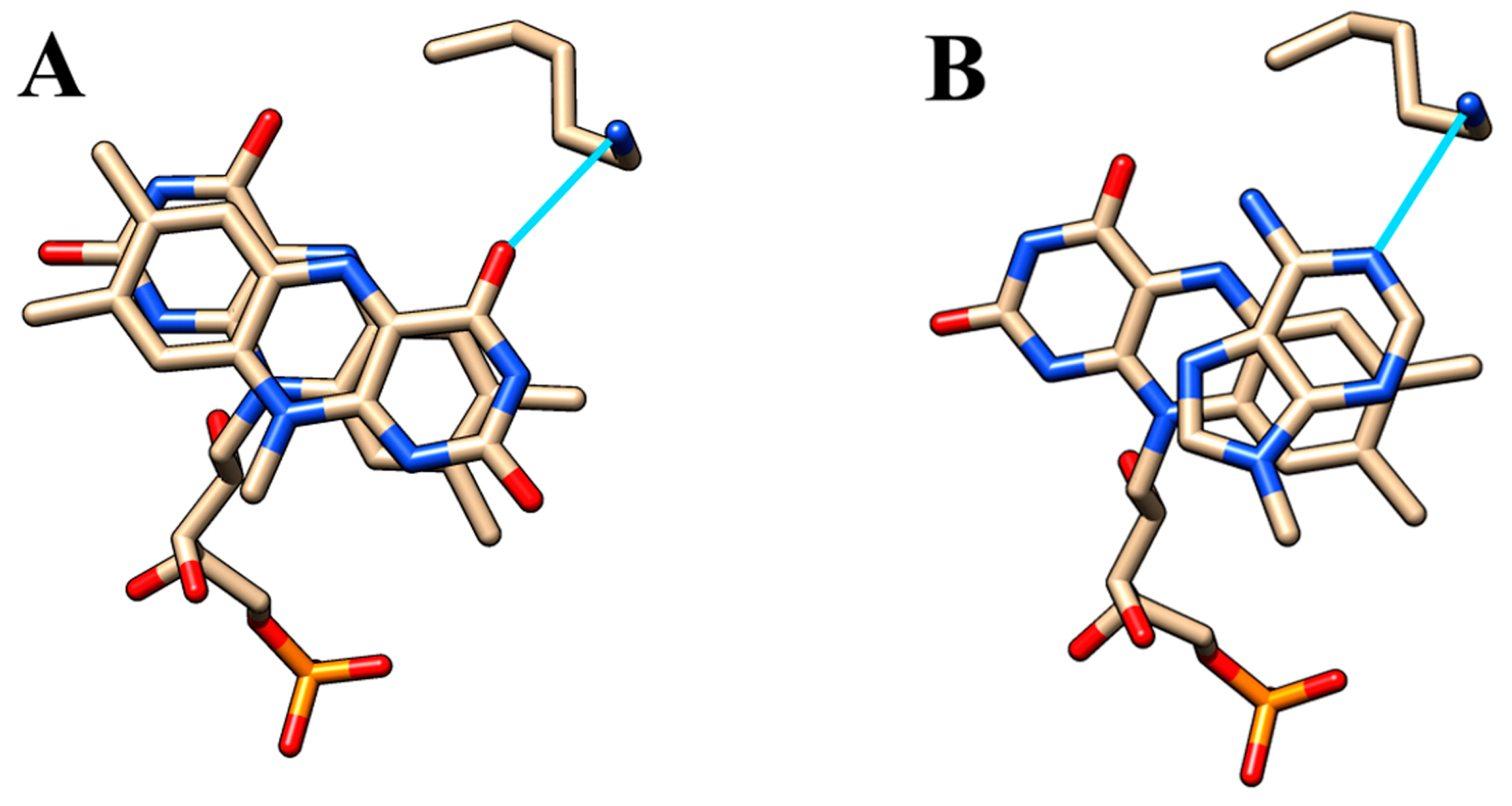
2.8. Effect of Arg-to-Lys Substitution at Position 116
3. Discussion
3.1. Histidine-117
3.2. Lysine-82
3.3. Arginine-116
4. Materials and Methods
4.1. Recombinant Native and Mutant Protein Production and Purification
4.2. Determination of Protein Concentration
4.3. Enzyme Activity Assay
4.4. Chemical Modification
4.5. Rapid Kinetics Measurements
4.6. Stereospecificity Determination
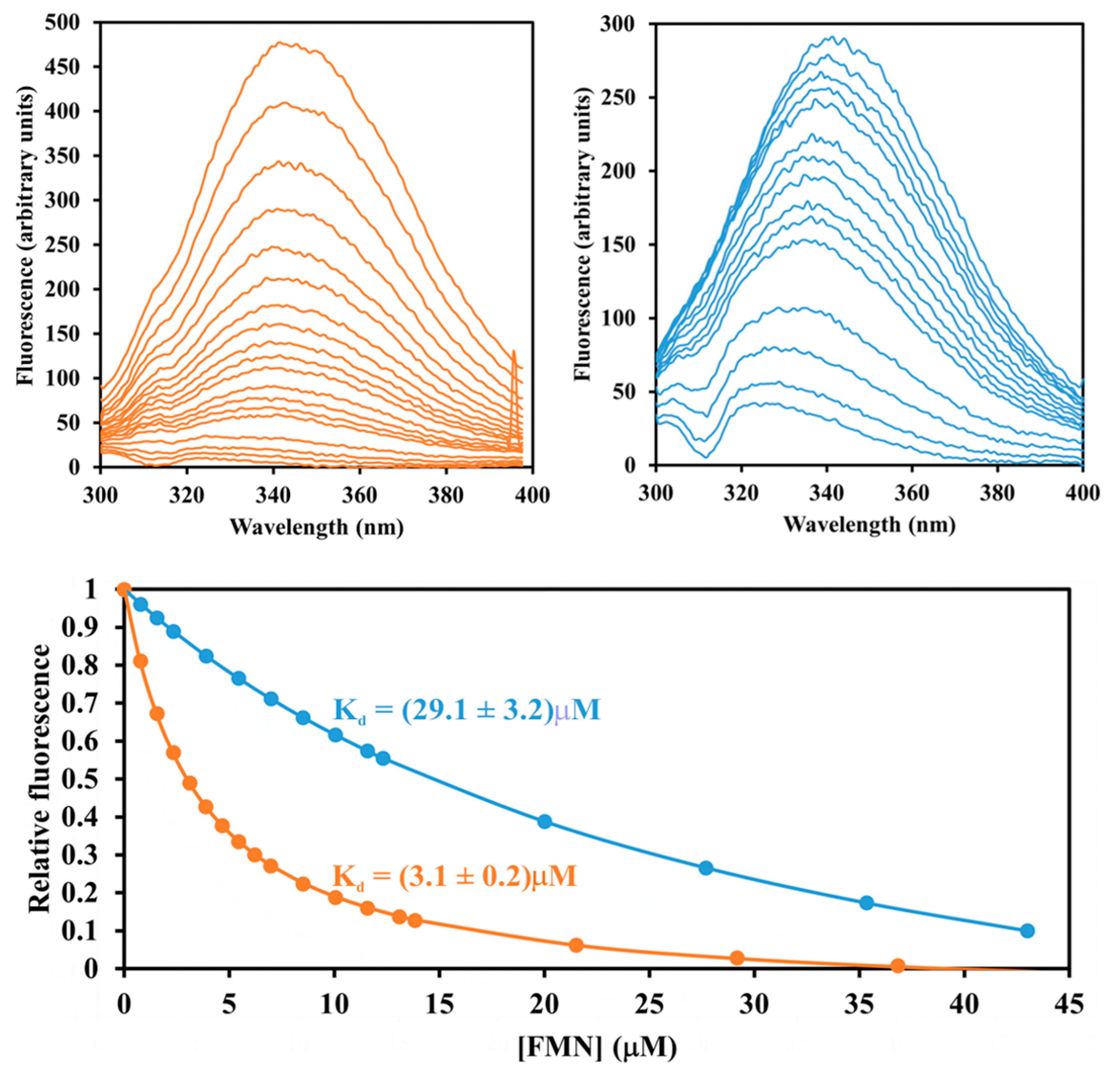
4.7. Dissociation Constants
4.8. Crystallization, X-ray Data Collection, and Structure Refinement
4.9. Small-Angle X-ray Scattering (SAXS)
4.10. In Silico Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Mazoch, J.; Tesarik, R.; Sedlacek, V.; Kucera, I.; Turanek, J. Isolation and biochemical characterization of two soluble iron(III) reductases from Paracoccus denitrificans. Eur. J. Biochem. 2004, 271, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, V.; van Spanning, R.J.M.; Kucera, I. Characterization of the quinone reductase activity of the ferric reductase B protein from Paracoccus denitrificans. Arch. Biochem. Biophys. 2009, 483, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, V.; Ptackova, N.; Rejmontova, P.; Kucera, I. The flavoprotein FerB of Paracoccus denitrificans binds to membranes, reduces ubiquinone and superoxide, and acts as an in vivo antioxidant. FEBS J. 2015, 282, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, V.; Kucera, I. Arginine-95 is important for recruiting superoxide to the active site of the FerB flavoenzyme of Paracoccus denitrificans. FEBS Lett. 2019, 593, 697–702. [Google Scholar] [CrossRef]
- Sedlacek, V.; Klumpler, T.; Marek, J.; Kucera, I. The structural and functional basis of catalysis mediated by NAD(P)H:acceptor oxidoreductase (FerB) of Paracoccus denitrificans. PLoS ONE 2014, 9, e96262. [Google Scholar] [CrossRef]
- Pernikarova, V.; Sedlacek, V.; Potesil, D.; Prochazkova, I.; Zdrahal, Z.; Bouchal, P.; Kucera, I. Proteomic responses to a methyl viologen-induced oxidative stress in the wild type and FerB mutant strains of Paracoccus denitrificans. J. Proteom. 2015, 125, 68–75. [Google Scholar] [CrossRef]
