
Optimization of Biotinylated RNA or DNA Pull-Down Assays for Detection of Binding Proteins: Examples of IRP1, IRP2, HuR, AUF1, and Nrf2


Abstract
1. Introduction
2. Results
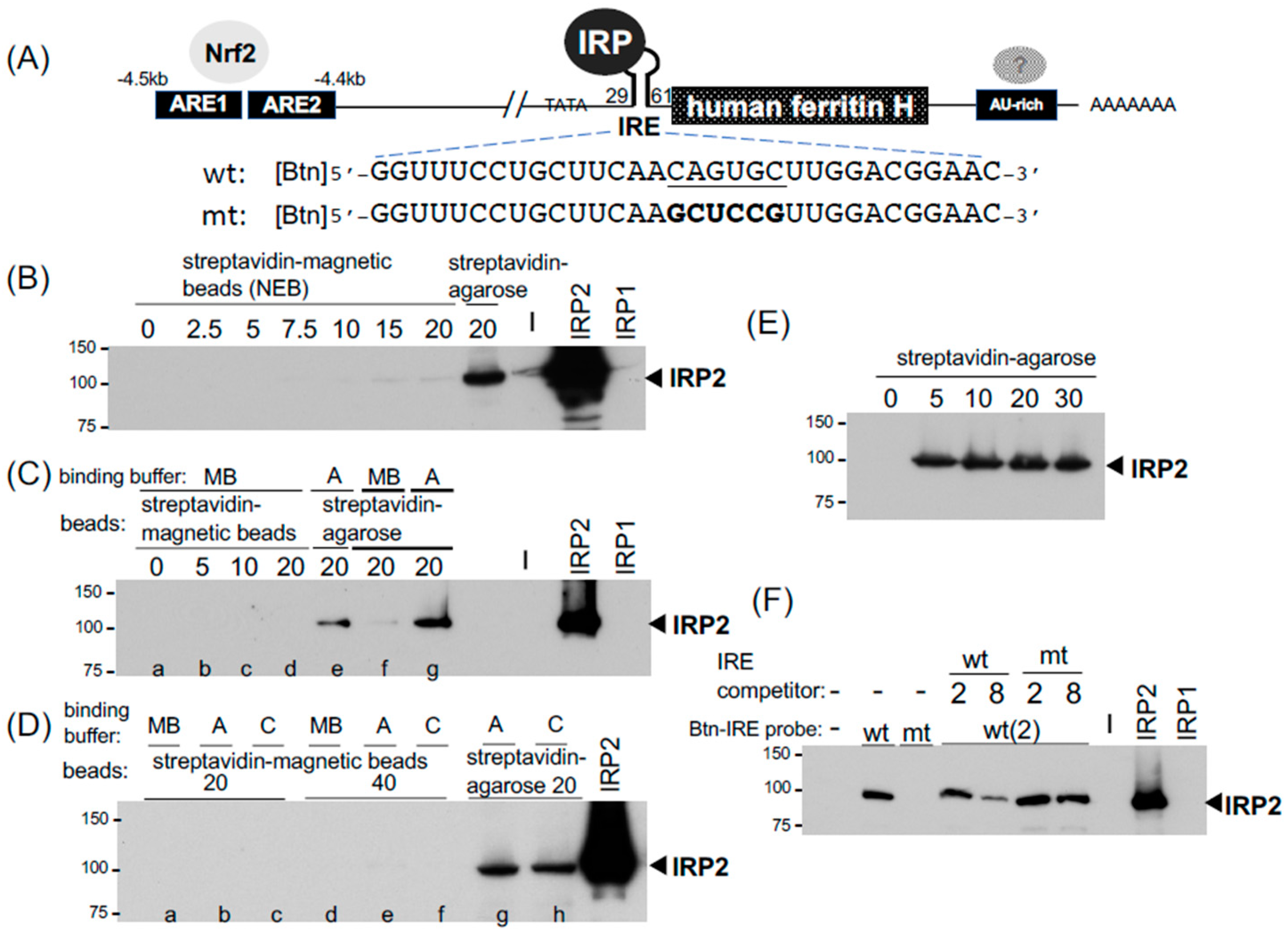
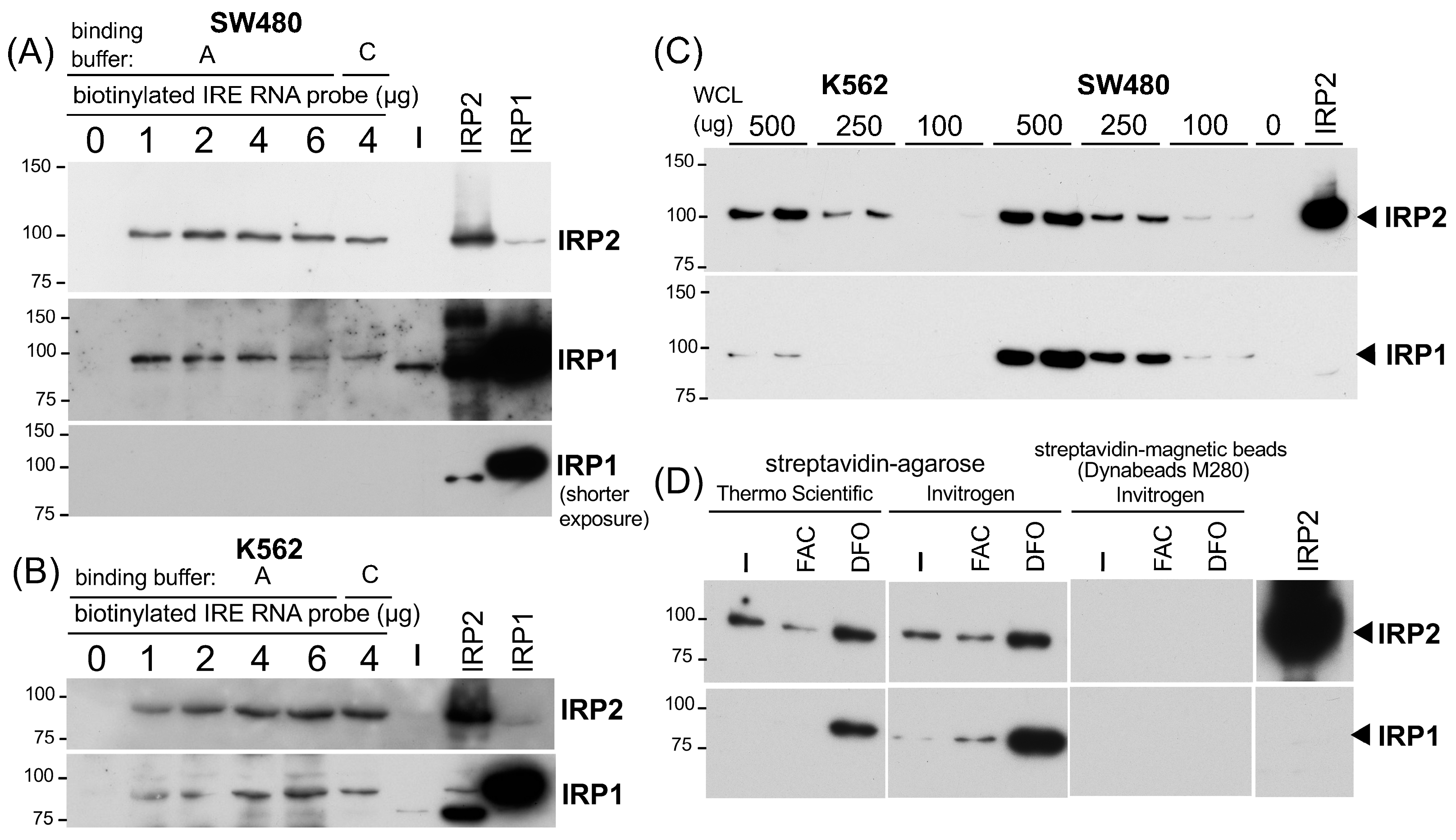
2.1. The IRP-IRE Pull-Down Set-Up and Comparison between Streptavidin-Agarose and Magnetic Beads
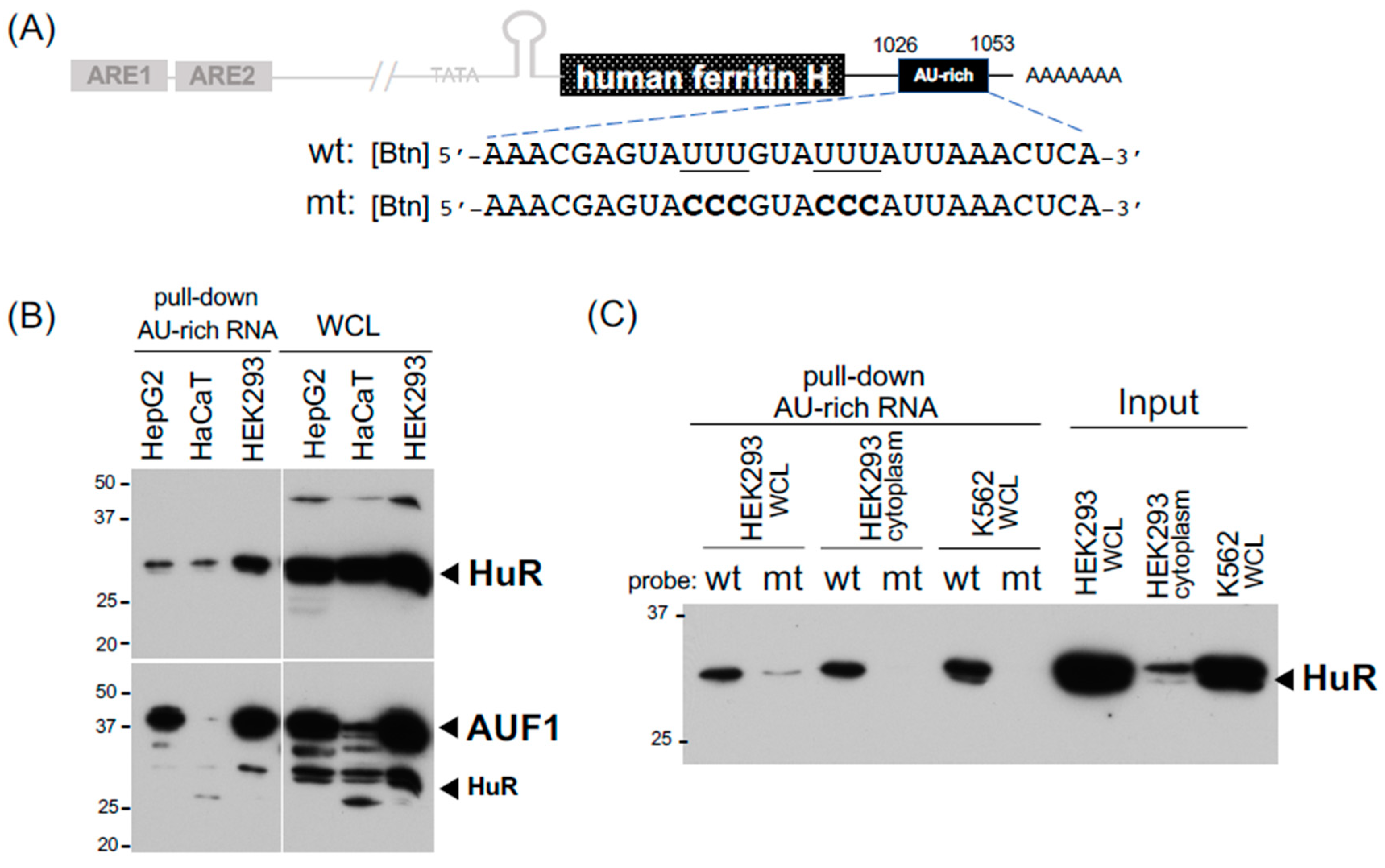
2.2. Detection of Binding Proteins to the 3′-UTR AU-Rich Element in the Human Ferritin H mRNA
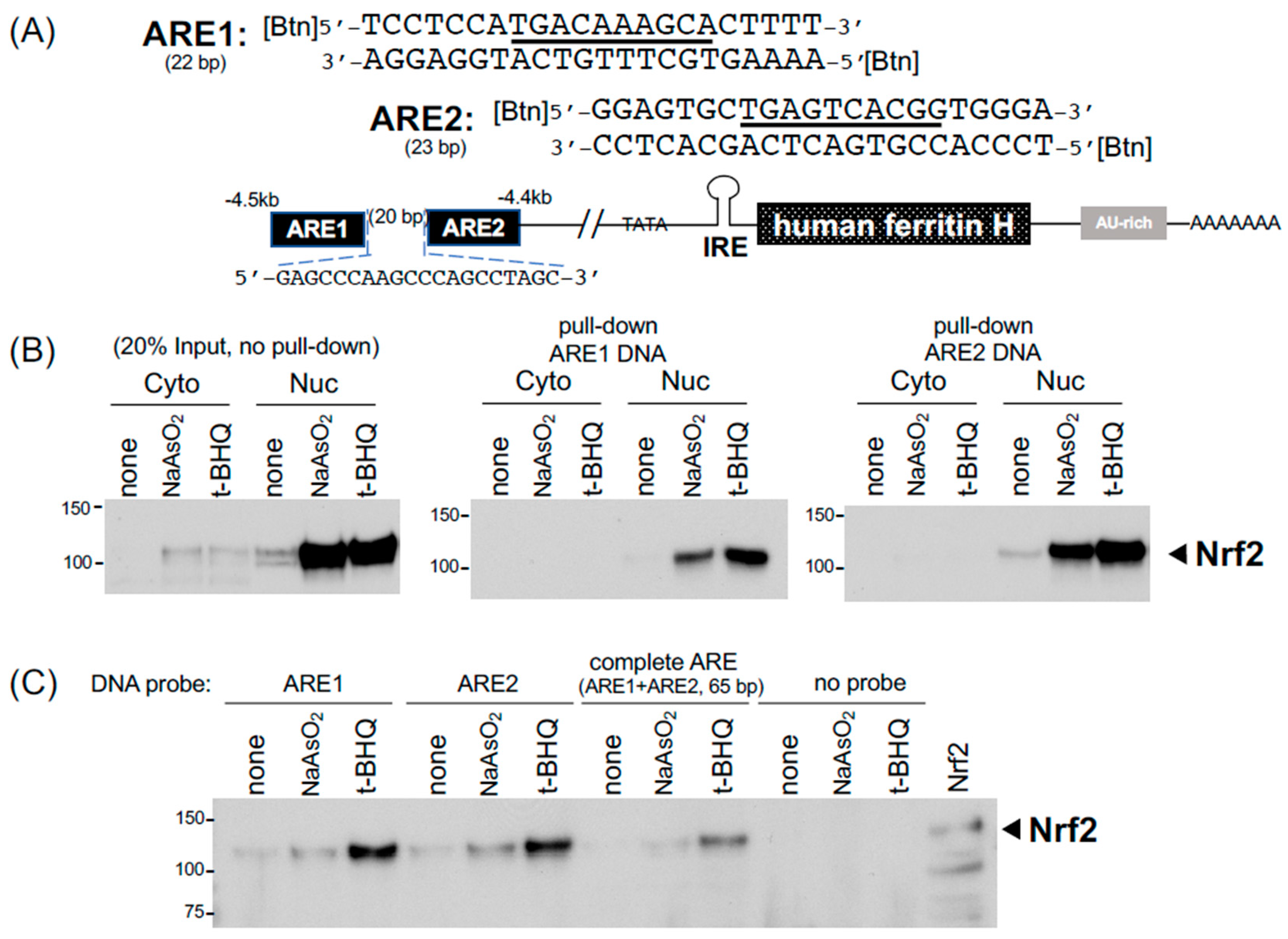
2.3. DNA Pull-Down for Detection of Nrf2 Bound to the Ferritin H Antioxidant-Responsive Element
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Chemicals, and Buffer
4.2. RNA and DNA Oligonucleotides
4.3. Streptavidin-Agarose and Streptavidin-Magnetic Beads
4.4. RNA and DNA Pull-Down Procedure
4.5. Western Blotting
4.6. Antibodies
4.7. Isolation of Cytoplasmic and Nuclear Fractions
4.8. Plasmid DNA and Transfection
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gebauer, F.; Schwarzl, T.; Valcarcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, A.; O’Malley, B.W. Mechanisms of enhancer action: The known and the unknown. Genome Biol. 2021, 22, 108. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.H. Post-transcriptional regulation of gene expression and human disease. Curr. Opin. Cell. Biol. 2018, 52, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczyk, M.; Kasprowicz, M.K.; Kasprzyk, M.E.; Wrzesinski, J. Human Long Noncoding RNA Interactome: Detection, Characterization and Function. Int. J. Mol. Sci. 2020, 21, 1027. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer. 2020, 19, 79. [Google Scholar] [CrossRef]
- Hellman, L.M.; Fried, M.G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef]
- Verweij, E.W.E.; Bosma, R.; Gao, M.; van den Bor, J.; Al Araaj, B.; de Munnik, S.M.; Ma, X.; Leurs, R.; Vischer, H.F. BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H(1) Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and beta-Arrestins. Int. J. Mol. Sci. 2022, 23, 3184. [Google Scholar] [CrossRef]
- Perez-Dominguez, S.; Caballero-Mancebo, S.; Marcuello, C.; Martinez-Julvez, M.; Medina, M.; Lostao, A. Nanomechanical Study of Enzyme: Coenzyme Complexes: Bipartite Sites in Plastidic Ferredoxin-NADP(+) Reductase for the Interaction with NADP+. Antioxidants 2022, 11, 537. [Google Scholar] [CrossRef]
