
Unveiling the Machinery behind Chromosome Folding by Polymer Physics Modeling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
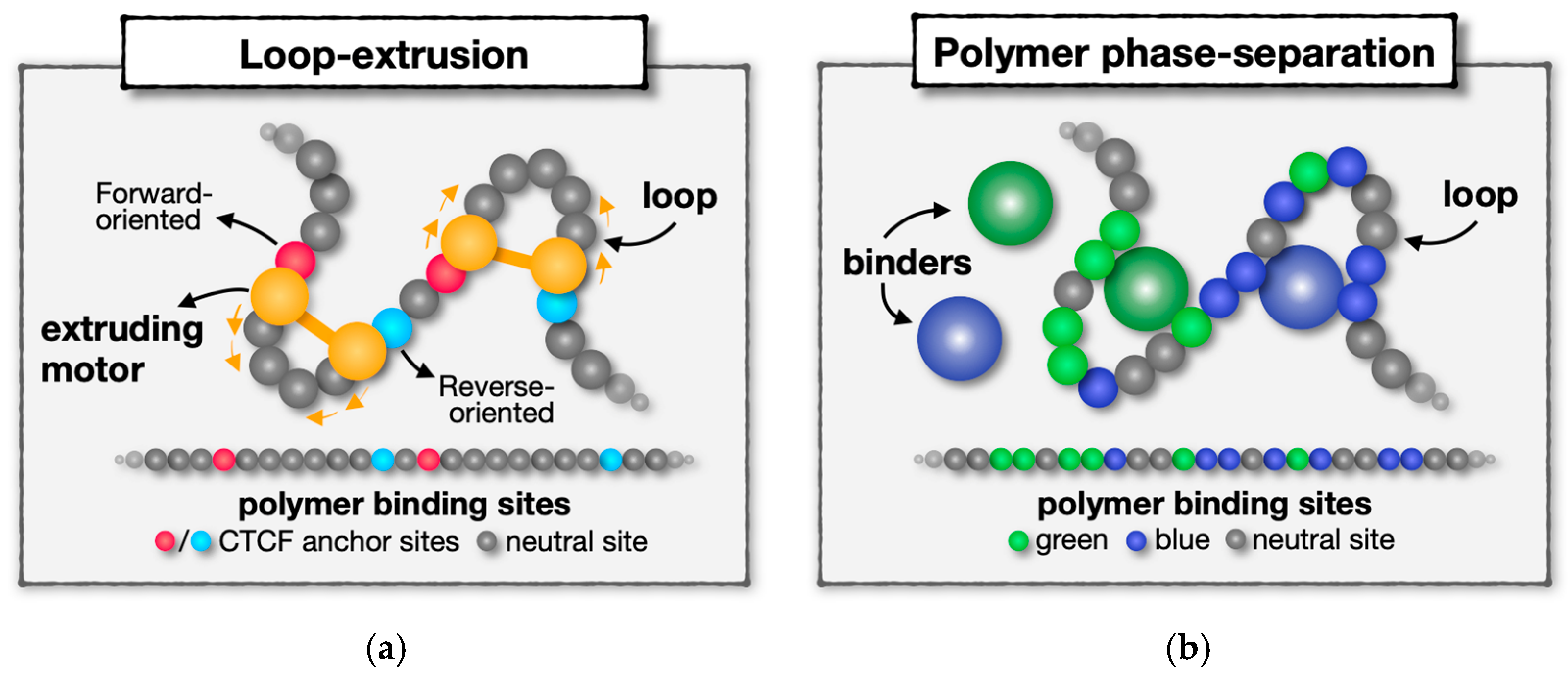
2.1. Loop-Extrusion and Polymer Phase-Separation as Mechanisms of DNA Contact Formation
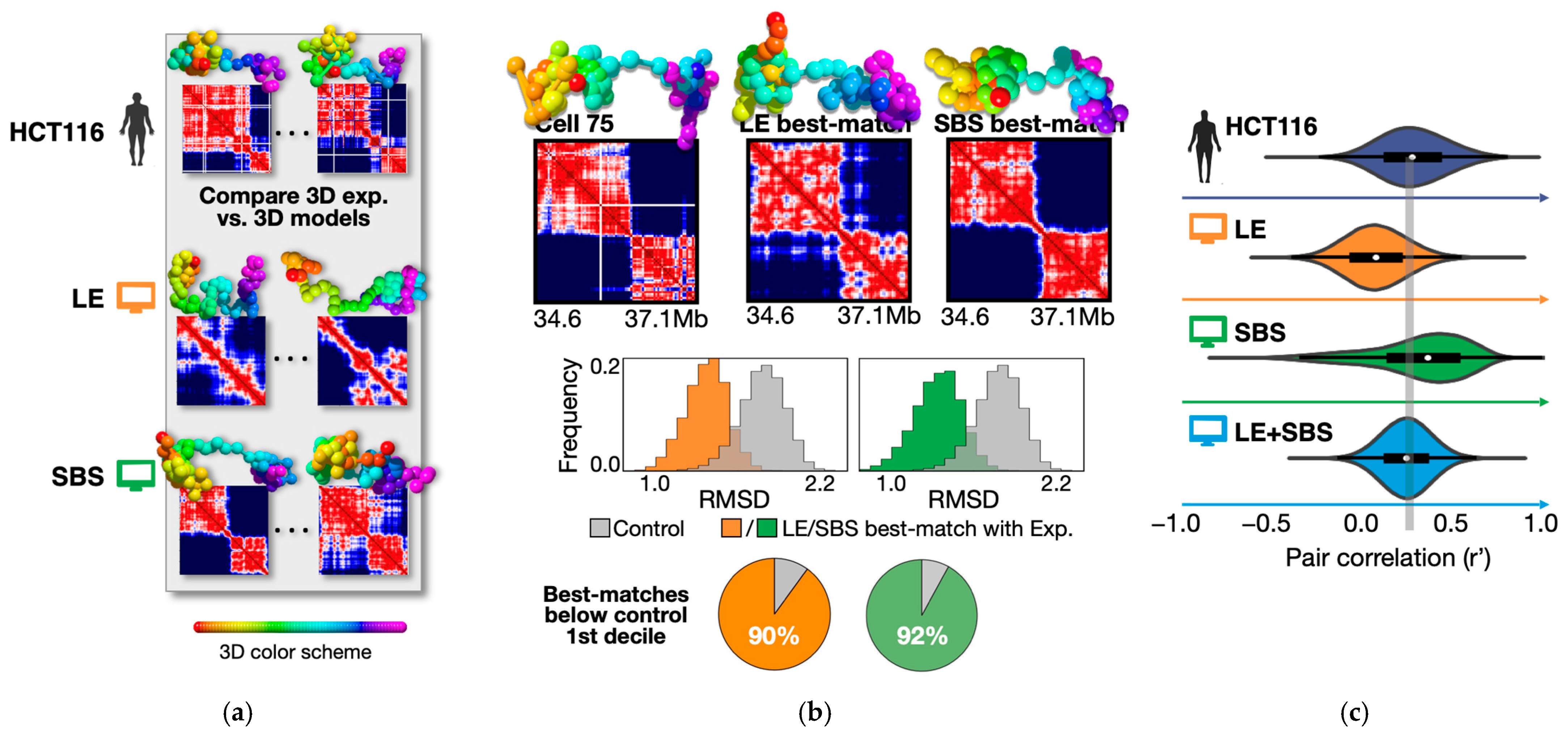
2.1.1. The Polymer Models Are Consistent with the Structure of Chromatin at the Population-Average Level
2.1.2. Loop-Extrusion and Polymer Phase-Separation Cooperate in Determining Heterogeneity of Chromatin Architectures in Single Cells
2.2. Further Validation and Key Applications of Chromatin Polymer Models
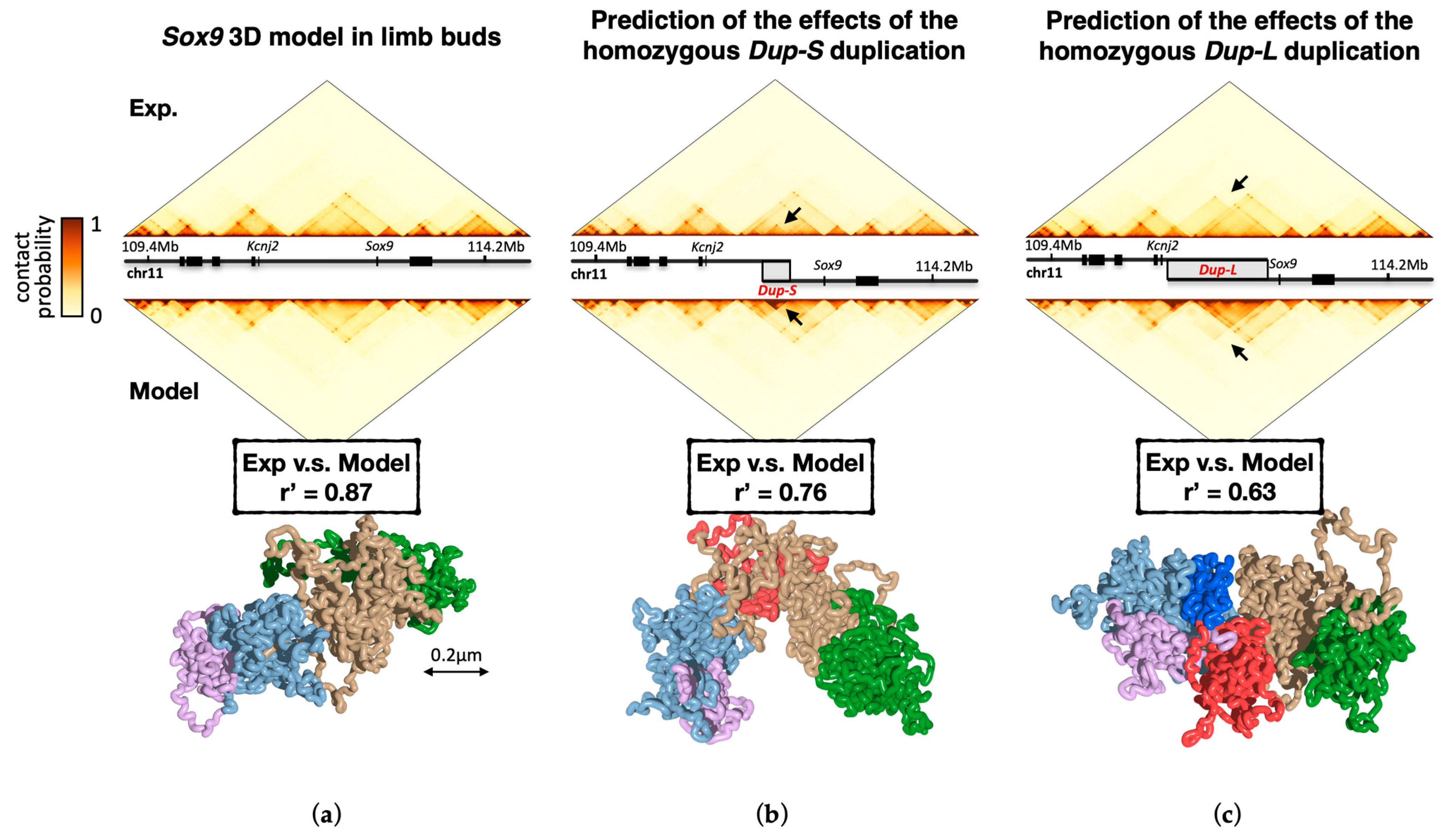
2.2.1. Prediction of Genomic Mutations Effects
2.2.2. A Combinatorial Epigenetic Code Linking 1D and 3D Chromatin Organization
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, W.A.; Van Steensel, B. Genome Architecture: Domain Organization of Interphase Chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Kempfer, R.; Pombo, A. Methods for Mapping 3D Chromosome Architecture. Nat. Rev. Genet. 2020, 21, 207–226. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Cavalli, G. The Role of Chromosome Domains in Shaping the Functional Genome. Cell 2015, 160, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and Function of the 3D Genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 26, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.A.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; De Santiago, I.; Lavitas, L.M.; Branco, M.R.; et al. Complex Multi-Enhancer Contacts Captured by Genome Architecture Mapping. Nature 2017, 543, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Winick-Ng, W.; Kukalev, A.; Harabula, I.; Zea-Redondo, L.; Szabó, D.; Meijer, M.; Serebreni, L.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. Cell-Type Specialization Is Encoded by Specific Chromatin Topologies. Nature 2021, 599, 684–691. [Google Scholar] [CrossRef]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-Chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757.e24. [Google Scholar] [CrossRef]
- Arrastia, M.V.; Jachowicz, J.W.; Ollikainen, N.; Curtis, M.S.; Lai, C.; Quinodoz, S.A.; Selck, D.A.; Ismagilov, R.F.; Guttman, M. Single-Cell Measurement of Higher-Order 3D Genome Organization with ScSPRITE. Nat. Biotechnol. 2021, 40, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-Resolution Chromatin Tracing Reveals Domains and Cooperative Interactions in Single Cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-Resolution Imaging Reveals Distinct Chromatin Folding for Different Epigenetic States. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef]
- Szabo, Q.; Donjon, A.; Jerković, I.; Papadopoulos, G.L.; Cheutin, T.; Bonev, B.; Nora, E.P.; Bruneau, B.G.; Bantignies, F.; Cavalli, G. Regulation of Single-Cell Genome Organization into TADs and Chromatin Nanodomains. Nat. Genet. 2020, 52, 1151–1157. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; Chattoraj, S.; Castillo, D.; Nguyen, S.C.; Nir, G.; Lioutas, A.; Hershberg, E.A.; Martins, N.M.C.; Reginato, P.L.; Hannan, M.; et al. 3D Mapping and Accelerated Super-Resolution Imaging of the Human Genome Using in Situ Sequencing. Nat. Methods 2020, 17, 822–832. [Google Scholar] [CrossRef]
- Wang, S.; Su, J.H.; Beliveau, B.J.; Bintu, B.; Moffitt, J.R.; Wu, C.T.; Zhuang, X. Spatial Organization of Chromatin Domains and Compartments in Single Chromosomes. Science 2016, 353, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, P1502–P1515.E10. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Zheng, S.; Yun, J.; Shah, S.; Pierson, N.; White, J.; Schindler, S.; Tischbirek, C.H.; Yuan, G.C.; Cai, L. Single-Cell Nuclear Architecture across Cell Types in the Mouse Brain. Science 2021, 374, 586–594. [Google Scholar] [CrossRef]
- Takei, Y.; Yun, J.; Zheng, S.; Ollikainen, N.; Pierson, N.; White, J.; Shah, S.; Thomassie, J.; Suo, S.; Eng, C.H.L.; et al. Integrated Spatial Genomics Reveals Global Architecture of Single Nuclei. Nature 2021, 590, 344–350. [Google Scholar] [CrossRef]