- Sedlacek, V.; Kucera, I. Functional and mechanistic characterization of an atypical flavin reductase encoded by the pden_5119 gene in Paracoccus denitrificans. Mol. Microbiol. 2019, 112, 166–183. [Google Scholar] [CrossRef]
- Sedlacek, V.; Kryl, M.; Kucera, I. The ArsH protein product of the Paracoccus denitrificans ars operon has an activity of organoarsenic reductase and is regulated by a redox-responsive repressor. Antioxidants 2022, 11, 902. [Google Scholar] [CrossRef]
- Edgcomb, S.P.; Murphy, K.P. Variability in the pKa of histidine side-chains correlates with burial within proteins. Proteins 2002, 49, 1–6. [Google Scholar] [CrossRef]
- Miles, E.W. Modification of histidyl residues in proteins by diethylpyrocarbonate. Methods Enzymol. 1977, 47, 431–442. [Google Scholar] [PubMed]
- Chen, S.S.; Engel, P.C. Equilibrium position of reaction of bovine liver glutamate dehydrogenase with pyridoxal 5′-phosphate. Demonstration that covalent modification with this reagent completely abolishes catalytic activity. Biochem. J. 1975, 147, 351–358. [Google Scholar] [CrossRef]
- Simpson, R.T.; Riordan, J.F.; Vallee, B.L. Functional tyrosyl residues in active center of bovine pancreatic carboxypeptidase A. Biochemistry 1963, 2, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K. Reaction of phenylglyoxal with arginine residues in proteins. J. Biol. Chem. 1968, 243, 6171–6179. [Google Scholar] [CrossRef]
- Nissen, M.S.; Youn, B.Y.; Knowles, B.D.; Ballinger, J.W.; Jun, S.Y.; Belchik, S.M.; Xun, L.Y.; Kang, C.H. Crystal structures of NADH:FMN oxidoreductase (EmoB) at different stages of catalysis. J. Biol. Chem. 2008, 283, 28710–28720. [Google Scholar] [CrossRef]