- Marcuello, C.; de Miguel, R.; Lostao, A. Molecular Recognition of Proteins through Quantitative Force Maps at Single Molecule Level. Biomolecules 2022, 12, 594. [Google Scholar] [CrossRef]
- Jakobowska, I.; Becker, F.; Minguzzi, S.; Hansen, K.; Henke, B.; Epalle, N.H.; Beitz, E.; Hannus, S. Fluorescence Cross-Correlation Spectroscopy Yields True Affinity and Binding Kinetics of Plasmodium Lactate Transport Inhibitors. Pharmaceuticals 2021, 14, 757. [Google Scholar] [CrossRef]
- Wang, F.; Yao, T.; Yang, W.; Wu, P.; Liu, Y.; Yang, B. Protocol to detect nucleotide-protein interaction in vitro using a non-radioactive competitive electrophoretic mobility shift assay. STAR Protoc. 2022, 3, 101730. [Google Scholar] [CrossRef]
- Yook, J.S.; You, M.; Kim, Y.; Zhou, M.; Liu, Z.; Kim, Y.C.; Lee, J.; Chung, S. The thermogenic characteristics of adipocytes are dependent on the regulation of iron homeostasis. J. Biol. Chem. 2021, 296, 100452. [Google Scholar] [CrossRef]
- Bai, Q.; Bai, Z.; Sun, L. Detection of RNA-binding Proteins by In Vitro RNA Pull-down in Adipocyte Culture. J. Vis. Exp. 2016, 113, e54207. [Google Scholar]
- Zheng, X.; Cho, S.; Moon, H.; Loh, T.J.; Jang, H.N.; Shen, H. Detecting RNA-Protein Interaction Using End-Labeled Biotinylated RNA Oligonucleotides and Immunoblotting. Methods Mol. Biol. 2016, 1421, 35–44. [Google Scholar] [PubMed]
- Panda, A.C.; Martindale, J.L.; Gorospe, M. Affinity Pulldown of Biotinylated RNA for Detection of Protein-RNA Complexes. Bio. Protoc. 2016, 6, e2062. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C.; Matzapetakis, M.; Liu, X. Ferritins: Iron/oxygen biominerals in protein nanocages. J. Biol. Inorg. Chem. 2006, 11, 803–810. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D. Iron transport proteins: Gateways of cellular and systemic iron homeostasis. J. Biol. Chem. 2017, 292, 12735–12743. [Google Scholar] [CrossRef]
- Dunn, L.L.; Suryo Rahmanto, Y.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell. Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef]
- Theil, E.C. Iron regulatory elements (IREs): A family of mRNA non-coding sequences. Biochem. J. 1994, 304, 1. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, L.C. Iron regulatory proteins and their role in controlling iron metabolism. Met. Integr. Biometal Sci. 2015, 7, 232–243. [Google Scholar] [CrossRef]
