

Role of Dopamine in the Heart in Health and Disease


Abstract
1. Introduction
2. Dopamine Synthesis
3. Dopamine Levels and Metabolism in the Heart
4. Dopamine and D1-Dopamine Receptors, Especially in the Animal Heart: Signal Transduction and Regulation
4.1. General Dopamine Receptor Classification
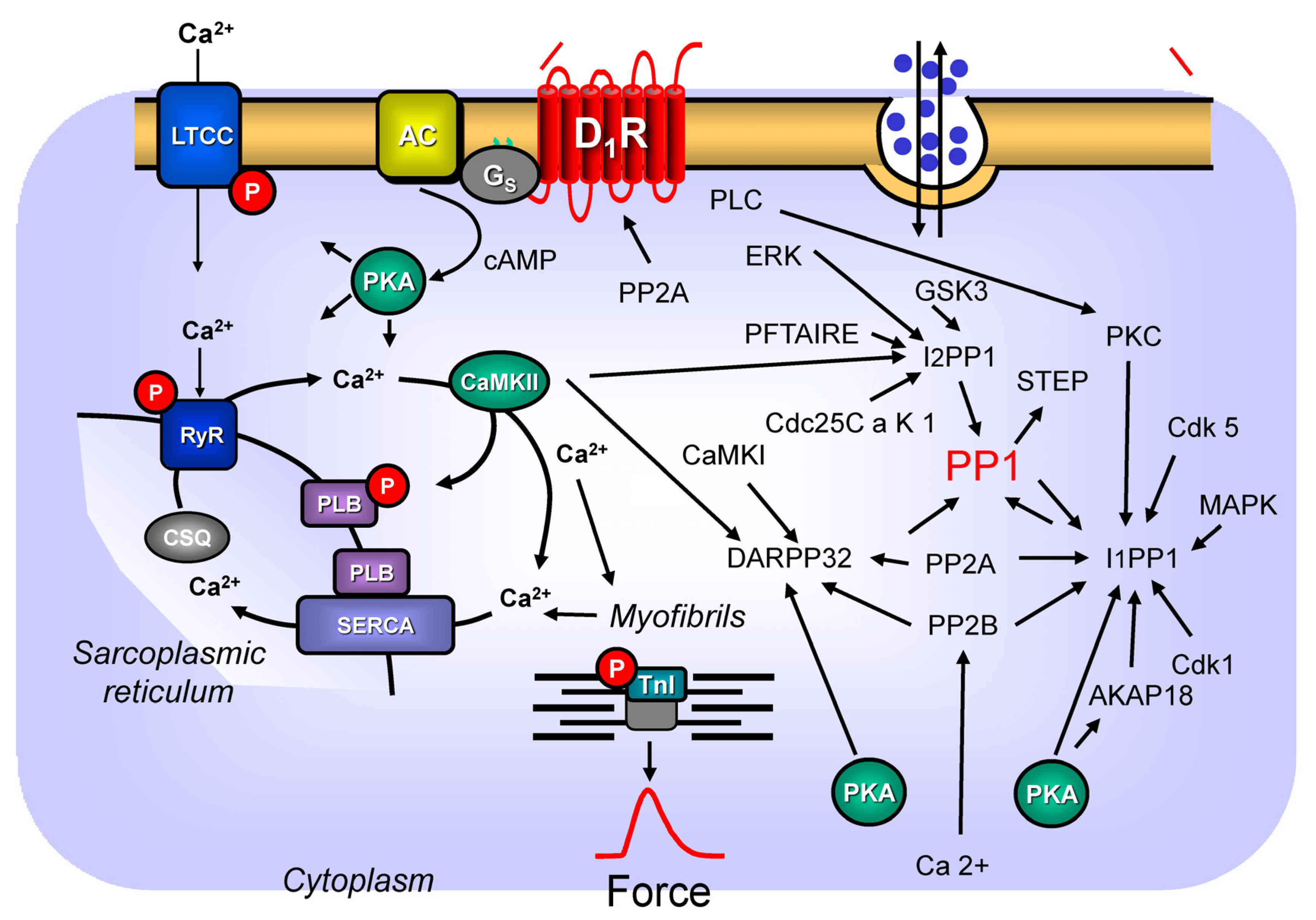
4.2. Signal Transduction of Dopamine Receptors via PP2A
4.3. Dopamine Receptors and DARPP32
4.4. Dopamine Receptors and Inhibitor-1 of PP1
4.5. Dopamine Receptors and Inhibitor-2 of PP1
4.6. Dopamine Receptors and Inositol Trisphosphate (IP3) Levels
4.7. Upregulation, Downregulation, and Desensitization
4.8. Dimerization of Dopamine Receptors
5. Inotropic and Chronotropic Effects of Dopamine in Animal Hearts (Table 2)
5.1. Right Atrium
5.2. Left Atrium
5.3. Perfused Heart
5.4. Papillary Muscles
5.5. Ventricular Strips
5.6. Anaesthetized Animals
5.7. Cardiomyocytes
5.8. Electrophysiological Studies
5.9. Age Dependence of Dopamine-Induced Effects in the Heart
6. Effects of Dopamine on Isolated Vessels
7. The Human Heart
7.1. Dopamine Receptors and Their Action in Human Hearts
7.2. L-DOPA in Human Hearts
8. Dopamine in Disease
8.1. Hypertension
8.1.1. The D1-Dopamine Receptor
8.1.2. The D2-Dopamine-Receptor
8.1.3. The D3-Dopamine Receptor
8.1.4. The D4-Dopamine-Receptor
8.1.5. The D5-Dopamine Receptor
8.1.6. Drug-Induced Hypertension and Dopamine Receptors
8.1.7. Genetically Induced Hypertension
8.2. Ischemia
8.3. Genetically Induced Heart Failure
8.4. Sepsis
9. Clinical Relevance
10. Outlook
11. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Myslivecek, J. Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors. Life 2022, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Wiegerinck, R.F.; Shen, M.; Cojoc, A.; Zeidenweber, C.M.; Wagner, M.B. Dopamine increases L-type calcium current more in newborn than adult rabbit cardiomyocytes via D1 and β2 receptors. Am. J. Physiol. Circ. Physiol. 2008, 294, H2327–H2335. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, I.; Harmon, S.K.; Todd, R.D.; O’Malley, K.L. The Rat D4 Dopamine Receptor Couples to Cone Transducin (Gαt2) to Inhibit Forskolin-stimulated cAMP Accumulation. J. Biol. Chem. 1997, 272, 16599–16602. [Google Scholar] [CrossRef]
- Piercey, M.F.; Hoffmann, W.E.; Smith, M.W.; Hyslop, D.K. Inhibition of dopamine neuron firing by pramipexole, a dopamine D3 receptor-preferring agonist: Comparison to other dopamine receptor agonists. Eur. J. Pharmacol. 1996, 312, 35–44. [Google Scholar] [CrossRef]
- DeNinno, M.P.; Schoenleber, R.; Asin, K.E.; MacKenzie, R.; Kebabian, J.W. (1R,3S)-1-(Aminomethyl)-3,4-dihydro-5,6-dihydroxy-3-phenyl-1H-2-benzopyran: A potent and selective D1 agonist. J. Med. Chem. 1990, 33, 2948–2950. [Google Scholar] [CrossRef]
- Neumeyer, J.L.; Baindur, N.; Niznik, H.B.; Guan, H.C.; Seeman, P. (+/−)-3-Allyl-6-bromo-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine, a new high affinity D1 dopamine receptor ligand: Synthesis and structure-activity relationship. J. Med. Chem. 1991, 34, 3366–3371. [Google Scholar] [CrossRef]
- Salmi, P.; Isacson, R.; Kull, B. Dihydrexidine—The First Full Dopamine D1 Receptor Agonist. CNS Drug Rev. 2006, 10, 230–242. [Google Scholar] [CrossRef]
- Zhao, R.-R.; Fennell, W.H.; Abel, F.L. Effects of doparaine D1 and dopamine D2 receptor agonists on coronary and peripheral tiemodynamics. Eur. J. Pharmacol. 1990, 190, 193–202. [Google Scholar] [CrossRef]
- Rashid, A.J.; So, C.H.; Kong, M.M.C.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O’Dowd, B.F.; George, S.R. D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of G q /11 in the striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Kvernmo, T.; Houben, J.; Sylte, I. Receptor-binding and pharmacokinetic properties of dopaminergic agonists. Curr. Top. Med. Chem. 2008, 8, 1049–1067. [Google Scholar] [CrossRef]
- Kopia, G.A.; Valocik, R.E. Demonstration of specific dopamine-1 receptor-mediated coronary vasodilation in the anesthetized dog. J. Pharmacol. Exp. Ther. 1989, 248, 215–221. [Google Scholar]
- Andersen, P.H.; Grønvald, F.C.; Hohlweg, R.; Hansen, L.B.; Guddal, E.; Braestrup, C.; Nielsen, E.B. NNC-112, NNC-687 and NNC-756, new selective and highly potent dopamine D1 receptor antagonists. Eur. J. Pharmacol. 1992, 219, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lumley, P.; Broadley, K.J.; Levy, G.P. Analysis of the inotropic: Chronotropic selectivity of dobutamine and dopamine in anaesthetized dogs and guinea-pig isolated atria. Cardiovasc Res. 1977, 11, 17–25. [Google Scholar] [CrossRef]
- Vettel, C.; Hottenrott, M.C.; Spindler, R.; Benck, U.; Schnuelle, P.; Tsagogiorgas, C.; Krämer, B.K.; Hoeger, S.; El-Armouche, A.; Wieland, T.; et al. Dopamine and Lipophilic Derivates Protect Cardiomyocytes against Cold Preservation Injury. Experiment 2013, 348, 77–78. [Google Scholar] [CrossRef]
- Lencesova, L.; Szadvari, I.; Babula, P.; Kubickova, J.; Chovancova, B.; Lopusna, K.; Rezuchova, I.; Novakova, Z.; Krizanova, O. Disruption of dopamine D1/D2 receptor complex is involved in the function of haloperidol in cardiac H9c2 cells. Life Sci. 2017, 191, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.-E. Physiology and pharmacology of cardiovascular catecholamine receptors: Implications for treatment of chronic heart failure. Am. Heart J. 1990, 120, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E. Dopamine, cardiovascular dopamine receptors and chronic heart failure. Cardiologia 1995, 40 (Suppl. S1), 125–129. [Google Scholar]
- Holtz, P.; Credner, K.; Koepp, W. Die enzymatische Entstehung von Oxytyramin im Organismus und die physiologische Bedeutung der Dopadecarboxylase. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1942, 200, 356–388. [Google Scholar] [CrossRef]
- Bucolo, C.; Leggio, G.M.; Drago, F.; Salomone, S. Dopamine outside the brain: The eye, cardiovascular system and endocrine pancreas. Pharmacol. Ther. 2019, 203, 107392. [Google Scholar] [CrossRef]
- Sonne, J.; Lopez-Ojeda, W. Dopamine; StatPearls Publishing: Treasure Island, FL, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535451/ (accessed on 10 December 2022).