- Driggers, C.M.; Dayal, P.V.; Ellis, H.R.; Karplus, P.A. Crystal structure of Escherichia coli SsuE: Defining a general catalytic cycle for FMN reductases of the flavodoxin-like superfamily. Biochemistry 2014, 53, 3509–3519. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.R. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys. 2010, 497, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.M.; Ellis, H.R. Investigations of two-component flavin-dependent monooxygenase systems. Methods Enzymol. 2019, 620, 399–422. [Google Scholar]
- Li, G.F.; Glusac, K.D. Light-triggered proton and electron transfer in flavin cofactors. J. Phys. Chem. A 2008, 112, 4573–4583. [Google Scholar] [CrossRef]
- Rippa, M.; Spanio, L.; Pontremo, S. A specific interaction of pyridoxal 5′-phosphate and 6-phosphogluconic dehydrogenase. Arch. Biochem. Biophys. 1967, 118, 48–57. [Google Scholar] [CrossRef]
- Gould, K.G.; Engel, P.C. Modification of mouse testicular lactate dehydrogenase by pyridoxal 5′-phosphate. Biochem. J. 1980, 191, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Lilley, K.S.; Engel, P.C. The essential active-site lysines of clostridial glutamate dehydrogenase. A study with pyridoxal-5′-phosphate. Eur. J. Biochem. 1992, 207, 533–540. [Google Scholar] [CrossRef]
- Olano, J.; Soler, J.; Busto, F.; de Arriaga, D. Chemical modification of NADP-isocitrate dehydrogenase from Cephalosporium acremonium. Evidence of essential histidine and lysine groups at the active site. Eur. J. Biochem. 1999, 261, 640–649. [Google Scholar] [CrossRef]
- Vazquez, M.A.; Munoz, F.; Donoso, J.; Garcia Blanco, F. Stability of Schiff bases of amino acids and pyridoxal-5′-phosphate. Amino Acids 1992, 3, 81–94. [Google Scholar] [CrossRef]
- Gillet, N.; Levy, B.; Moliner, V.; Demachy, I.; de la Lande, A. Electron and hydrogen atom transfers in the hydride carrier protein EmoB. J. Chem. Theory Comput. 2014, 10, 5036–5046. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Gomez Castellanos, J.R.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same substrate, many reactions: Oxygen activation in flavoenzymes. Chem. Rev. 2018, 118, 1742–1769. [Google Scholar] [CrossRef]