- Tsuji, Y.; Ayaki, H.; Whitman, S.P.; Morrow, C.S.; Torti, S.V.; Torti, F.M. Coordinate transcriptional and translational regulation of ferritin inresponse to oxidative stress. Mol. Cell. Biol. 2000, 20, 5818–5827. [Google Scholar] [CrossRef]
- Tsuji, Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 2005, 24, 7567–7578. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef]
- Goossen, B.; Caughman, S.W.; Harford, J.B.; Klausner, R.D.; Hentze, M.W. Translational repression by a complex between the iron-responsive element of ferritin mRNA and its specific cytoplasmic binding protein is position-dependent in vivo. EMBO J. 1990, 9, 4127–4133. [Google Scholar] [CrossRef]
- Mullner, E.W.; Neupert, B.; Kuhn, L.C. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell 1989, 58, 373–382. [Google Scholar] [CrossRef]
- Miyazawa, M.; Bogdan, A.R.; Hashimoto, K.; Tsuji, Y. Regulation of transferrin receptor-1 mRNA by the interplay between IRE-binding proteins and miR-7/miR-141 in the 3’-IRE stem-loops. Rna 2018, 24, 468–479. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.S.; Chau, L.Y. Post-transcriptional regulation of H-ferritin mRNA. Identification of a pyrimidine-rich sequence in the 3′-untranslated region associated with message stability in human monocytic THP-1 cells. J. Biol. Chem. 1999, 274, 30209–30214. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, E.L.; Tsuji, Y. Elevated intracellular calcium increases ferritin H expression through an NFAT-independent post-transcriptional mechanism involving mRNA stabilization. Biochem. J. 2008, 411, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Mackenzie, E.L.; Hailemariam, K.; Sakamoto, K.; Tsuji, Y. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol. Cell. Biol. 2006, 26, 2845–2856. [Google Scholar] [CrossRef]
- Huang, B.W.; Ray, P.D.; Iwasaki, K.; Tsuji, Y. Transcriptional regulation of the human ferritin gene by coordinated regulation of Nrf2 and protein arginine methyltransferases PRMT1 and PRMT4. FASEB J 2013, 27, 3763–3774. [Google Scholar] [CrossRef]
- Zealy, R.W.; Wrenn, S.P.; Davila, S.; Min, K.W.; Yoon, J.H. microRNA-binding proteins: Specificity and function. Wiley Interdiscip. Rev. RNA 2017, 8, e1414. [Google Scholar] [CrossRef]
- Ciafre, S.A.; Galardi, S. microRNAs and RNA-binding proteins: A complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013, 10, 935–942. [Google Scholar] [CrossRef]