- Goldstein, D.S.; Holmes, C. Neuronal source of plasma dopamine. Clin. Chem. 2008, 54, 1864–1871. [Google Scholar] [CrossRef]
- Czibik, G.; Mezdari, Z.; Altintas, D.M.; Bréhat, J.; Pini, M.; D’Humières, T.; Delmont, T.; Radu, C.; Breau, M.; Liang, H.; et al. Dysregulated Phenylalanine Catabolism Plays a Key Role in the Trajectory of Cardiac Aging. Circulation 2021, 144, 559–574. [Google Scholar] [CrossRef]
- Niwa, T.; Sugimoto, S. Inhibitory and Stimulatory Effects of Selective Serotonin Reuptake Inhibitors on Cytochrome P450 2D6-mediated Dopamine Formation from p-Tyramine. J. Pharm. Pharm. Sci. 2019, 22, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Borlak, J. Gene expression in distinct regions of the heart. Lancet 2000, 355, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Cicali, E.J.; Smith, D.M.; Duong, B.Q.; Kovar, L.; Cavallari, L.H.; Johnson, J.A. A Scoping Review of the Evidence Behind Cytochrome P450 2D6 Isoenzyme Inhibitor Classifications. Clin. Pharmacol. Ther. 2020, 108, 116–125. [Google Scholar] [CrossRef]
- Gavrilovic, L.; Spasojevic, N.; Zivkovic, M.; Dronjak, S. Effect of immobilization stress on gene expression of catecholamine biosynthetic enzymes in heart auricles of socially isolated rats. Braz. J. Med. Biol. Res. 2009, 42, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Tang, B.-P.; Gan, T.; Xu, G.; Zhou, X.; Li, H.; Guo, X.; Mahemuti, A.; Sun, Q.; et al. Expression of tyrosine hydroxylase and growth-associated protein 43 in aging atrial fibrillation patients of Xinjiang Uygur and Han nationality. Genet. Mol. Res. 2013, 12, 5257–5266. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, P.R.; Dickson, P.W. Tyrosine hydroxylase phosphorylation in vivo. J. Neurochem. 2019, 149, 706–728. [Google Scholar] [CrossRef]
- Cahill, A.L.; Perlman, R.L. Phosphorylation of tyrosine hydroxylase in the superior cervical ganglion. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 1984, 805, 217–226. [Google Scholar] [CrossRef]
- Endoh, M. Effects of dopamine on sinus rate and ventricular contractile force of the dog heart in vitro and in vivo. Br. J. Pharmacol. 1975, 55, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Lund, D.D.; Subieta, A.R.; Prall, J.L.; Schmid, P.G.; Roskoski, R., Jr. Regional distribution of tyrosine hydroxylase and dopamine beta-hydroxylase activities in guinea pig heart. J. Auton. Nerv. Syst. 1981, 4, 319–326. [Google Scholar] [CrossRef]
- Locke, T.M.; Fujita, H.; Hunker, A.; Johanson, S.S.; Darvas, M.; du Lac, S.; Zweifel, L.S.; Carlson, E.S. Purkinje Cell-Specific Knockout of Tyrosine Hydroxylase Impairs Cognitive Behaviors. Front. Cell. Neurosci. 2020, 14, 228. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, K.; Kawamoto, C.; Hara, S.; Mori-Kojima, M.; Ohye, T.; Sumi-Ichinose, C.; Saito, N.; Sasaoka, T.; Metzger, D.; Ichinose, H. Tyrosine hydroxylase conditional KO mice reveal peripheral tissue-dependent differences in dopamine biosynthetic pathways. J. Biol. Chem. 2021, 296, 100544. [Google Scholar] [CrossRef] [PubMed]
- Soares-Da-Silva, P.; Davidson, R. Effects of 6-hydroxydopamine on dopamine and noradrenaline content in dog blood vessels and heart. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1985, 329, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.D.; Lewis, M.J.; Williams, J. Proceedings: The effect of delta-1-tetrahydrocannabinol on the noradrenaline and dopamine content of the brain and heart of the rat. Br. J. Pharmacol. 1974, 52, 446P. [Google Scholar]
- Regitz, V.; Bossaller, C.; Strasser, R.; Schüler, S.; Hetzer, R.; Fleck, E. Myocardial catecholamine content after heart transplantation. Circulation 1990, 82, 620–623. [Google Scholar] [CrossRef]
- Elayan, H.; Kennedy, B.; Ziegler, M.G. Propranolol reduces rat dopamine-β-hydroxylase activity and catecholamine levels. Eur. J. Pharmacol. 1992, 212, 259–262. [Google Scholar] [CrossRef]
- Brodde, O.-E.; Vogelsang, M.; Broede, A.; Michel-Reher, M.; Beisenbusch-Schäfer, E.; Hakim, K.; Zerkowski, H.-R. Diminished Responsiveness of Gs-Coupled Receptors in Severely Failing Human Hearts: No Difference in Dilated Versus Ischemic Cardiomyopathy. J. Cardiovasc. Pharmacol. 1998, 31, 585–594. [Google Scholar] [CrossRef]
- Goodman, L.S.; Gilman, A. The Pharmacological Basis of Therapeutics, 13th ed.; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; McGraw-Hill: New York, NY, USA, 2018. [Google Scholar]
- Cubells, J.F.; Schroeder, J.P.; Barrie, E.S.; Manvich, D.F.; Sadee, W.; Berg, T.; Mercer, K.; Stowe, T.A.; Liles, L.C.; Squires, K.E.; et al. Human Bacterial Artificial Chromosome (BAC) Transgenesis Fully Rescues Noradrenergic Function in Dopamine β-Hydroxylase Knockout Mice. PLoS ONE 2016, 11, e0154864. [Google Scholar] [CrossRef]
- Fornai, F.; Chen, K.; Giorgi, F.S.; Gesi, M.; Alessandri, M.G.; Shih, J.C. Striatal dopamine metabolism in monoamine oxidase B-deficient mice: A brain dialysis study. J. Neurochem. 2002, 73, 2434–2440. [Google Scholar] [CrossRef]
- Cuevas, S.; Villar, V.A.; Jose, P.A.; Armando, I. Renal Dopamine Receptors, Oxidative Stress, and Hypertension. Int. J. Mol. Sci. 2013, 14, 17553–17572. [Google Scholar] [CrossRef] [PubMed]
- Palomar, A.R.; Larios, B.N.; De Sánchez, V.C.; Pérez, L.M.; López, F.D.L.C.; Flores, G.; Gómez-Villalobos, M.D.J. Expression and Distribution of Dopamine Transporter in Cardiac Tissues of the Guinea Pig. Neurochem. Res. 2010, 36, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Asghar, M.; Tayebati, S.K.; Lokhandwala, M.F.; Hussain, T. Potential Dopamine-1 Receptor Stimulation in Hypertension Management. Curr. Hypertens. Rep. 2011, 13, 294–302. [Google Scholar] [CrossRef]
- Ozono, R.; O’Connell, D.P.; Vaughan, C.; Botkin, S.J.; Walk, S.F.; Felder, R.A.; Carey, R.M. Expression of the Subtype 1A Dopamine Receptor in the Rat Heart. Hypertension 1996, 27, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Osnes, J.-B.; Øye, I. Adenosine 3‘, 5’-cyclic Monophosphate in Perfused Rat Hearts Exposed to Isoprenaline and Dopamine. Acta Physiol. Scand. 1976, 96, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Functional Selectivity of Dopamine D1 Receptor Signaling: Retrospect and Prospect. Int. J. Mol. Sci. 2021, 22, 11914. [Google Scholar] [CrossRef]
- Deighton, N.M.; Motomura, S.; Bals, S.; Zerkowski, H.R.; E Brodde, O. Characterization of the beta adrenoceptor subtype(s) mediating the positive inotropic effects of epinine, dopamine, dobutamine, denopamine and xamoterol in isolated human right atrium. Experiment 1992, 262, 532–538. [Google Scholar]
- Wagner, J.; Schümann, H.J.; Knorr, A.; Rohm, N.; Reidemeister, J.C. Stimulation by adrenaline and dopamine but not by noradrenaline of myocardial α-adrenoceptors mediating positive inotropic effects in human atrial preparations. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980, 312, 99–102. [Google Scholar] [CrossRef]