- Pandeswari, P.B.; Sabareesh, V.; Vijayalakshmi, M.A. Insights into stoichiometry of arginine modification by phenylglyoxal and 1,2-cyclohexanedione probed by LC-ESI-MS. J. Prot. Proteom. 2016, 7, 323–347. [Google Scholar]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; p. 263. [Google Scholar]
- Hu, W.; Xie, J.; Chau, H.W. Evaluation of parameter uncertainties in nonlinear regression using Microsoft Excel Spreadsheet. Environ. Syst. Res. 2015, 4, 4. [Google Scholar] [CrossRef]
- Melchior, W.B.; Fahrney, D. Ethoxyformylation of proteins. Reaction of ethoxyformic anhydride with alpha-chymotrypsin, pepsin, and pancreatic ribonuclease at pH 4. Biochemistry 1970, 9, 251–258. [Google Scholar] [CrossRef]
- Kitz, R.; Wilson, I.B. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [Google Scholar] [CrossRef] [PubMed]
- Gomi, T.; Fujioka, M. Evidence for an essential histidine residue in S-adenosylhomocysteinase from rat liver. Biochemistry 1983, 22, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lostao, A.; El Harrous, M.; Daoudi, F.; Romero, A.; Parody-Morreale, A.; Sancho, J. Dissecting the energetics of the apoflavodoxin-FMN complex. J. Biol. Chem. 2000, 275, 9518–9526. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Kovalevskiy, O.; Nicholls, R.A.; Murshudov, G.N. Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr. D Struct. Biol. 2016, 72, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Salzemann, J.; Bloch, V.; Stockinger, H.; Berglund, A.C.; Blanchet, C.; Bongcam-Rudloff, E.; Combet, C.; Da Costa, A.L.; Deleage, G.; et al. PDB_REDO: Automated re-refinement of X-ray structure models in the PDB. J. Appl. Crystallogr. 2009, 42, 376–384. [Google Scholar] [CrossRef]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL–a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Liu, N.; Xu, Z. Using LeDock as a docking tool for computational drug design. IOP Conf. Ser. Earth Environ. Sci. 2018, 218, 012143. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Expect Value | Identities | Z-Score | rmsd |
|---|---|---|---|---|
| 4C76 | 2 × 10−42 | 76/165 (46%) | 23.1 | 1.5 |