- Smith, E.S.; Whitty, E.; Yoo, B.; Moore, A.; Sempere, L.F.; Medarova, Z. Clinical Applications of Short Non-Coding RNA-Based Therapies in the Era of Precision Medicine. Cancers 2022, 14, 1588. [Google Scholar] [CrossRef]
- Fillebeen, C.; Wilkinson, N.; Pantopoulos, K. Electrophoretic mobility shift assay (EMSA) for the study of RNA-protein interactions: The IRE/IRP example. J. Vis. Exp. 2014, 94, 52230. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiu, B.; Zhang, L.; Chen, M.; Chi, W.; Li, L.; Guo, R.; Xue, J.; Yang, B.; Huang, X.; et al. Immunosuppressive lncRNA LINC00624 promotes tumor progression and therapy resistance through ADAR1 stabilization. J. Immunother. Cancer 2022, 10, e004666. [Google Scholar] [CrossRef]
- Li, Q.; Mo, W.; Ding, Y.; Ding, X. Study of lncRNA TPA in Promoting Invasion and Metastasis of Breast Cancer Mediated by TGF-beta Signaling Pathway. Front. Cell. Dev. Biol. 2021, 9, 688751. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Caughman, S.W.; Rouault, T.A.; Barriocanal, J.G.; Dancis, A.; Harford, J.B.; Klausner, R.D. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987, 238, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Hentze, M.W.; Caughman, S.W.; Harford, J.B.; Klausner, R.D. Binding of a cytosolic protein to the iron-responsive element of human ferritin messenger RNA. Science 1988, 241, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Leibold, E.A.; Munro, H.N. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy and light-subunit mRNAs. Proc. Natl. Acad. Sci. USA 1988, 85, 2171–2175. [Google Scholar] [CrossRef]
- Tsuji, Y.; Akebi, N.; Lam, T.K.; Nakabeppu, Y.; Torti, S.V.; Torti, F.M. FER-1, an enhancer of the ferritin H gene and a target of E1A-mediated transcriptional repression. Mol. Cell. Biol. 1995, 15, 5152–5164. [Google Scholar] [CrossRef]
- Misquitta, C.M.; Chen, T.; Grover, A.K. Control of protein expression through mRNA stability in calcium signalling. Cell Calcium 2006, 40, 329–346. [Google Scholar] [CrossRef]
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef]
- Tsuji, Y.; Torti, S.V.; Torti, F.M. Activation of the ferritin H enhancer, FER-1, by the cooperative action of the members of the AP1 and Sp1 transcription factor families. J. Biol. Chem. 1998, 273, 2984–2992. [Google Scholar] [CrossRef]