- Mateo, L.J.; Sinnott-Armstrong, N.; Boettiger, A.N. Tracing DNA Paths and RNA Profiles in Cultured Cells and Tissues with ORCA. Nat. Protoc. 2021, 16, 1647–1713. [Google Scholar] [CrossRef] [PubMed]
- Cardozo Gizzi, A.M.; Cattoni, D.I.; Fiche, J.B.; Espinola, S.M.; Gurgo, J.; Messina, O.; Houbron, C.; Ogiyama, Y.; Papadopoulos, G.L.; Cavalli, G.; et al. Microscopy-Based Chromosome Conformation Capture Enables Simultaneous Visualization of Genome Organization and Transcription in Intact Organisms. Mol. Cell 2019, 74, 212–222.e5. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020, 182, 1641–1659.e26. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.J.; Murphy, S.E.; Hafner, A.; Cinquini, I.S.; Walker, C.A.; Boettiger, A.N. Visualizing DNA Folding and RNA in Embryos at Single-Cell Resolution. Nature 2019, 568, 49–54. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e24. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial Partitioning of the Regulatory Landscape of the X-Inactivation Centre. Nature 2012, 5, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of Genome Folding into Topologically Associating Domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef]
- Fraser, J.; Ferrai, C.; Chiariello, A.M.; Schueler, M.; Rito, T.; Laudanno, G.; Barbieri, M.; Moore, B.L.; Kraemer, D.C.; Aitken, S.; et al. Hierarchical Folding and Reorganization of Chromosomes Are Linked to Transcriptional Changes in Cellular Differentiation. Mol. Syst. Biol. 2015, 11, 852. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, C. Chromosome Territories, Nuclear Architecture and Gene Regulation in Mammalian Cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef]
- Kubo, N.; Ishii, H.; Xiong, X.; Bianco, S.; Meitinger, F.; Hu, R.; Hocker, J.D.; Conte, M.; Gorkin, D.; Yu, M.; et al. Promoter-Proximal CTCF Binding Promotes Distal Enhancer-Dependent Gene Activation. Nat. Struct. Mol. Biol. 2021, 28, 152–161. [Google Scholar] [CrossRef]
- Furlong, E.E.M.; Levine, M. Developmental Enhancers and Chromosome Topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef]
- Huang, H.; Zhu, Q.; Jussila, A.; Han, Y.; Bintu, B.; Kern, C.; Conte, M.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. CTCF Mediates Dosage- and Sequence-Context-Dependent Transcriptional Insulation by Forming Local Chromatin Domains. Nat. Genet. 2021, 53, 1064–1074. [Google Scholar] [CrossRef]
- Zuin, J.; Roth, G.; Zhan, Y.; Cramard, J.; Redolfi, J.; Piskadlo, E.; Mach, P.; Kryzhanovska, M.; Tihanyi, G.; Kohler, H.; et al. Nonlinear Control of Transcription through Enhancer–Promoter Interactions. Nature 2022, 604, 571–577. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural Variation in the 3D Genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Valton, A.L.; Dekker, J. TAD Disruption as Oncogenic Driver. Curr. Opin. Genet. Dev. 2016, 36, 34–40. [Google Scholar] [CrossRef]
- Wang, X.; Xu, J.; Zhang, B.; Hou, Y.; Song, F.; Lyu, H.; Yue, F. Genome-Wide Detection of Enhancer-Hijacking Events from Chromatin Interaction Data in Rearranged Genomes. Nat. Methods 2021, 18, 661–668. [Google Scholar] [CrossRef]
- Finn, E.H.; Misteli, T. Molecular Basis and Biological Function of Variability in Spatial Genome Organization. Science 2019, 365, eaaw9498. [Google Scholar] [CrossRef]
- Gabriele, M.; Brandão, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.-H.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and Cohesin-Mediated Chromatin Looping Revealed by Live-Cell Imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef]
- Mach, P.; Kos, P.I.; Zhan, Y.; Cramard, J.; Gaudin, S.; Tünnermann, J.; Marchi, E.; Eglinger, J.; Zuin, J.; Kryzhanovska, M.; et al. Cohesin and CTCF Control the Dynamics of Chromosome Folding. Nat. Genet. 2022, 54, 1907–1918. [Google Scholar] [CrossRef]
- Tjong, H.; Li, W.; Kalhor, R.; Dai, C.; Hao, S.; Gong, K.; Zhou, Y.; Li, H.; Zhou, X.J.; Le Gros, M.A.; et al. Population-Based 3D Genome Structure Analysis Reveals Driving Forces in Spatial Genome Organization. Proc. Natl. Acad. Sci. USA 2016, 113, E1663–E1672. [Google Scholar] [CrossRef]