- Rump, L.C.; Riera-Knorrenschild, G.; Schwertfeger, E.; Bohmann, C.; Spillner, G.; Schollmeyer, P. Dopaminergic and α-Adrenergic Control of Neurotransmission in Human Right Atrium. J. Cardiovasc. Pharmacol. 1995, 26, 462–470. [Google Scholar] [CrossRef]
- Bravo, G.; Ghysel-Burton, J.; Jaumin, P.; Godfraind, T. A comparison of the inotropic effects of dopamine and epinine in human isolated cardiac preparations. Experiment 1991, 257, 439–443. [Google Scholar]
- Brown, L.; Lorenz, B.; Erdmann, E. The inotropic effects of dopamine and its precursor levodopa on isolated human ventricular myocardium. J. Mol. Med. 1985, 63, 1117–1123. [Google Scholar] [CrossRef]
- Port, J.D.; Gilbert, E.M.; Larrabee, P.; Mealey, P.; Volkman, K.; Ginsburg, R.; E Hershberger, R.; Murray, J.; Bristow, M.R. Neurotransmitter depletion compromises the ability of indirect-acting amines to provide inotropic support in the failing human heart. Circulation 1990, 81, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Erdmann, E. Concentration-response curves of positive inotropic agents before and after ouabain pretreatment. Cardiovasc. Res. 1985, 19, 288–298. [Google Scholar] [CrossRef]
- Martinez-Mir, I.; Morales-Olivas, F.J.; Rubio, E. The lack of the effect of DA-1 and DA-2 dopamine agonists on the isolated guineapig atria. J. Auton. Pharmacol. 1987, 7, 111–118. [Google Scholar] [CrossRef]
- Einstein, R.; Barrett, A.M. A comparison of the cardiac actions of dopamine and noradrenaline in anaesthetized dogs and guinea-pig atria. Clin. Exp. Pharmacol. Physiol. 1977, 4, 143–151. [Google Scholar] [CrossRef]
- Tsai, T.H.; Langer, S.Z.; Trendelenburg, U. Effects of dopamine and alpha-methyl-dopamine on smooth muscle and on the cardiac pacemaker. Experiment 1967, 156, 310–324. [Google Scholar]
- Brown, L. Pharmacological responses to dopamine in isolated guinea-pig cardiovascular tissues: Mechanisms of action. Arch. Int. De Pharmacodyn. Et De Ther. 1990, 308, 47–62. [Google Scholar]
- Furukawa, T.; Ono, N.; Maeda, Y.; Nakahara, T.; Yoshihara, K. A possible mode of cardiovascular actions of dopamine in dogs. Jpn. J. Pharmacol. 1976, 26, 481–492. [Google Scholar] [CrossRef]
- Habuchi, Y.; Tanaka, H.; Nishio, M.; Yamamoto, T.; Komori, T.; Morikawa, J.; Yoshimura, M. Dopamine stimulation of cardiac beta-adrenoceptors: The involvement of sympathetic amine transporters and the effect of SKF38393. Br. J. Pharmacol. 1997, 122, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Matsuoka, S.; Fujioka, Y.; Noma, A. Effects of dopamine on L-type Ca2+current in single atrial and ventricular myocytes of the rat. Br. J. Pharmacol. 1997, 121, 1247–1254. [Google Scholar] [CrossRef]
- Shi, Q.; Li, M.; Mika, D.; Fu, Q.; Kim, S.; Phan, J.; Shen, A.; Vandecasteele, G.; Xiang, Y.K. Heterologous desensitization of cardiac β-adrenergic signal via hormone-induced βAR/arrestin/PDE4 complexes. Cardiovasc. Res. 2017, 113, 656–670. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.-H.; Parikh, K.S.; Ryan, J.J.; D’Souza, K.M.; Theccanat, T.; Toth, P.; Pogoriler, J.; Paul, J.; Blaxall, B.C.; et al. GRK2-Mediated Inhibition of Adrenergic and Dopaminergic Signaling in Right Ventricular Hypertrophy. Circulation 2012, 126, 2859–2869. [Google Scholar] [CrossRef] [PubMed]
- James, T.N.; Bear, E.S.; Lang, K.F.; Green, E.W.; Winkler, H.H. Adrenergic mechanisms in the sinus node. Arch. Intern. Med. 1970, 125, 512–547. [Google Scholar] [CrossRef]
- Black, W.L.; Rolett, E.L. Cardiovascular adrenergic activity of dopamine in the dog. Am. Heart J. 1968, 75, 233–239. [Google Scholar] [CrossRef]
- Endoh, M.; Schümann, H.; Krappitz, N.; Hillen, B. α-adrenoceptors mediating positive inotropic effects on the ventricular myocardium: Some aspects of structure-activity relationship of sympathomimetic amines. Jpn. J. Pharmacol. 1976, 26, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.-E.; Inui, J.; Motomura, S.; Schümann, H.-J. The Mode of Direct Action of Dopamine on the Rabbit Heart. J. Cardiovasc. Pharmacol. 1980, 2, 567–582. [Google Scholar] [CrossRef]
- Schümann, H.J.; Motomura, S.; Endoh, M.; Brodde, O.E. Comparison of the mechanisms underlying the positive inotropic actions of dopamine, adrenaline and isoprenaline on the isolated rabbit papillary muscle. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1977, 297, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Motomura, S.; Brodde, O.-E.; Schümann, H.J. No evidence for involvement of dopaminergic receptors in the positive inotropic action of dopamine on the isolated rabbit papillary muscle. Jpn. J. Pharmacol. 1978, 28, 145–153. [Google Scholar] [CrossRef]
- Wakita, Y. Inotropic, chronotropic, and arrhythmogenic effects of dopamine on the isolated working heart of rabbit. J. Physiol. Sci. 2007, 57, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, E.V.; Nielsen-Kudsk, F. Myocardial pharmacodynamics of dopamine, dobutamine, amrinone and isoprenaline compared in the isolated rabbit heart. Eur. J. Pharmacol. 1986, 124, 51–57. [Google Scholar] [CrossRef]
- Van Woerkens, L.; Duncker, D.; Boer, M.D.; McFalls, E.; Sassen, L.; Saxena, P.; Verdouw, P. Evidence against a role for dopamine D1 receptors in the myocardium of the pig. Br. J. Pharmacol. 1991, 104, 246–250. [Google Scholar] [CrossRef]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.L.; Allen, J.A.; Mente, S.; O’Connor, R.E.; DeMarco, G.J.; Efremov, I.; Tierney, P.; Volfson, D.; Davoren, J.; Guilmette, E.; et al. Impaired β-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun. 2018, 9, 674. [Google Scholar] [CrossRef]
- Matsuyama, N.; Shibata, S.; Matoba, A.; Kudo, T.-A.; Danielsson, J.; Kohjitani, A.; Masaki, E.; Emala, C.W.; Mizuta, K. The dopamine D1 receptor is expressed and induces CREB phosphorylation and MUC5AC expression in human airway epithelium. Respir. Res. 2018, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.U.; Lewin, G.; Baba, H.A.; Bokník, P.; Fabritz, L.; Kirchhefer, U.; Kirchhof, P.; Loser, K.; Matus, M.; Neumann, J.; et al. Heart-directed Expression of a Human Cardiac Isoform of cAMP-Response Element Modulator in Transgenic Mice. J. Biol. Chem. 2005, 280, 6906–6914. [Google Scholar] [CrossRef]
- García-Pedraza, J.; Morán, A.; Martín, M.L.; Ollauri-Ibáñez, C.; Rodríguez-Barbero, A.; Villalón, C.M.; García-Domingo, M. Dopamine D4 receptor subtype activation reduces the rat cardiac parasympathetic discharge. Pflugers Arch. 2020, 472, 1693–1703. [Google Scholar] [CrossRef]
- Li, H.-Z.; Han, L.-P.; Jiang, C.-M.; Zhao, Y.-J.; Gao, J.; Lin, Y.; Ma, S.-X.; Tian, Y.; Yang, B.-F.; Xu, C.-Q.; et al. Effect of Dopamine Receptor 1 on Apoptosis of Cultured Neonatal Rat Cardiomyocytes in Simulated Ischaemia/Reperfusion. Basic Clin. Pharmacol. Toxicol. 2008, 102, 329–336. [Google Scholar] [CrossRef]
- Li, H.-Z.; Guo, J.; Gao, J.; Han, L.-P.; Jiang, C.-M.; Bai, S.-Z.; Zhang, W.-H.; Li, G.-W.; Wang, L.-N.; Zhao, Y.-J.; et al. Role of dopamine D2 receptors in ischemia/reperfusion induced apoptosis of cultured neonatal rat cardiomyocytes. J. Biomed. Sci. 2011, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sumida, T.S.; Nomura, S.; Satoh, M.; Higo, T.; Ito, M.; Ko, T.; Fujita, K.; Sweet, M.E.; Sanbe, A.; et al. Cardiac dopamine D1 receptor triggers ventricular arrhythmia in chronic heart failure. Nat. Commun. 2020, 11, 4364. [Google Scholar] [CrossRef]