- Kresoja-Rakic, J.; Felley-Bosco, E. Desthiobiotin-Streptavidin-Affinity Mediated Purification of RNA-Interacting Proteins in Mesothelioma Cells. J. Vis. Exp. 2018, 134, e57516. [Google Scholar]
- Berg Luecke, L.; Gundry, R.L. Assessment of Streptavidin Bead Binding Capacity to Improve Quality of Streptavidin-based Enrichment Studies. J. Proteome Res. 2021, 20, 1153–1164. [Google Scholar] [CrossRef]
- Sui, H.; Chen, Q.; Imamichi, T. A pull-down assay using DNA/RNA-conjugated beads with a customized competition strategy: An effective approach to identify DNA/RNA binding proteins. MethodsX 2020, 7, 100890. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.; Kanhere, A. Identification of RNA-Protein Interactions Through In Vitro RNA Pull-Down Assays. Methods Mol. Biol. 2016, 1480, 99–113. [Google Scholar] [PubMed]
- Zhou, H.; Huang, X.; Shi, W.; Xu, S.; Chen, J.; Huang, K.; Wang, Y. LncRNA RP3-326I13.1 promotes cisplatin resistance in lung adenocarcinoma by binding to HSP90B and upregulating MMP13. Cell Cycle 2022, 21, 1391–1405. [Google Scholar] [CrossRef]
- Du, W.W.; Zhang, C.; Yang, W.; Yong, T.; Awan, F.M.; Yang, B.B. Identifying and Characterizing circRNA-Protein Interaction. Theranostics 2017, 7, 4183–4191. [Google Scholar] [CrossRef] [PubMed]
- Poria, D.K.; Guha, A.; Nandi, I.; Ray, P.S. RNA-binding protein HuR sequesters microRNA-21 to prevent translation repression of proinflammatory tumor suppressor gene programmed cell death 4. Oncogene 2016, 35, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, K.; Ghoshal, B.; Ghosh, S.; Chakrabarty, Y.; Shwetha, S.; Das, S.; Bhattacharyya, S.N. Reversible HuR-microRNA binding controls extracellular export of miR-122 and augments stress response. EMBO Rep. 2016, 17, 1184–1203. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Jo, M.H.; White, E.J.; De, S.; Hafner, M.; Zucconi, B.E.; Abdelmohsen, K.; Martindale, J.L.; Yang, X.; Wood, W.H., 3rd; et al. AUF1 promotes let-7b loading on Argonaute 2. Genes Dev. 2015, 29, 1599–1604. [Google Scholar] [CrossRef]
- Gemmill, D.; D’Souza, S.; Meier-Stephenson, V.; Patel, T.R. Current approaches for RNA-labelling to identify RNA-binding proteins. Biochem. Cell Biol. 2020, 98, 31–41. [Google Scholar] [CrossRef]
- Barra, J.; Leucci, E. Probing Long Non-coding RNA-Protein Interactions. Front. Mol. Biosci. 2017, 4, 45. [Google Scholar] [CrossRef]