- Lesne, A.; Riposo, J.; Roger, P.; Cournac, A.; Mozziconacci, J. 3D Genome Reconstruction from Chromosomal Contacts. Nat. Methods 2014, 11, 1141–1143. [Google Scholar] [CrossRef]
- Lin, D.; Bonora, G.; Yardimci, G.G.; Noble, W.S. Computational Methods for Analyzing and Modeling Genome Structure and Organization. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 11, e1435. [Google Scholar] [CrossRef]
- Nir, G.; Farabella, I.; Pérez Estrada, C.; Ebeling, C.G.; Beliveau, B.J.; Sasaki, H.M.; Lee, S.H.; Nguyen, S.C.; McCole, R.B.; Chattoraj, S.; et al. Walking along Chromosomes with Super-Resolution Imaging, Contact Maps, and Integrative Modeling. PLoS Genet. 2018, 14, e1007872. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Baù, D.; Goodstadt, M.; Castillo, D.; Filion, G.; Marti-Renom, M.A. Automatic Analysis and 3D-Modelling of Hi-C Data Using TADbit Reveals Structural Features of the Fly Chromatin Colors. PLoS Comput. Biol. 2017, 13, e1005665. [Google Scholar] [CrossRef]
- Kim, H.J.; Yardımcı, G.G.; Bonora, G.; Ramani, V.; Liu, J.; Qiu, R.; Lee, C.; Hesson, J.; Ware, C.B.; Shendure, J.; et al. Capturing Cell Type-Specific Chromatin Compartment Patterns by Applying Topic Modeling to Single-Cell Hi-C Data. PLoS Comput. Biol. 2020, 16, e1008173. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, B. Predicting Three-Dimensional Genome Organization with Chromatin States. PLoS Comput. Biol. 2019, 15, e1007024. [Google Scholar] [CrossRef]
- Boninsegna, L.; Yildirim, A.; Polles, G.; Zhan, Y.; Quinodoz, S.A.; Finn, E.H.; Guttman, M.; Zhou, X.J.; Alber, F. Integrative Genome Modeling Platform Reveals Essentiality of Rare Contact Events in 3D Genome Organizations. Nat. Methods 2022, 19, 938–949. [Google Scholar] [CrossRef]
- Yildirim, A.; Boninsegna, L.; Zhan, Y.; Alber, F. Uncovering the Principles of Genome Folding by 3D Chromatin Modeling. Cold Spring Harb. Perspect. Biol. 2022, 14, a039693. [Google Scholar] [CrossRef]
- International Nucleome Consortium; Belokopytova, P.; Viesná, E.; Chiliński, M.; Qi, Y.; Salari, H.; Di Stefano, M.; Esposito, A.; Conte, M.; Chiariello, A.M.; et al. 3DGenBench: A Web-Server to Benchmark Computational Models for 3D Genomics. Nucleic Acids Res. 2022, 50, W4–W12. [Google Scholar] [CrossRef]
- Fudenberg, G.; Kelley, D.R.; Pollard, K.S. Predicting 3D Genome Folding from DNA Sequence with Akita. Nat. Methods 2020, 17, 1111–1117. [Google Scholar] [CrossRef]
- Zhang, S.; Chasman, D.; Knaack, S.; Roy, S. In Silico Prediction of High-Resolution Hi-C Interaction Matrices. Nat. Commun. 2019, 10, 5449. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Zhang, R.; van Schaik, T.; Zhang, L.; Sasaki, T.; Peric-Hupkes, D.; Chen, Y.; Gilbert, D.M.; van Steensel, B.; et al. SPIN Reveals Genome-Wide Landscape of Nuclear Compartmentalization. Genome Biol. 2021, 22, 36. [Google Scholar] [CrossRef]
- Schwessinger, R.; Gosden, M.; Downes, D.; Brown, R.C.; Oudelaar, A.M.; Telenius, J.; Teh, Y.W.; Lunter, G.; Hughes, J.R. DeepC: Predicting 3D Genome Folding Using Megabase-Scale Transfer Learning. Nat. Methods 2020, 17, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Thirumalai, D. From Hi-C Contact Map to Three-Dimensional Organization of Interphase Human Chromosomes. Phys. Rev. X 2021, 11, 011051. [Google Scholar] [CrossRef]
- Li, Q.; Tjong, H.; Li, X.; Gong, K.; Zhou, X.J.; Chiolo, I.; Alber, F. The Three-Dimensional Genome Organization of Drosophila Melanogaster through Data Integration. Genome Biol. 2017, 18, 145. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Paulsen, J.; Lien, T.G.; Hovig, E.; Micheletti, C. Hi-C-Constrained Physical Models of Human Chromosomes Recover Functionally-Related Properties of Genome Organization. Sci. Rep. 2016, 6, 35985. [Google Scholar] [CrossRef]