| 3K1Y | 6 × 10−32 | 62/171(36%) | 21.9 | 2.1 |
| 4LTD | 6 × 10−26 | 63/172 (37%) | 21.3 | 2.1 |
| 4PTY | 8 × 10−21 | 58/179 (32%) | 21.2 | 1.9 |
| KdFMN (µM) | KNADH (µM) | kred (s−1) | KO2 (µM) | kox (s−1) | kcat (s−1) | |
|---|---|---|---|---|---|---|
| Wild-type | 3.8 ± 0.4 | 19 ± 1 | 41 ± 1 | 30 ± 3 | 24 ± 1 | 21 ± 1 |
| K82A | 6.8 ± 3.4 | 53 ± 18 | 0.78 ± 0.05 | 18 ± 5 | 5.8 ± 0.4 | 0.62 ± 0.05 |
| R116A | 3.5 ± 1.5 | 39 ± 17 | 6.5 ± 0.5 | 75 ± 2 | 0.91 ± 0.01 | 0.79 ± 0.05 |
| H117A | 9.1 ± 0.6 | 40 ± 7 | 2.5 ± 0.1 | 70 ± 16 | 0.79 ± 0.05 | 0.74 ± 0.06 |
| R116K | 0.6 ± 0.1 | 20 ± 4 | 41 ± 2 | 34 ± 2 | 26 ± 6 | 17 ± 2 |
| Wild-Type (µmol·s−1·mg−1) | K82A (µmol·s−1·mg−1) | R116A (µmol·s−1·mg−1) | H117A (µmol·s−1·mg−1) | |
|---|---|---|---|---|
| NADH | 1.039 ± 0.051 | 0.0303 ± 0.0024 | 0.0396 ± 0.0024 | 0.036 ± 0.003 |
| R-NADD | 1.012 ± 0.036 | 0.0309 ± 0.028 | 0.0372 ± 0.0035 | 0.034 ± 0.007 |
| S-NADD | 0.422 ± 0.010 | 0.0296 ± 0.0005 | 0.0144 ± 0.0010 | 0.009 ± 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryl, M.; Sedláček, V.; Kučera, I. Structural Insight into Catalysis by the Flavin-Dependent NADH Oxidase (Pden_5119) of Paracoccus denitrificans. Int. J. Mol. Sci. 2023, 24, 3732. https://doi.org/10.3390/ijms24043732
Kryl M, Sedláček V, Kučera I. Structural Insight into Catalysis by the Flavin-Dependent NADH Oxidase (Pden_5119) of Paracoccus denitrificans. International Journal of Molecular Sciences. 2023; 24(4):3732. https://doi.org/10.3390/ijms24043732
Chicago/Turabian StyleKryl, Martin, Vojtěch Sedláček, and Igor Kučera. 2023. "Structural Insight into Catalysis by the Flavin-Dependent NADH Oxidase (Pden_5119) of Paracoccus denitrificans" International Journal of Molecular Sciences 24, no. 4: 3732. https://doi.org/10.3390/ijms24043732
APA StyleKryl, M., Sedláček, V., & Kučera, I. (2023). Structural Insight into Catalysis by the Flavin-Dependent NADH Oxidase (Pden_5119) of Paracoccus denitrificans. International Journal of Molecular Sciences, 24(4), 3732. https://doi.org/10.3390/ijms24043732