- Hashimoto, K.; Tsuji, Y. Arsenic-Induced Activation of the Homeodomain-Interacting Protein Kinase 2 (HIPK2) to cAMP-Response Element Binding Protein (CREB) Axis. J. Mol. Biol. 2017, 429, 64–78. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER (Catalog Number) | LOT Number |
|---|---|---|---|
| Primary Antibodies | |||
| rabbit monoclonal anti-Aconitase 1 (IRP1) | Abcam | ab126595 | YI071316CS |
| rabbit monoclonal anti-IRP2 | Cell Signaling Technology | 37135 | 1 |
| rabbit monoclonal anti-HuRELAVL1 | Cell Signaling Technology | 12582 | 1 |
| rabbit monoclonal anti-AUF1/hnRNP D | Cell Signaling Technology | 12382 | 1 |
| rabbit polyclonal anti-Nrf2 | Santa Cruz Biotechnology | SC-13032x | I1311 |
| rabbit monoclonal anti-Nrf2 | Cell Signaling Technology | 12721 | 8 |
| Secondary Antibodies | |||
| horse anti-mouse IgG-peroxidase conjugated | Cell Signaling Technology | 7076S | 31 |
| goat anti-rabbit IgG-peroxidase conjugated | MilliporeSigma | AP132P | |
| Streptavidin-resins | |||
| high capacity streptavidin agarose | ThermoFisher Scientific | 20359 | QB213813, XB342916 |
| streptavidin agarose | Invitrogen | 15942-050 | 1404248 |
| streptavidin magnetic beads | New England BioLabs | S1420S | 10128395 |
| Dyna beads M-280 Streptavidin | Invitrogen | 11205D | 348667 |
| Dyna beads MyOne Streptavidin T1 | Invitrogen | 65801D | 127408950 |
| Culture Media and Supplementals | |||
| Dulbecco’s modified Eagle Medium (DMEM) | Corning | 50-003-PC | |
| Minimum Essential Medium (MEM) | Corning | 50-011-PC | |
| RPMI1640 | Corning | 50-020-PC | |
| Opti-MEM | Life Technologies | 22600-134 | |
| Penicillin Streptomycin solution, 100X | Corning | 30-002-CI | |
| Sodium pyruvate | Corning | MT25000CI | |
| Non-essential amino acid solution, 100X | Corning | 25-025-CI | |
| Trypsin, 10X | Corning | 25-054-CI | |
| Fetal Bovine Serum | Seradigm | 1400-500 | |
| Cell Lines | Source/Culture Media(containing 1x Penicillin Streptomycin) | ||
| HaCaT immortalized human keratinocyte | NE Fusenig/ DMEM high glucose (4.5 g/L) +10%FBS | ||
| HepG2 human hepatocellular carcinoma | ATCC/MEM +10% FBS | HB-8065 | |
| HEK293 immortalized human embryonic kidney cells | ATCC/MEM +10% FBS | CRL-1573 | |
| K562 human erythroleukemia | ATCC/RPMI1640, 25 mM HEPES +10%FBS | CCL-243 | |
| SW480 human colon adenocarcinoma | ATCC/DMEM high glucose (4.5 g/L) +10%FBS | CCL-228 | |