- Gildea, J.J.; Xu, P.; Kemp, B.A.; Carey, R.M.; Jose, P.A.; Felder, R.A. The Dopamine D 1 Receptor and Angiotensin II Type-2 Receptor are Required for Inhibition of Sodium Transport Through a Protein Phosphatase 2A Pathway. Hypertension 2019, 73, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Asico, L.; Luo, Y.; Andrews, P.; Eisner, G.; Hopfer, U.; Felder, R.; Jose, P. D1 dopamine receptor hyperphosphorylation in renal proximal tubules in hypertension. Kidney Int. 2006, 70, 1072–1079. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Quiñones, H.; Gisler, S.M.; Di Sole, F.; Hu, M.-C.; Shi, M.; Zhang, J.; Fuster, D.G.; Wright, N.; Mumby, M.; et al. Acute regulation of renal Na+/H+ exchanger NHE3 by dopamine: Role of protein phosphatase 2A. Am. J. Physiol. Physiol. 2010, 298, F1205–F1213. [Google Scholar] [CrossRef]
- Herzig, S.; Neumann, J. Effects of Serine/Threonine Protein Phosphatases on Ion Channels in Excitable Membranes. Physiol. Rev. 2000, 80, 173–210. [Google Scholar] [CrossRef]
- Neumann, J.; Boknik, P.; Kirchhefer, U.; Gergs, U. The role of PP5 and PP2C in cardiac health and disease. Cell. Signal. 2021, 85, 110035. [Google Scholar] [CrossRef]
- Götz, J.; Probst, A.; Ehler, E.; Hemmings, B.; Kues, W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Cα. Proc. Natl. Acad. Sci. USA 1998, 95, 12370–12375. [Google Scholar] [CrossRef]
- Hemmings, H.C., Jr.; Greengard, P. DARPP-32, a dopamine-regulated phosphoprotein. Prog Brain Res. 1986, 69, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Shuto, T. Potential for targeting dopamine/DARPP-32 signaling in neuropsychiatric and neurodegenerative disorders. Expert Opin. Ther. Targets 2017, 21, 259–272. [Google Scholar] [CrossRef]
- Sirenko, S.T.; Zahanich, I.; Li, Y.; Lukyanenko, Y.O.; Lyashkov, A.E.; Ziman, B.D.; Tarasov, K.V.; Younes, A.; Riordon, D.R.; Tarasova, Y.S.; et al. Phosphoprotein Phosphatase 1 but Not 2A Activity Modulates Coupled-Clock Mechanisms to Impact on Intrinsic Automaticity of Sinoatrial Nodal Pacemaker Cells. Cells 2021, 10, 3106. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Hou, X.; Fang, H. Structure, Function and Modulation of Striatal-enriched Protein Tyrosine Phosphatase (STEP). Curr. Med. Chem. 2021, 28, 7714–7728. [Google Scholar] [CrossRef]
- Neumann, J.; Gupta, R.C.; Schmitz, W.; Scholz, H.; Nairn, A.C.; Watanabe, A.M. Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circ. Res. 1991, 69, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Patil, P.; Neumann, J.; Staschen, C.; Yue, D. Mechanisms of beta-adrenergic stimulation of cardiac Ca2+ channels revealed by discrete-time Markov analysis of slow gating. Biophys. J. 1993, 65, 1599–1612. [Google Scholar] [CrossRef]
- Gupta, R.C.; Neumann, J.; Watanabe, A.M.; Lesch, M.; Sabbah, H.N. Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am. J. Physiol. Circ. Physiol. 1996, 270, H1159–H1164. [Google Scholar] [CrossRef]
- Gergs, U.; Bernhardt, G.; Buchwalow, I.B.; Edler, H.; Fröba, J.; Keller, M.; Kirchhefer, U.; Köhler, F.; Mißlinger, N.; Wache, H.; et al. Initial Characterization of Transgenic Mice Overexpressing Human Histamine H2Receptors. Experiment 2019, 369, 129–141. [Google Scholar] [CrossRef]
- Bibb, J.A.; Nishi, A.; O’Callaghan, J.P.; Ule, J.; Lan, M.; Snyder, G.L.; Horiuchi, A.; Saito, T.; Hisanaga, S.-I.; Czernik, A.J.; et al. Phosphorylation of Protein Phosphatase Inhibitor-1 by Cdk5. J. Biol. Chem. 2001, 276, 14490–14497. [Google Scholar] [CrossRef]
- Nguyen, C.; Nishi, A.; Kansy, J.W.; Fernandez, J.; Hayashi, K.; Gillardon, F.; Hemmings, H.C.; Nairn, A.C.; Bibb, J.A. Regulation of Protein Phosphatase Inhibitor-1 by Cyclin-dependent Kinase 5. J. Biol. Chem. 2007, 282, 16511–16520. [Google Scholar] [CrossRef] [PubMed]
- Sahin, B.; Shu, H.; Fernandez, J.; El-Armouche, A.; Molkentin, J.D.; Nairn, A.C.; Bibb, J.A. Phosphorylation of Protein Phosphatase Inhibitor-1 by Protein Kinase C. J. Biol. Chem. 2006, 281, 24322–24335. [Google Scholar] [CrossRef]
- Rodriguez, P.; Mitton, B.; Waggoner, J.; Kranias, E.G. Identification of a Novel Phosphorylation Site in Protein Phosphatase Inhibitor-1 as a Negative Regulator of Cardiac Function. J. Biol. Chem. 2006, 281, 38599–38608. [Google Scholar] [CrossRef]
- Singh, A.; Redden, J.M.; Kapiloff, M.S.; Dodge-Kafka, K.L. The Large Isoforms of A-Kinase Anchoring Protein 18 Mediate the Phosphorylation of Inhibitor-1 by Protein Kinase A and the Inhibition of Protein Phosphatase 1 Activity. Mol. Pharmacol. 2010, 79, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Brüchert, N.; Mavila, N.; Boknik, P.; Baba, H.A.; Fabritz, L.; Gergs, U.; Kirchhefer, U.; Kirchhof, P.; Matus, M.; Schmitz, W.; et al. Inhibitor-2 prevents phosphatase 1 induced cardiac hypertrophy and mortality. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1539–H1546. [Google Scholar] [CrossRef]
- Grote-Wessels, S.; Baba, H.A.; Fabritz, L.; Gillmann, H.J.; Matus, M.; Müller, F.U.; Neumann, J.; Schmitz, W.; Theilmeier, G.; Kirchhefer, U. Inhibition of protein phosphatase 1 by an active form of inhibitor-2 exacerbates progression of cardiac failure in a model with pressure overload. Cardiovas Res. 2008, 79, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Roach, P.; Bondor, J.; Fox, S.P.; DePaoli-Roach, A.A. Molecular mechanism of the synergistic phosphorylation of phosphatase inhibitor-2. Cloning, expression, and site-directed mutagenesis of inhibitor-2. J. Biol. Chem. 1994, 269, 944–954. [Google Scholar] [CrossRef]
- Krause, T.; Grote-Wessels, S.; Balzer, F.; Boknik, P.; Gergs, U.; Kirchhefer, U.; Buchwalow, I.B.; Müller, F.U.; Schmitz, W.; Neumann, J. Successful overexpression of wild-type inhibitor-2 of PP1 in cardiovascular cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Guan, K.-L.; Roach, P.J.; DePaoli-Roach, A.A. Phosphorylation and Activation of the ATP-Mg-dependent Protein Phosphatase by the Mitogen-activated Protein Kinase. J. Biol. Chem. 1995, 270, 18352–18358. [Google Scholar] [CrossRef]
- Jose, P.; Yu, P.-Y.; Yamapchi, I.; Eisner, G.M.; Mouradian, M.M.; Felder, C.C.; Felder, R.A. Dopamine D1 Receptor Regulation of Phospholipase C. Hypertens. Res. 1995, 18, S39–S42. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Mavrikaki, M.; Ullrich, C.; Albarran-Zeckler, R.; Brantley, A.F.; Smith, R.G. Hippocampal Dopamine/DRD1 Signaling Dependent on the Ghrelin Receptor. Cell 2015, 163, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharmacol. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rankin, M.L.; Sibley, D.R. Constitutive phosphorylation by protein kinase C regulates D1 dopamine receptor signaling. J. Neurochem. 2010, 115, 1655–1667. [Google Scholar] [CrossRef]