- Di Stefano, M.; Paulsen, J.; Jost, D.; Marti-Renom, M.A. 4D Nucleome Modeling. Curr. Opin. Genet. Dev. 2021, 67, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin Extrusion Explains Key Features of Loop and Domain Formation in Wild-Type and Engineered Genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Brown, J.M.; Waithe, D.; Babbs, C.; Davies, J.; Hughes, J.R.; Buckle, V.J.; Marenduzzo, D. Predicting the Three-Dimensional Folding of Cis-Regulatory Regions in Mammalian Genomes Using Bioinformatic Data and Polymer Models. Genome Biol. 2016, 17, 59. [Google Scholar] [CrossRef]
- Conte, M.; Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Esposito, A.; Nicodemi, M. Polymer Physics Indicates Chromatin Folding Variability across Single-Cells Results from State Degeneracy in Phase Separation. Nat. Commun. 2020, 11, 3289. [Google Scholar] [CrossRef]
- Conte, M.; Irani, E.; Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Nicodemi, M. Loop-Extrusion and Polymer Phase-Separation Can Co-Exist at the Single-Molecule Level to Shape Chromatin Folding. Nat. Commun. 2022, 13, 4070. [Google Scholar] [CrossRef]
- Fiorillo, L.; Bianco, S.; Esposito, A.; Conte, M.; Sciarretta, R.; Musella, F.; Chiariello, A.M. A Modern Challenge of Polymer Physics: Novel Ways to Study, Interpret, and Reconstruct Chromatin Structure. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1454. [Google Scholar] [CrossRef]
- Di Pierro, M.; Zhang, B.; Aiden, E.L.; Wolynes, P.G.; Onuchic, J.N. Transferable Model for Chromosome Architecture. Proc. Natl. Acad. Sci. USA 2016, 113, 12168–12173. [Google Scholar] [CrossRef]
- Barbieri, M.; Xie, S.Q.S.Q.; Torlai Triglia, E.; Chiariello, A.M.A.M.; Bianco, S.; De Santiago, I.; Branco, M.R.M.R.; Rueda, D.; Nicodemi, M.; Pombo, A. Active and Poised Promoter States Drive Folding of the Extended HoxB Locus in Mouse Embryonic Stem Cells. Nat. Struct. Mol. Biol. 2017, 24, 515–524. [Google Scholar] [CrossRef]
- Shi, G.; Liu, L.; Hyeon, C.; Thirumalai, D. Interphase Human Chromosome Exhibits out of Equilibrium Glassy Dynamics. Nat. Commun. 2018, 9, 3161. [Google Scholar] [CrossRef] [PubMed]
- Salari, H.; Di Stefano, M.; Jost, D. Spatial Organization of Chromosomes Leads to Heterogeneous Chromatin Motion and Drives the Liquid- or Gel-like Dynamical Behavior of Chromatin. Genome Res. 2022, 32, 28–43. [Google Scholar] [CrossRef]
- Bianco, S.; Annunziatella, C.; Andrey, G.; Chiariello, A.M.; Esposito, A.; Fiorillo, L.; Prisco, A.; Conte, M.; Campanile, R.; Nicodemi, M. Modeling Single-Molecule Conformations of the HoxD Region in Mouse Embryonic Stem and Cortical Neuronal Cells. Cell Rep. 2019, 28, 1574–1583.e4. [Google Scholar] [CrossRef]
- Plewczynski, D.; Kadlof, M. Computational Modelling of Three-Dimensional Genome Structure. Methods 2020, 181–182, 1–4. [Google Scholar] [CrossRef]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin Organization by an Interplay of Loop Extrusion and Compartmental Segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Nicodemi, M.; Pombo, A. Models of Chromosome Structure. Curr. Opin. Cell Biol. 2014, 28, 90–95. [Google Scholar] [CrossRef]
- Nicodemi, M.; Prisco, A. Thermodynamic Pathways to Genome Spatial Organization in the Cell Nucleus. Biophys. J. 2009, 96, 2168–2177. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.; Heermann, D.W. Diffusion-Driven Looping Provides a Consistent Provides a Consistent Framework for Chromatin Organization. PLoS ONE 2010, 5, e12218. [Google Scholar] [CrossRef]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.-M.L.M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of Chromatin Folding Is Captured by the Strings and Binders Switch Model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Lupiáñez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schöpflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer Physics Predicts the Effects of Structural Variants on Chromatin Architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Annunziatella, C.; Bianco, S.; Esposito, A.; Nicodemi, M. Polymer Physics of Chromosome Large-Scale 3D Organisation. Sci. Rep. 2016, 6, 29775. [Google Scholar] [CrossRef]