| Chemicals | |||
| Acrylamide | J.T. Baker | 4081-01 | |
| Ammonium iron(III) citrate (FAC) | Sigma-Aldrich | F5879 | |
| Ammonium persulfate | Thermo Scientific | 17874 | |
| Bis-Acrylamide | EMD Millipore | 2620 | |
| Bovine Serum Albumin (BSA) Fraction V | EMD Millipore (Calbiochem) | 2930 | |
| Bromophenol blue | Sigma-Aldrich | B-8026 | |
| Clarity Max Western ECL Substrate | Bio-Rad | 170-5062 | |
| Clarity Western ECL Substrate | Bio-Rad | 170-5061 | |
| Deferoxamine mesylate salt (DFO) | Sigma-Aldrich | D9533 | |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycine | J.T. Baker | 4057-06 | |
| 2-mercaptoethanol | EMD Millipore | 6050 | |
| Nonidet-P40 (NP40) | US Biological | N-3500 | |
| Pierce ECL Western Blotting substrate | Thermo Scientific | 32106 | |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | P-7626 | |
| Phosphatase Inhibitors (Simple Stop I) | Gold Biotechnology | GB-450-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Potassium Phosphate, Monobasic | EM Science | B10203-34 | |
| Precision Plus Protein Dual Color Standards | Bio-Rad | 161-0374 | |
| Protease Inhibitor Cocktail Set I | Calbiochem/Millipore | 539131 | |
| Protein Assay Dye Reagent Concentrate | Bio-Rad | 5000006 | |
| Skim milk powder | DIFCO, Beckton-Dickinson | 232100 | 8032502 |
| Sodium Arsenite | Fisher Scientific | S225I-100 | 51093 |
| Sodium Chloride | BDH | 7647-14-5 | |
| Sodium Deoxycholate | Sigma-Aldrich | D-6750 | |
| Sodium Dodecyl Sulfate (SDS) | Calbiochem | 7910 | |
| Sodium Phosphate, Dibasic | Fisher Scientific | BP332-500 | |
| TEMED | Fisher Scientific | BP150-100 | |
| tert-Butylhydroquinone (t-BHQ) | Sigma-Aldrich | 112941-100G | 18830PIHO |
| Tris (Hydroxymethyl) Aminomethane | J.T. Baker | 4109-06 | |
| Tween 20 | Fisher Scientific | BP337-500 | |
| Binding and Washing Buffer | |||
| Binding Buffer A | 20 mM Tris pH7.4, 300 mM KCl, 0.2 mM EDTA. 1.5 mM MgCl2, and 0.5 mM PMSF | ||
| Binding Buffer C | 20 mM Hepes, pH 7.4, 100 mM KCl, 0.5 mM EDTA, 1.5 mM MgCl2, 20% glycerol, 1 mM DTT | ||
| Dyna Beads Buffer | 5 mM Tris, pH7.5, 1M NaCl, and 0.5 mM EDTA | ||
| Magnetic Beads Buffer (NEB) | 20 mM Tris, pH7.5, 0.5M NaCl, and 1 mM EDTA | ||
| Phosphate Buffered Saline (PBS) | 137 mM NaCl, 27 mM KCl, 15 mM KH2PO4, 81 mM Na2HPO4 | ||
| Washing Buffer | 25 mM Tris, pH7.4, 15 mM NaCl, 1% NP40, and 0.5% sodium deoxycholate | ||