- Kim, O.-J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.-M.; Sibley, D.R. The Role of Phosphorylation in D1 Dopamine Receptor Desensitization. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef]
- Banday, A.A.; Lokhandwala, M.F. Transcriptional Regulation of Renal Dopamine D1 Receptor Function During Oxidative Stress. Hypertension 2015, 65, 1064–1072. [Google Scholar] [CrossRef]
- Moreno, E.; Hoffmann, H.; Gonzalez-Sepúlveda, M.; Navarro, G.; Casadó, V.; Cortés, A.; Mallol, J.; Vignes, M.; McCormick, P.J.; Canela, E.I.; et al. Dopamine D1-histamine H3 Receptor Heteromers Provide a Selective Link to MAPK Signaling in GABAergic Neurons of the Direct Striatal Pathway. J. Biol. Chem. 2011, 286, 5846–5854. [Google Scholar] [CrossRef]
- Moreno-Delgado, D.; Puigdellívol, M.; Moreno, E.; Rodríguez-Ruiz, M.; Botta, J.; Gasperini, P.; Chiarlone, A.; Howell, L.A.; Scarselli, M.; Casadó, V.; et al. Modulation of dopamine D1 receptors via histamine H3 receptors is a novel therapeutic target for Huntington’s disease. Elife 2020, 9, e51093. [Google Scholar] [CrossRef]
- McCaffrey, S.L.; Lim, G.; Bullock, M.; Kasparian, A.O.; Clifton-Bligh, R.; Campbell, W.B.; Widiapradja, A.; Levick, S.P. The Histamine 3 Receptor Is Expressed in the Heart and Its Activation Opposes Adverse Cardiac Remodeling in the Angiotensin II Mouse Model. Int. J. Mol. Sci. 2020, 21, 9757. [Google Scholar] [CrossRef] [PubMed]
- Pekcec, A.; Schülert, N.; Stierstorfer, B.; Deiana, S.; Dorner-Ciossek, C.; Rosenbrock, H. Targeting the dopamine D1receptor or its downstream signalling by inhibiting phosphodiesterase-1 improves cognitive performance. Br. J. Pharmacol. 2017, 175, 3021–3033. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Pharmacological Analysis of Dopamine Action on the Isolated Dog Atrium. Tohoku J. Exp. Med. 1975, 115, 355–360. [Google Scholar] [CrossRef]
- Taghi, S.; Matsuo, T.; Fujiwara, M.; Toda, N. Effects of dopamine and DOPA on the isolated rabbit’s atrium treated with reserpine. Jpn. J. Pharmacol. 1962, 12, 197–207. [Google Scholar] [CrossRef]
- Schümann, H.J. What role do alpha- and beta-adrenoceptors play in the regulation of the heart? Eur. Heart J. 1983, 4 (Suppl. A), 55–60. [Google Scholar] [CrossRef] [PubMed]
- Habuchi, Y.; Tanaka, H.; Yamamoto, T.; Komori, T.; Nishio, M.; Yoshimura, M. The mechanisms underlying heart stimulation by dopamine, with special reference to direct and indirect β adrenoceptor stimulation. Clin. Exp. Hypertens. 1997, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Baumann, G.; Schrader, J.; Gerlach, E. Inhibitory action of adenosine on histamine- and dopamine-stimulated cardiac contractility and adenylate cyclase in guinea pigs. Circ. Res. 1981, 48, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, R.; Miragoli, G. Activity of ibopamine on some isolated organs. Arzneimittel-forschung 1986, 36, 312–317. [Google Scholar] [PubMed]
- Mugelli, A.; Ledda, F.; Mantelli, L.; Torrini, M.; Maccioni, T. Studies on the positive inotropic effect of dopamine in the guinea-pig heart. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1977, 301, 49–55. [Google Scholar] [CrossRef]
- Ouedraogo, L.; Magnon, M.; Sawadogo, L.; Tricoche, R. Receptors involved in the positive inotropic action induced by dopamine on the ventricle of a 7-day-old chick embryo heart. Fundam. Clin. Pharmacol. 1998, 12, 133–142. [Google Scholar] [CrossRef]
- Goldman, S.; Olajos, M.; Morkin, E. Effects of verapamil on positive inotropic stimulation in the left atrium and ventricle of conscious dogs. Experiment 1982, 222, 270–275. [Google Scholar]
- Kecskeméti, V.; Kelemen, K. Effect of dopamine on the transmembrane potentials of guinea-pig heart preparations. Pol. J. Pharmacol. Pharm. 1985, 37, 411–419. [Google Scholar]
- Aronson, R.S.; Gelles, J.M. Electrophysiologic effects of dopamine on sheep cardiac Purkinje fibers. J. Pharmacol. Exp. Ther. 1974, 188, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Perlitz, F.; Mohr, F.-W. An in vitro model for assessment of drug-induced torsade de pointes arrhythmia. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 378, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Tarabová, B.; Nováková, M.; Lacinová, L. Haloperidol moderately inhibits cardiovascular L-type calcium current. Gen. Physiol. Biophys. 2009, 28, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Bébarová, M.; Matejovič, P.; Pásek, M.; Nováková, M. Effect of haloperidol on transient outward potassium current in rat ventricular myocytes. Eur. J. Pharmacol. 2006, 550, 15–23. [Google Scholar] [CrossRef]
- Suessbrich, H.; Schönherr, R.; Heinemann, S.H.; Attali, B.; Lang, F.; Busch, A.E. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br. J. Pharmacol. 1997, 120, 968–974. [Google Scholar] [CrossRef]
- Simkó, J.; Szentandrássy, N.; Harmati, G.; Bárándi, L.; Horváth, B.; Magyar, J.; Bányász, T.; Lőrincz, I.; Nánási, P.P. Effects of ropinirole on action potential characteristics and the underlying ion currents in canine ventricular myocytes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2010, 382, 213–220. [Google Scholar] [CrossRef]
- Hurst, R.S.; Higdon, N.R.; Lawson, J.A.; Clark, M.A.; Rutherford-Root, K.L.; McDonald, W.G.; Haas, J.V.; McGrath, J.P.; Meglasson, M.D. Dopamine receptor agonists differ in their actions on cardiac ion channels. Eur. J. Pharmacol. 2003, 482, 31–37. [Google Scholar] [CrossRef]
- Matsumoto, T.; Ozono, R.; Sasaki, N.; Oshima, T.; Matsuura, H.; Kajiyama, G.; Carey, R.M.; Kambe, M. Type 1A dopamine receptor expression in the heart is not altered in spontaneously hypertensive rats. Am. J. Hypertens. 2000, 13, 673–677. [Google Scholar] [CrossRef]
- Driscoll, D.J.; Gillette, P.C.; Ezrailson, E.G.; Schwartz, A. Inotropic Response of the Neonatal Canine Myocardium to Dopamine. Pediatr. Res. 1978, 12, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Bilalova, G.A.; Kazanchikova, L.M.; Zefirov, T.L.; Sitdikov, F.G. Inotropic Effect of Dopamine on Rat Heart during Postnatal Ontogeny. Bull. Exp. Biol. Med. 2013, 156, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Pfeil, U.; Kuncova, J.; Brüggmann, D.; Paddenberg, R.; Rafiq, A.; Henrich, M.; Weigand, M.A.; Schlüter, K.-D.; Mewe, M.; Middendorff, R.; et al. Intrinsic vascular dopamine—A key modulator of hypoxia-induced vasodilatation in splanchnic vessels. J. Physiol. 2014, 592, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Tonnarini, G.; Parlapiano, C.; Cavallotti, D.; Tego, A.; Curione, M.; Giancaspro, G.; Vincentelli, G.M.; Leone, S.; Cavallotti, C. Dopamine receptor subtypes in the human coronary vessels of healthy subjects. J. Recept. Signal Transduct. 2010, 31, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, F.E.; Drago, J.; A Felder, R.; Printz, M.P.; Eisner, G.M.; Robillard, J.E.; Sibley, D.R.; Westphal, H.J.; Jose, P.A. Role of the D1A dopamine receptor in the pathogenesis of genetic hypertension. J. Clin. Investig. 1996, 97, 2283–2288. [Google Scholar] [CrossRef]