- Esposito, A.; Annunziatella, C.; Bianco, S.; Chiariello, A.M.; Fiorillo, L.; Nicodemi, M. Models of Polymer Physics for the Architecture of the Cell Nucleus. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1444. [Google Scholar] [CrossRef]
- Buckle, A.; Brackley, C.A.; Boyle, S.; Marenduzzo, D.; Gilbert, N. Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Mol. Cell 2018, 72, 786–797.e11. [Google Scholar] [CrossRef]
- Brackley, C.A.; Taylor, S.; Papantonis, A.; Cook, P.R.; Marenduzzo, D. Nonspecific Bridging-Induced Attraction Drives Clustering of DNA-Binding Proteins and Genome Organization. Proc. Natl. Acad. Sci. USA 2013, 110, E3605–E3611. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Fiorillo, L.; Annunziatella, C.; Esposito, A.; Musella, F.; Abraham, A.; Bianco, S.; Chiariello, A.M. Dynamic and Equilibrium Properties of Finite-Size Polymer Models of Chromosome Folding. Phys. Rev. E 2021, 104, 054402. [Google Scholar] [CrossRef] [PubMed]
- Racko, D.; Benedetti, F.; Dorier, J.; Stasiak, A. Transcription-Induced Supercoiling as the Driving Force of Chromatin Loop Extrusion during Formation of TADs in Interphase Chromosomes. Nucleic Acids Res. 2018, 46, 1648–1660. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Chiariello, A.M.; Conte, M.; Esposito, A.; Fiorillo, L.; Musella, F.; Nicodemi, M. Computational Approaches from Polymer Physics to Investigate Chromatin Folding. Curr. Opin. Cell Biol. 2020, 64, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Bianco, S.; Oudelaar, A.M.; Esposito, A.; Annunziatella, C.; Fiorillo, L.; Conte, M.; Corrado, A.; Prisco, A.; Larke, M.S.C.; et al. A Dynamic Folded Hairpin Conformation Is Associated with α-Globin Activation in Erythroid Cells. Cell Rep. 2020, 30, 2125–2135.e5. [Google Scholar] [CrossRef]
- Esposito, A.; Abraham, A.; Conte, M.; Vercellone, F.; Prisco, A.; Bianco, S.; Chiariello, A.M. The Physics of DNA Folding: Polymer Models and Phase-Separation. Polymers 2022, 14, 1918. [Google Scholar] [CrossRef]
- Nicodemi, M.; Panning, B.; Prisco, A. A Thermodynamic Switch for Chromosome Colocalization. Genetics 2008, 179, 717–721. [Google Scholar] [CrossRef]
- Banigan, E.J.; Mirny, L.A. Loop Extrusion: Theory Meets Single-Molecule Experiments. Curr. Opin. Cell Biol. 2020, 64, 124–138. [Google Scholar] [CrossRef]
- Goloborodko, A.; Marko, J.F.; Mirny, L.A. Chromosome Compaction by Active Loop Extrusion. Biophys. J. 2016, 110, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Johnson, J.; Michieletto, D.; Morozov, A.N.; Nicodemi, M.; Cook, P.R.; Marenduzzo, D. Nonequilibrium Chromosome Looping via Molecular Slip Links. Phys. Rev. Lett. 2017, 119, 138101. [Google Scholar] [CrossRef]
- Chiariello, A.M.; Bianco, S.; Esposito, A.; Fiorillo, L.; Conte, M.; Irani, E.; Musella, F.; Abraham, A.; Prisco, A.; Nicodemi, M. Physical Mechanisms of Chromatin Spatial Organization. FEBS J. 2021, 289, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Jost, D.; Carrivain, P.; Cavalli, G.; Vaillant, C. Modeling Epigenome Folding: Formation and Dynamics of Topologically Associated Chromatin Domains. Nucleic Acids Res. 2014, 42, 9553–9561. [Google Scholar] [CrossRef]
- Zhang, B.; Wolynes, P.G. Topology, Structures, and Energy Landscapes of Human Chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 6062–6067. [Google Scholar] [CrossRef]
- Esposito, A.; Bianco, S.; Chiariello, A.M.; Abraham, A.; Fiorillo, L.; Conte, M.; Campanile, R.; Nicodemi, M. Polymer Physics Reveals a Combinatorial Code Linking 3D Chromatin Architecture to 1D Chromatin States. Cell Rep. 2022, 38, 110601. [Google Scholar] [CrossRef]
- Crippa, M.; Zhan, Y.; Tiana, G. Effective Model of Loop Extrusion Predicts Chromosomal Domains. Phys. Rev. E 2020, 102, 032414. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A Pathway for Mitotic Chromosome Formation. Science 2018, 359, eaao6135. [Google Scholar] [CrossRef]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two Independent Modes of Chromatin Organization Revealed by Cohesin Removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Ganji, M.; Shaltiel, I.A.; Bisht, S.; Kim, E.; Kalichava, A.; Haering, C.H.; Dekker, C. Real-Time Imaging of DNA Loop Extrusion by Condensin. Science 2018, 360, 102–105. [Google Scholar] [CrossRef]