| Lysis Buffer (WCLs) | |||
| IP Lysis Buffer | 25 mM Tris, pH7.4, 150 mM NaCl, 1 mM EDTA, 1% NP40, and 5% glycerol | ||
| RIPA Buffer | 25 mM Tris, pH7.4, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, and 0.1% SDS | ||
| Lysis Buffer (Cytoplasm/Nucleus) | |||
| Hypotonic Buffer | 20 mM Trisl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 | ||
| Nuclear Extraction Buffer | 100 mM Tris, pH 7.4, 2 mM Na3VO4, 100 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 1 mM EGTA, 0.1% SDS, 1 mM NaF, | ||
| 0.5% deoxycholate, 20 mM Na4P2O7, and 1X proteinase inhibitor cocktail | |||
| Western Blotting Buffer | |||
| PVDF Membrane Stripping Solution | 1.5% glycine, 0.1% SDS, 1% Tween 20, pH 2.2 adjusted with HCl | ||
| SDS PAGE Running Buffer | 25 mM Tris base (no pH adjustment), 192 mM Glycine, and 0.1% SDS, | ||
| 2X SDS PAGE Sample Buffer | 62.5 mM Tris, pH 6.8, 25% Glycerol, 2% SDS, 0.01% bromophenol blue, and 5% β-mercaptoethanol | ||
| Tris Buffered Saline (TBS) | 20 mM Tris, pH 7.6 and 137 mM NaCl | ||
| Western Blotting Transfer Buffer | 25 mM Tris pH 8.3, 192 mM Glycine, and 20% methanol | ||
| plasmid DNA | |||
| pCMVSPORT6 IRP1 | Open Biosystems | ||
| pCR4 IRP2 | Open Biosystems | ||
| pRK5IRP2 | this work | ||
| pRK5Nrf2 | this work | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuji, Y. Optimization of Biotinylated RNA or DNA Pull-Down Assays for Detection of Binding Proteins: Examples of IRP1, IRP2, HuR, AUF1, and Nrf2. Int. J. Mol. Sci. 2023, 24, 3604. https://doi.org/10.3390/ijms24043604
Tsuji Y. Optimization of Biotinylated RNA or DNA Pull-Down Assays for Detection of Binding Proteins: Examples of IRP1, IRP2, HuR, AUF1, and Nrf2. International Journal of Molecular Sciences. 2023; 24(4):3604. https://doi.org/10.3390/ijms24043604
Chicago/Turabian StyleTsuji, Yoshiaki. 2023. "Optimization of Biotinylated RNA or DNA Pull-Down Assays for Detection of Binding Proteins: Examples of IRP1, IRP2, HuR, AUF1, and Nrf2" International Journal of Molecular Sciences 24, no. 4: 3604. https://doi.org/10.3390/ijms24043604
APA StyleTsuji, Y. (2023). Optimization of Biotinylated RNA or DNA Pull-Down Assays for Detection of Binding Proteins: Examples of IRP1, IRP2, HuR, AUF1, and Nrf2. International Journal of Molecular Sciences, 24(4), 3604. https://doi.org/10.3390/ijms24043604