- Drago, J.; Gerfen, C.R.; Lachowicz, J.E.; Steiner, H.; Hollon, T.R.; Love, P.E.; Ooi, G.T.; Grinberg, A.; Lee, E.J.; Huang, S.P. Altered striatal function in a mutant mouse lacking D1A dopamine receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 12564–12568. [Google Scholar] [CrossRef] [PubMed]
- Mussa, S.; Guzik, T.J.; Black, E.; Dipp, M.A.; Channon, K.M.; Taggart, D.P. Comparative efficacies and durations of action of phenoxybenzamine, verapamil/nitroglycerin solution, and papaverine as topical antispasmodics for radial artery coronary bypass grafting. J. Thorac. Cardiovasc. Surg. 2003, 126, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Enokibori, M.; Matsumoto, T.; Okamura, T. Responsiveness to Dopamine of Isolated Epicardial Coronary Arteries from Humans, Monkeys, and Dogs. Obstet. Anesth. Dig. 1993, 77, 526–532. [Google Scholar] [CrossRef]
- Katai, R.; Tsuneyoshi, I.; Hamasaki, J.; Onomoto, M.; Suehiro, S.; Sakata, R.; Kanmura, Y. The Variable Effects of Dopamine Among Human Isolated Arteries Commonly Used for Coronary Bypass Grafts. Obstet. Anesth. Dig. 2004, 98, 915–920. [Google Scholar] [CrossRef]
- Hughes, A.; Sever, P. Action of Fenoldopam, a Selective Dopamine (DA1 Receptor Agonist, on Isolated Human Arteries. J. Vasc. Res. 1989, 26, 119–127. [Google Scholar] [CrossRef]
- Cavallotti, C.; Mancone, M.; Bruzzone, P.; Sabbatini, M.; Mignini, F. Dopamine receptor subtypes in the native human heart. Heart Vessel. 2010, 25, 432–437. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, M.; Asico, L.D.; Eisner, G.M.; Jose, P.A. The dopaminergic system in hypertension. Clin. Sci. 2007, 112, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.B.; Murray, C.; Shorten, G.D. Fenoldopam—A Selective Peripheral Dopamine-Receptor Agonist for the Treatment of Severe Hypertension. N. Engl. J. Med. 2001, 345, 1548–1557. [Google Scholar] [CrossRef]
- Amenta, F.; Gallo, P.; Rossodivita, A.; Ricci, A. Radioligand binding and autoradiographic analysis of dopamine receptors in the human heart. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1993, 347, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Yang, Z.; Li, Y.; Lan, H.; Cyganek, L.; Yuecel, G.; Lang, S.; Bieback, K.; El-Battrawy, I.; Zhou, X.; et al. Dopamine D1/D5 Receptor Signaling Is Involved in Arrhythmogenesis in the Setting of Takotsubo Cardiomyopathy. Front. Cardiovasc. Med. 2022, 8, 777463. [Google Scholar] [CrossRef] [PubMed]
- Rayo Abella, L.M.; Gergs, U.; Pockes, S.; Hofmann, B.; Neumann, J. Effect of dopamine-1 receptor agonists in D1-dopamine receptor overexpressing mouse atrial preparations. Naunyn-Schmiedeberg’s Arch. Pharmacol. (Abstract) 2023, 396 (Suppl. S1), S43–S44. [Google Scholar]
- Kaumann, A.J.; Lemoine, H.; Schwederski-Menke, U.; Ehle, B. Relations between β-adrenoceptor occupancy and increases of contractile force and adenylate cyclase activity induced by catecholamines in human ventricular myocardium. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 339, 99–112. [Google Scholar] [CrossRef]
- Lim, J.Y.; Park, S.J.; Kim, H.J.; Kim, H.J.; Choo, S.J.; Chung, C.H.; Lee, J.W.; Park, D.; Kim, J.B. Comparison of dopamine versus norepinephrine in circulatory shock after cardiac surgery: A randomized controlled trial. J. Card. Surg. 2021, 36, 3711–3718. [Google Scholar] [CrossRef]
- Hiemstra, B.; Koster, G.; Wetterslev, J.; Gluud, C.; Jakobsen, J.C.; Scheeren, T.W.L.; Keus, F.; Van Der Horst, I.C.C. Dopamine in critically ill patients with cardiac dysfunction: A systematic review with meta-analysis and trial sequential analysis. Acta Anaesthesiol. Scand. 2018, 63, 424–437. [Google Scholar] [CrossRef]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.W.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 2015, 105, 83–89. [Google Scholar] [CrossRef]
- Hampton, J.; van Veldhuisen, D.; Kleber, F.; Cowley, A.; Ardia, A.; Block, P.; Cortina, A.; Cserhalmi, L.; Follath, F.; Jensen, G.; et al. Randomised study of effect of ibopamine on survival in patients with advanced severe heart failure. Lancet 1997, 349, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Böhm, M.; Schulz, C.; Schmidt, U.; Schmid, B.; Dienemann, H.; Reichart, B.; Erdmann, E. Cardiac inotropic as well as coronary and pulmonary artery actions of epinine in human isolated tissues. Experiment 1993, 265, 346–357. [Google Scholar]
- Rajfer, S.I.; Anton, A.H.; Rossen, J.D.; Goldberg, L.I. Beneficial Hemodynamic Effects of Oral Levodopa in Heart Failure. N. Engl. J. Med. 1984, 310, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.M.; Johnson, C.E.; Brown, C.E.; Chance, W.T.; Beekman, R.H. Hemodynamic and Clinical Effects of Oral Levodopa in Children With Congestive Heart Failure. J. Am. Coll. Cardiol. 1997, 30, 237–242. [Google Scholar] [CrossRef]
- Goldberg, L.I.; Whitsett, T.L. Cardiovascular effects of levodopa. Clin. Pharmacol. Ther. 1971, 12, 376–382. [Google Scholar] [CrossRef]
- Nakano, M.; Koga, M.; Hashimoto, T.; Matsushita, N.; Masukawa, D.; Mizuno, Y.; Uchimura, H.; Niikura, R.; Miyazaki, T.; Nakamura, F.; et al. Right ventricular overloading is attenuated in monocrotaline-induced pulmonary hypertension model rats with a disrupted Gpr143 gene, the gene that encodes the 3,4-l-dihydroxyphenyalanine (l-DOPA) receptor. J. Pharmacol. Sci. 2021, 148, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Moratalla, R.; Gold, L.H.; Hiroi, N.; Koob, G.F.; Graybiel, A.M.; Tonegawa, S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell 1994, 79, 729–742. [Google Scholar] [CrossRef]
- Kelly, M.A.; Rubinstein, M.; Phillips, T.J.; Lessov, C.N.; Burkhart-Kasch, S.; Zhang, G.; Bunzow, J.R.; Fang, Y.; Gerhardt, G.A.; Grandy, D.K.; et al. Locomotor Activity in D2 Dopamine Receptor-Deficient Mice Is Determined by Gene Dosage, Genetic Background, and Developmental Adaptations. J. Neurosci. 1998, 18, 3470–3479. [Google Scholar] [CrossRef]
- Li, X.X.; Bek, M.; Asico, L.D.; Yang, Z.; Grandy, D.K.; Goldstein, D.S.; Rubinstein, M.; Eisner, G.M.; Jose, P.A. Adrenergic and Endothelin B Receptor-Dependent Hypertension in Dopamine Receptor Type-2 Knockout Mice. Hypertension 2001, 38, 303–308. [Google Scholar] [CrossRef]
- Konkalmatt, P.R.; Asico, L.D.; Zhang, Y.; Yang, Y.; Drachenberg, C.; Zheng, X.; Han, F.; Jose, P.A.; Armando, I. Renal rescue of dopamine D2 receptor function reverses renal injury and high blood pressure. J. Clin. Investig. 2016, 1, e85888. [Google Scholar] [CrossRef]
- Bello, E.P.; Casas-Cordero, R.; Galiñanes, G.L.; Casey, E.; A Belluscio, M.; Rodríguez, V.; Noaín, D.; Murer, M.G.; Rubinstein, M. Inducible ablation of dopamine D2 receptors in adult mice impairs locomotion, motor skill learning and leads to severe parkinsonism. Mol. Psychiatry 2016, 22, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.L.; Tulis, D.A.; Keeler, B.E.; Virag, J.A.; Lust, R.M.; Clemens, S. The Dopamine D3 Receptor Knockout Mouse Mimics Aging-Related Changes in Autonomic Function and Cardiac Fibrosis. PLoS ONE 2013, 8, e74116. [Google Scholar] [CrossRef]