- Kim, Y.; Shi, Z.; Zhang, H.; Finkelstein, I.J.; Yu, H. Human Cohesin Compacts DNA by Loop Extrusion. Science 2019, 366, 1345–1349. [Google Scholar] [CrossRef]
- Kong, M.; Cutts, E.E.; Pan, D.; Beuron, F.; Kaliyappan, T.; Xue, C.; Morris, E.P.; Musacchio, A.; Vannini, A.; Greene, E.C. Human Condensin I and II Drive Extensive ATP-Dependent Compaction of Nucleosome-Bound DNA. Mol. Cell 2020, 79, 99–114.e9. [Google Scholar] [CrossRef]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.-M. DNA Loop Extrusion by Human Cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef]
- Cremer, M.; Brandstetter, K.; Maiser, A.; Rao, S.S.P.; Schmid, V.J.; Guirao-Ortiz, M.; Mitra, N.; Mamberti, S.; Klein, K.N.; Gilbert, D.M.; et al. Cohesin Depleted Cells Rebuild Functional Nuclear Compartments after Endomitosis. Nat. Commun. 2020, 11, 6146. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.; Sikorska, N.; Shchuka, V.M.; Chahar, S.; Ji, C.; Macpherson, N.N.; Moorthy, S.D.; de Kort, M.A.C.; Mullany, S.; Khader, N.; et al. Transcriptional Regulation and Chromatin Architecture Maintenance Are Decoupled Functions at the Sox2 Locus. Genes Dev. 2022, 36, 699–717. [Google Scholar] [CrossRef]
- Aljahani, A.; Hua, P.; Karpinska, M.A.; Quililan, K.; Davies, J.O.J.; Oudelaar, A.M. Analysis of Sub-Kilobase Chromatin Topology Reveals Nano-Scale Regulatory Interactions with Variable Dependence on Cohesin and CTCF. Nat. Commun. 2022, 13, 2139. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kopitchinski, N.; Eraso, A.; Awasthi, P.; Chari, R.; Rocha, P.P. High Affinity Enhancer-Promoter Interactions Can Bypass CTCF/Cohesin-Mediated Insulation and Contribute to Phenotypic Robustness. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.H.; Keeley, D.P.; et al. Phase Separation Drives Aberrant Chromatin Looping and Cancer Development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef]
- Ryu, J.K.; Bouchoux, C.; Liu, H.W.; Kim, E.; Minamino, M.; de Groot, R.; Katan, A.J.; Bonato, A.; Marenduzzo, D.; Michieletto, D.; et al. Bridging-Induced Phase Separation Induced by Cohesin SMC Protein Complexes. Sci. Adv. 2021, 7, eabe5905. [Google Scholar] [CrossRef]
- Erdel, F.; Rippe, K. Formation of Chromatin Subcompartments by Phase Separation. Biophys. J. 2018, 114, 2262–2270. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA Polymerase II Clusters Associate in Transcription-Dependent Condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging Dynamic and Selective Low-Complexity Domain Interactions That Control Gene Transcription. Science 2018, 361, eaar2555. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Manteiga, J.C.; Henninger, J.E.; Sabari, B.R.; Dall’Agnese, A.; Hannett, N.M.; Spille, J.-H.; Afeyan, L.K.; Zamudio, A.V.; Shrinivas, K.; et al. Pol II Phosphorylation Regulates a Switch between Transcriptional and Splicing Condensates. Nature 2019, 572, 543–548. [Google Scholar] [CrossRef]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid Droplet Formation by HP1α Suggests a Role for Phase Separation in Heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase Separation Drives Heterochromatin Domain Formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer Physics of Intracellular Phase Transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Liebchen, B.; Michieletto, D.; Mouvet, F.; Cook, P.R.; Marenduzzo, D. Ephemeral Protein Binding to DNA Shapes Stable Nuclear Bodies and Chromatin Domains. Biophys. J. 2017, 112, 1085–1093. [Google Scholar] [CrossRef]
- Chiariello, A.M.; Corberi, F.; Salerno, M. The Interplay between Phase Separation and Gene-Enhancer Communication: A Theoretical Study. Biophys. J. 2020, 119, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.S.; Sielaff, H.; Zhao, Z.W. Phase Separation-Mediated Chromatin Organization and Dynamics: From Imaging-Based Quantitative Characterizations to Functional Implications. Int. J. Mol. Sci. 2022, 23, 8039. [Google Scholar] [CrossRef]
- Ryu, J.K.; Hwang, D.E.; Choi, J.M. Current Understanding of Molecular Phase Separation in Chromosomes. Int. J. Mol. Sci. 2021, 22, 10736. [Google Scholar] [CrossRef]
- Brackey, C.A.; Marenduzzo, D.; Gilbert, N. Mechanistic Modeling of Chromatin Folding to Understand Function. Nat. Methods 2020, 17, 767–775. [Google Scholar] [CrossRef] [PubMed]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar] [CrossRef]
- Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Barbieri, M.; Esposito, A.; Annunziatella, C.; Conte, M.; Corrado, A.; Prisco, A.; Pombo, A.; et al. Inference of Chromosome 3D Structures from GAM Data by a Physics Computational Approach. Methods 2019, 181–182, 70–79. [Google Scholar] [CrossRef]
- Conte, M.; Esposito, A.; Fiorillo, L.; Campanile, R.; Annunziatella, C.; Corrado, A.; Chiariello, M.G.; Bianco, S.; Chiariello, A.M. Efficient Computational Implementation of Polymer Physics Models to Explore Chromatin Structure. Int. J. Parallel Emergent Distrib. Syst. 2019, 37, 91–102. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of Entangled Linear Polymer Melts: A Molecular-Dynamics Simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Rosa, A.; Everaers, R. Structure and Dynamics of Interphase Chromosomes. PLoS Comput. Biol. 2008, 4, e1000153. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Esposito, A.; Fiorillo, L.; Annunziatella, C.; Corrado, A.; Musella, F.; Sciarretta, R.; Chiariello, A.M.; Bianco, S. Hybrid Machine Learning and Polymer Physics Approach to Investigate 3D Chromatin Structure. In Proceedings of the Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer Nature: Cham, Switzerland, 2020; Volume 11997 LNCS. [Google Scholar]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Anderson, J.A.; Glaser, J.; Glotzer, S.C. HOOMD-Blue: A Python Package for High-Performance Molecular Dynamics and Hard Particle Monte Carlo Simulations. Comput. Mater. Sci. 2020, 173, 109363. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Conte, M.; Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Nicodemi, M.; Matteuzzi, T.; Vercellone, F. Polymer Models of Chromatin Imaging Data in Single Cells. Algorithms 2022, 15, 330. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Theobald, D.L. Rapid Calculation of RMSDs Using a Quaternion-Based Characteristic Polynomial. Acta Crystallogr. Sect. A Found. Crystallogr. 2005, 61, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, L.; Musella, F.; Conte, M.; Kempfer, R.; Chiariello, A.M.; Bianco, S.; Kukalev, A.; Irastorza-Azcarate, I.; Esposito, A.; Abraham, A.; et al. Comparison of the Hi-C, GAM and SPRITE Methods Using Polymer Models of Chromatin. Nat. Methods 2021, 18, 482–490. [Google Scholar] [CrossRef]
- Götz, M.; Messina, O.; Espinola, S.; Fiche, J.-B.; Nollmann, M. Multiple Parameters Shape the 3D Chromatin Structure of Single Nuclei at the Doc Locus in Drosophila. Nat. Commun. 2022, 13, 5375. [Google Scholar] [CrossRef] [PubMed]
- Kragesteen, B.K.; Spielmann, M.; Paliou, C.; Heinrich, V.; Schöpflin, R.; Esposito, A.; Annunziatella, C.; Bianco, S.; Chiariello, A.M.; Jerković, I.; et al. Dynamic 3D Chromatin Architecture Contributes to Enhancer Specificity and Limb Morphogenesis. Nat. Genet. 2018, 50, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Chiariello, A.M.; Conte, M.; Fiorillo, L.; Musella, F.; Sciarretta, R.; Bianco, S. Higher-Order Chromosome Structures Investigated by Polymer Physics in Cellular Morphogenesis and Differentiation. J. Mol. Biol. 2020, 432, 701–711. [Google Scholar] [CrossRef]
- Conte, M.; Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Esposito, A.; Musella, F.; Flora, F.; Abraham, A.; Nicodemi, M. A Polymer Physics Model to Dissect Genome Organization in Healthy and Pathological Phenotypes. In Methods in Molecular Biology; Springer Nature: Cham, Switzerland, 2022; Volume 2301. [Google Scholar]