- Gaweda, G.; Iyer, R.P.; Shaver, P.R.; Grilo, G.A.; Dinkins, M.-L.; Stoffel, H.J.; Clemens, S.; Brás, L.E.D.C. Dopamine receptor D3 agonist (Pramipexole) reduces morphine-induced cardiac fibrosis. Biochem. Biophys. Res. Commun. 2020, 529, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Garoffolo, G.; Pesce, M. From dissection of fibrotic pathways to assessment of drug interactions to reduce cardiac fibrosis and heart failure. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100036. [Google Scholar] [CrossRef]
- Rubinstein, M.; Phillips, T.J.; Bunzow, J.R.; Falzone, T.L.; Dziewczapolski, G.; Zhang, G.; Fang, Y.; Larson, J.L.; McDougall, J.A.; Chester, J.A.; et al. Mice Lacking Dopamine D4 Receptors Are Supersensitive to Ethanol, Cocaine, and Methamphetamine. Cell 1997, 90, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Bek, M.J.; Wang, X.; Asico, L.D.; Jones, J.E.; Zheng, S.; Li, X.; Eisner, G.M.; Grandy, D.K.; Carey, R.M.; Soares-Da-Silva, P.; et al. Angiotensin-II Type 1 Receptor–Mediated Hypertension in D4 Dopamine Receptor–Deficient Mice. Hypertension 2006, 47, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, Y.; Liu, X.; Wang, W.; Wang, Z.; Hu, Y.; Zhang, Y.; Zhang, Y.; Jose, P.A.; Wei, Q.; et al. Over-expression of a cardiac-specific human dopamine D5 receptor mutation in mice causes a dilated cardiomyopathy through ROS over-generation by NADPH oxidase activation and Nrf2 degradation. Redox Biol. 2018, 19, 134–146. [Google Scholar] [CrossRef]
- Hollon, T.R.; Bek, M.J.; Lachowicz, J.E.; Ariano, M.A.; Mezey, E.; Ramachandran, R.; Wersinger, S.R.; Soares-Da-Silva, P.; Liu, Z.F.; Grinberg, A.; et al. Mice Lacking D5Dopamine Receptors Have Increased Sympathetic Tone and Are Hypertensive. J. Neurosci. 2002, 22, 10801–10810. [Google Scholar] [CrossRef]
- Jiang, X.; Shao, M.; Liu, X.; Liu, X.; Zhang, X.; Wang, Y.; Yin, K.; Wang, S.; Hu, Y.; A Jose, P.; et al. Reversible Treatment of Pressure Overload-Induced Left Ventricular Hypertrophy through Drd5 Nucleic Acid Delivery Mediated by Functional Polyaminoglycoside. Adv. Sci. 2021, 8, 2003706. [Google Scholar] [CrossRef]
- Cops, J.; Haesen, S.; De Moor, B.; Mullens, W.; Hansen, D. Current animal models for the study of congestion in heart failure: An overview. Heart Fail. Rev. 2019, 24, 387–397. [Google Scholar] [CrossRef]
- Kuchel, O.; Racz, K.; Debinski, W.; Buu, N.T. A defective beta-hydroxylation of dopamine may precede the full development of hypertension in spontaneously hypertensive rats. Can. J. Cardiol. 1989, 5, 327–331. [Google Scholar]
- Kumar, G.K.; Overholt, J.L.; Bright, G.R.; Hui, K.Y.; Lu, H.; Gratzl, M.; Prabhakar, N.R. Release of dopamine and norepinephrine by hypoxia from PC-12 cells. Am. J. Physiol. Physiol. 1998, 274, C1592–C1600. [Google Scholar] [CrossRef] [PubMed]
- Orset, C.; Parrot, S.; Sauvinet, V.; Cottet-Emard, J.-M.; Bérod, A.; Pequignot, J.-M.; Denoroy, L. Dopamine transporters are involved in the onset of hypoxia-induced dopamine efflux in striatum as revealed by in vivo microdialysis. Neurochem. Int. 2005, 46, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Podzuweit, T.; Els, D.J.; McCarthy, J. Cyclic AMP mediated arrhythmias induced in the ischaemic pig heart. Basic Res. Cardiol. 1981, 76, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Chenliu, C.; Sheng, X.; Dan, P.; Qu, Y.; Claydon, V.E.; Lin, E.; Hove-Madsen, L.; Sanatani, S.; Tibbits, G.F. Ischemia–reperfusion destabilizes rhythmicity in immature atrioventricular pacemakers: A predisposing factor for postoperative arrhythmias in neonate rabbits. Heart Rhythm. 2016, 13, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Li, W.-L.; Xu, J.-J.; Zhu, S.-Q.; Long, X.; Che, J.-P. D2 dopamine receptor antagonist raclopride induces non-canonical autophagy in cardiac myocytes. J. Cell. Biochem. 2012, 114, 103–110. [Google Scholar] [CrossRef]
- Kuchel, O.; Cantin, M.; Buu, N.T.; Debinski, W.; Jasmin, G.; Genest, J. Catecholamine, dopamine-ß-hydroxylase and atrial natriuretic factor content in separate heart chambers of cardiomyopathic hamsters. Life Sci. 1987, 41, 2333–2338. [Google Scholar] [CrossRef]
- You, L.; Jiang, H. Cabergoline possesses a beneficial effect on blood-brain barrier (BBB) integrity against lipopolysaccharide (LPS). Bioengineered 2021, 12, 8358–8369. [Google Scholar] [CrossRef]
- Millan, M.J.; Maiofiss, L.; Cussac, D.; Audinot, V.; Boutin, J.-A.; Newman-Tancredi, A. Differential Actions of Antiparkinson Agents at Multiple Classes of Monoaminergic Receptor. I. A Multivariate Analysis of the Binding Profiles of 14 Drugs at 21 Native and Cloned Human Receptor Subtypes. Experiment 2002, 303, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.P.; Babu, P.P. Aberrant Dopamine Receptor Signaling Plays Critical Role in the Impairment of Striatal Neurons in Experimental Cerebral Malaria. Mol. Neurobiol. 2020, 57, 5069–5083. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, Y.; Wang, B.; Li, S.; Ma, C.; Liu, X.; Moynagh, P.N.; Zhou, J.; Yang, S. Dopamine Uses the DRD5-ARRB2-PP2A Signaling Axis to Block the TRAF6-Mediated NF-κB Pathway and Suppress Systemic Inflammation. Mol. Cell 2020, 78, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Zausig, Y.A.; Geilfus, D.; Missler, G.; Sinner, B.; Graf, B.M.; Zink, W. Direct cardiac effects of dobutamine, dopamine, epinephrine, and levosimendan in isolated septic rat hearts. Shock 2010, 34, 269–274. [Google Scholar] [CrossRef]
- Felsing, D.E.; Jain, M.K.; Allen, J.A. Advances in Dopamine D1 Receptor Ligands for Neurotherapeutics. Curr. Top. Med. Chem. 2019, 19, 1365–1380. [Google Scholar] [CrossRef]
- Stahl, S.M. Drugs for psychosis and mood: Unique actions at D3, D2, and D1 dopamine receptor subtypes. CNS Spectr. 2017, 22, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Hackman, B.B.; Griffin, B.; Mills, M.; Ramanathan, K.B. Comparative effects of fenoldopam mesylate and nitroprusside on left ventricular performance in severe systemic hypertension. Am. J. Cardiol. 1992, 69, 918–922. [Google Scholar] [CrossRef]
- Benck, U.; Hoeger, S.; Brinkkoetter, P.T.; Gottmann, U.; Doenmez, D.; Boesebeck, D.; Lauchart, W.; Gummert, J.; Karck, M.; Lehmkuhl, H.B.; et al. Effects of Donor Pre-Treatment With Dopamine on Survival After Heart Transplantation: A Cohort Study of Heart Transplant Recipients Nested in a Randomized Controlled Multicenter Trial. J. Am. Coll. Cardiol. 2011, 58, 1768–1777. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| (A) Dopamine Receptor Agonists | |||||
| D1 | D2 | D3 | D4 | D5 | Reference |
| dopamine pKi: 4.3–5.6 | dopamine pKi: 4.3–5.6 | dopamine pKi: 6.3–7.4 | dopamine pKi: 7.6 | dopamine pKi: 6.6 | Myslivecek 2022 [1] |
| 1 SKF-38393 | 2 MLS1547 | 1 SKF-38393 | 1 Ding et al. 2008 [2] 2 Myslivecek 2022 [1] | ||
| AT77636 | rotigotine | rotigotine | rotigotine | AT77636 | Myslivecek 2022 [1] |
| 1SKF-81297 | 1 ropinirole | 1 ropinirole | 2 quinpirole | 1 SKF-81297 | 1 Myslivecek 2022 [1] 2 Yamaguchi et al., 1997 [3] |