- Esposito, A.; Bianco, S.; Fiorillo, L.; Conte, M.; Abraham, A.; Musella, F.; Nicodemi, M.; Prisco, A.; Chiariello, A.M. Polymer Models Are a Versatile Tool to Study Chromatin 3d Organization. Biochem. Soc. Trans. 2021, 49, 1675–1684. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.L.; et al. Formation of New Chromatin Domains Determines Pathogenicity of Genomic Duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative Analysis of Metazoan Chromatin Organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and Analysis of Chromatin State Dynamics in Nine Human Cell Types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; et al. Lineage-Specific Genome Architecture Links Enhancers and Non-Coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef]
- Nicodemi, M.; Coniglio, A.; Herrmann, H.J. Compaction and Force Propagation in Granular Packings. Phys. A Stat. Mech. its Appl. 1997, 240, 405–418. [Google Scholar] [CrossRef]
- Nicodemi, M.; Jensen, H.J. Creep of Superconducting Vortices in the Limit of Vanishing Temperature: A Fingerprint of off-Equilibrium Dynamics. Phys. Rev. Lett. 2001, 86, 4378–4381. [Google Scholar] [CrossRef]
- Grebenkov, D.S.; Ciamarra, M.P.; Nicodemi, M.; Coniglio, A. Flow, Ordering, and Jamming of Sheared Granular Suspensions. Phys. Rev. Lett. 2008, 100, 078001. [Google Scholar] [CrossRef]
- Oliveira, L.P.; Jensen, H.J.; Nicodemi, M.; Sibani, P. Record Dynamics and the Observed Temperature Plateau in the Magnetic Creep-Rate of Type-II Superconductors. Phys. Rev. B-Condens. Matter Mater. Phys. 2005, 71, 104526. [Google Scholar] [CrossRef]
- Tarzia, M.; De Candia, A.; Fierro, A.; Nicodemi, M.; Coniglio, A. Glass Transition in Granular Media. Europhys. Lett. 2004, 66, 531–537. [Google Scholar] [CrossRef]
- Marti-Renom, M.A. Benchmarking Experiments with Polymer Modeling. Nat. Methods 2021, 18, 456–457. [Google Scholar] [CrossRef]
- Fulco, C.P.; Nasser, J.; Jones, T.R.; Munson, G.; Bergman, D.T.; Subramanian, V.; Grossman, S.R.; Anyoha, R.; Doughty, B.R.; Patwardhan, T.A.; et al. Activity-by-Contact Model of Enhancer–Promoter Regulation from Thousands of CRISPR Perturbations. Nat. Genet. 2019, 51, 1664–1669. [Google Scholar] [CrossRef]
- Karr, J.P.; Ferrie, J.J.; Tjian, R.; Darzacq, X. The Transcription Factor Activity Gradient (TAG) Model: Contemplating a Contact-Independent Mechanism for Enhancer–Promoter Communication. Genes Dev. 2022, 36, 7–16. [Google Scholar] [CrossRef]
- Iannone, F.; Ambrosino, F.; Bracco, G.; De Rosa, M.; Funel, A.; Guarnieri, G.; Migliori, S.; Palombi, F.; Ponti, G.; Santomauro, G.; et al. CRESCO ENEA HPC Clusters: A Working Example of a Multifabric GPFS Spectrum Scale Layout. In Proceedings of the 2019 International Conference on High Performance Computing and Simulation, HPCS 2019, Dublin, Ireland, 15–19 July 2019. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, M.; Esposito, A.; Vercellone, F.; Abraham, A.; Bianco, S. Unveiling the Machinery behind Chromosome Folding by Polymer Physics Modeling. Int. J. Mol. Sci. 2023, 24, 3660. https://doi.org/10.3390/ijms24043660
Conte M, Esposito A, Vercellone F, Abraham A, Bianco S. Unveiling the Machinery behind Chromosome Folding by Polymer Physics Modeling. International Journal of Molecular Sciences. 2023; 24(4):3660. https://doi.org/10.3390/ijms24043660
Chicago/Turabian StyleConte, Mattia, Andrea Esposito, Francesca Vercellone, Alex Abraham, and Simona Bianco. 2023. "Unveiling the Machinery behind Chromosome Folding by Polymer Physics Modeling" International Journal of Molecular Sciences 24, no. 4: 3660. https://doi.org/10.3390/ijms24043660
APA StyleConte, M., Esposito, A., Vercellone, F., Abraham, A., & Bianco, S. (2023). Unveiling the Machinery behind Chromosome Folding by Polymer Physics Modeling. International Journal of Molecular Sciences, 24(4), 3660. https://doi.org/10.3390/ijms24043660