| 1 SKF-83959 | 1,2 pramipexol | 1,2 pramipexol | 2 pramipexol | 1 SKF-83959 | 1 Myslivecek 2022 [1] 2 Piercey et al., 1996 [4] |
| fenoldopam (=SKF 82526) | 2 PD 128907 | PD 128907 | fenoldopam (=SKF 82526) | Myslivecek 2022 [1] | |
| 2A68930 | 1 PD16077 | 1PD16077 | 1 Myslivecek 2022 [1] 2 DeNinno et al., 1990 [5] | ||
| 2 chloro APB | 1 A412997 | 1A412997 | 1 A412997 | 1 Myslivecek 2022 [1] 2 Neumeyer et al., 1991 [6] | |
| dihydrexidine | Salmi et al., 2004 [7] | ||||
| apomorphine | Myslivecek 2022 [1] | ||||
| propylbutyl-dopamine | Zhao et al. 1990 [8] | ||||
| SKF81297 | Rashid et al., 2007 [9] | ||||
| quinpirole | Rashid et al., 2007 [9] | ||||
| bromocriptine 0.1 µM (as Ki) | bromocriptine | Kvernmo et al., 2008 [10] | |||
| pergolide 0.6 µM (as Ki) | pergolide | Kvernmo et al., 2008 [10] | |||
| (B) Dopamine Receptor Antagonists | |||||
| D1 | D2 | D3 | D4 | D5 | Reference |
| 2 haloperidol | 2 haloperidol | 2 S33084 | 2 L745870 | 2 Myslivecek 2022 [1] | |
| 1 SCH-38390 | 2 pipotiazine | 2 SB277011-A | 2 sonepiprazole | 1 SCH-39390 | 1 Ding et al. 2008 [2] |
| 2,3 SKF-83566 | 2 perospirone | 2 perospirone | 2 perospirone | 2 SKF-83566 | 2 Myslivecek 2022 [1] 3 Kopia and Valocik 1989 [11] |
| 2 ecopipam | 2 raclopride | 2 raclopride | 2 A-381393 | 2 ecopipam | 2 Myslivecek 2022 [1] |
| 2 SCH23982 | 2 ML321 | 2 sulpiride | 2 sulpiride | 2 Myslivecek 2022 [1] | |
| 1 odapipam (=NNC 756 Ki: 0.17 nM) | 1 odapipam (Ki: 942 nM) 2 prochlorperazine | 2 prochlorperazine | 2 prochlorper-azine | 1 Andersen et al., 1992 [12] 2 Myslivecek 2022 [1] | |
| NGB 2904 | NGB 2904 | NGB 2904 | Myslivecek 2022 [1] | ||
| metoclopramide | S14297 | S14297 | Myslivecek 2022 [1] | ||
| domperidon | Myslivecek 2022 [1] | ||||
| pimozide | pimozide | pimozide | Lumley et al., 1977 [13] | ||
| fluphenazine | fluphenazine | Vettel et al., 2014 [14] | |||
| melperone | Lencesova et al., 2017 [15] | ||||
| cabergoline | cabergoline | Myslivecek 2022 [1] | |||
| Receptor | α1-Adrenergic pKi: 5.6 | β1-Adrenergic pKi: 5.0 | β2-Adrenergic pKi: 4.3 | Other Effects | Reference Myslivecek 2022 [1] |
|---|---|---|---|---|---|
| Human right atrium | 2 | 1,4 | 1,4 | 3 D1-induced release of noradrenaline | 1 Deighton et al., 1992 [48] 2 Wagner et al., 1980 [49] 3 Rump et al., 1995 [50] 4 Bravo et al., 1991 [51] |
| Human ventricle | No 1 | 1,3,4 | 1,3,4 | release of noradrenaline 1,2 1 D1- or D2-mediated | 1 Brown et al., 1985 [52], 2 Port et al., 1990 [53] 3 Brown et al., 1985 [52] 4 Bravo et al., 1991 [51] |
| Cat right papillary muscle | 1 Receptor type not studied | 1 Brown und Erdmann 1985 [54] | |||
| Guinea pig right atrium | 1,2 propranolol | 1,2 propranolol | 1,3,4 PCE attenuated by reserpine 4 inhibition of noradrenaline synthesis reduced potency of dopamine, 4 poteniated by cocaine, 5 not attenuated by pimozide | 1 Martinez-Mir et al., 1987 [55] 2 Einstein and Barrett [56] 3 Tsai et al., 1967 [57] 4 Brown 1990 [58] 5 Lumley et al., 1977 [13] | |
| Guinea pig left atrium | 2 propranolol 4 | 2 propranolol 4 | 1 receptor type not studied, 2 attenuated by reserpine, 3 not attenuated by haloperidol, 4 inhibition of noradrenaline synthesis reduced potency of dopamine, 4 MAO-Inhibition increased potency of dopamine, 4 not altered by SCH23390, domperidone, cocaine | 1 Brown and Erdmann 1985 [54] 2 Martinez-Mir et al., 1987 [55] 3 Einstein and Barrett [56] 4 Brown 1990 [59] | |
| Guinea pig right papillary muscle | not involved | Receptor type not studied, attenuated by reserpine, not attenuated by haloperidol, inhibition of noradrenaline synthesis reduced potency of dopamine, MAO-inhibition increased potency of dopamine | Brown and Erdmann 1985 [54] | ||
| Isolated guinea pig heart | 1,2 noradrenaline release, 2 PCE | 1 Lumley et al., 1977 [13] 2 Habuchi et al., 1997 [60] | |||
| Guinea pig right atrial cardiomyocytes | LTCC increases | Habuchi et al., 1997 [60] | |||
| Rat neonatal cardiomyocytes | atenolol | increase in cAMP | Vettel et al., 2014 [14] | ||
| Rat atrium | Zhao et al., 1997 [61] | ||||
| Rat ventricular cardiomyocyte | 1,2 isoprenaline effect is attenuated by dopamine | 2 no PIE of dopamine | 1 Zhao et al., 1997 [61] 2 Shi et al., 2017 [62] | ||
| Isolated rat heart monocrotaline treated | PIE was SCH23390-sensitive | Piao et al., 2012 [63] | |||
| Dog | 1,2 propranolol 3 practolol | 1,2 propranolol | 1,3 PCE, 2,3 PIE | 1 James et al., 1970 [64] 2 Black and Rolett 1968 [65] 3 Lumley et al., 1977 [13] | |
| Rabbit left atrium | 1,2 pindolol | 1,2 pindolol | 2 PIE attenuated by cocaine and reserpine | 1 Endoh et al., 1976 [66], 2 Brodde et al., 1980 [67] | |
| Rabbit right ventricular papillary muscle | 1,2,3 | 1,2 pindolol | 1,2 pindolol | 2 PIE attenuated by cocaine, 2No effect of pimozide | 1 Schümann et al., 1977 [68] 2 Brodde et al., 1980 [67] Motomura et al., 1978 [69] |
| Rabbit isolated ventricle | Noradrenaline release | Wakita 2007 [70] | |||
| Rabbit ventricular cardiomyocyte | LTCC | SCH23390-sensitive LTCC | Ding et al., 2008 [2] | ||
| Isolated rabbit hearts | Vigholt Sørensen et al., 1986 [71] | ||||
| Anesthetized pig | PIE not D1 mediated | Van Woerkens et al., 1991 [72] |
| Tissue/Species | LTCC | Agonist | Antagonist | Force | |
|---|---|---|---|---|---|
| Neonatal rabbit cardiomyocyte | Large increase | SKF-38393, dopamine | SCH-39390 | n.d. | Ding et al., 2008 [2] |
| Adult rabbit cardiomyocyte | Small increase | SKF-38393, dopamine | SCH-39390 | n.d. | Ding et al., 2008 [2] |
| Isolated heart from monocrotaline- treated rat | n.d. | dopamine | SCH-39390 | small increase | Piao et al., 2012 [63] |
| pKi: | Reference | |
|---|---|---|
| α2-adrenoceptor | 6.01 | Myslivecek 2022 [1] |
| DAT: dopamine transporter | 5.3 | Myslivecek 2022 [1] |
| NET: noradrenaline transporter | 4.55 | Myslivecek 2022 [1] |
| SERT: serotonin transporter | 4.53 | Myslivecek 2022 [1] |
| Melatonin receptors MT1A | 5.15 | Myslivecek 2022 [1] |
| Melatonin receptors MT1B | 5.04 | Myslivecek 2022 [1] |
| TAAR: trace amine associated receptors | 6.38 | Borowsky et al., 2001 [73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, J.; Hofmann, B.; Dhein, S.; Gergs, U. Role of Dopamine in the Heart in Health and Disease. Int. J. Mol. Sci. 2023, 24, 5042. https://doi.org/10.3390/ijms24055042
Neumann J, Hofmann B, Dhein S, Gergs U. Role of Dopamine in the Heart in Health and Disease. International Journal of Molecular Sciences. 2023; 24(5):5042. https://doi.org/10.3390/ijms24055042
Chicago/Turabian StyleNeumann, Joachim, Britt Hofmann, Stefan Dhein, and Ulrich Gergs. 2023. "Role of Dopamine in the Heart in Health and Disease" International Journal of Molecular Sciences 24, no. 5: 5042. https://doi.org/10.3390/ijms24055042
APA StyleNeumann, J., Hofmann, B., Dhein, S., & Gergs, U. (2023). Role of Dopamine in the Heart in Health and Disease. International Journal of Molecular Sciences, 24(5), 5042. https://doi.org/10.3390/ijms24055042