

Expression of Epithelial and Mesenchymal Markers in Plasmatic Extracellular Vesicles as a Diagnostic Tool for Neoplastic Processes

 ,
,
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics of Patients
2.2. Characterization of Plasmatic Extracellular Vesicles
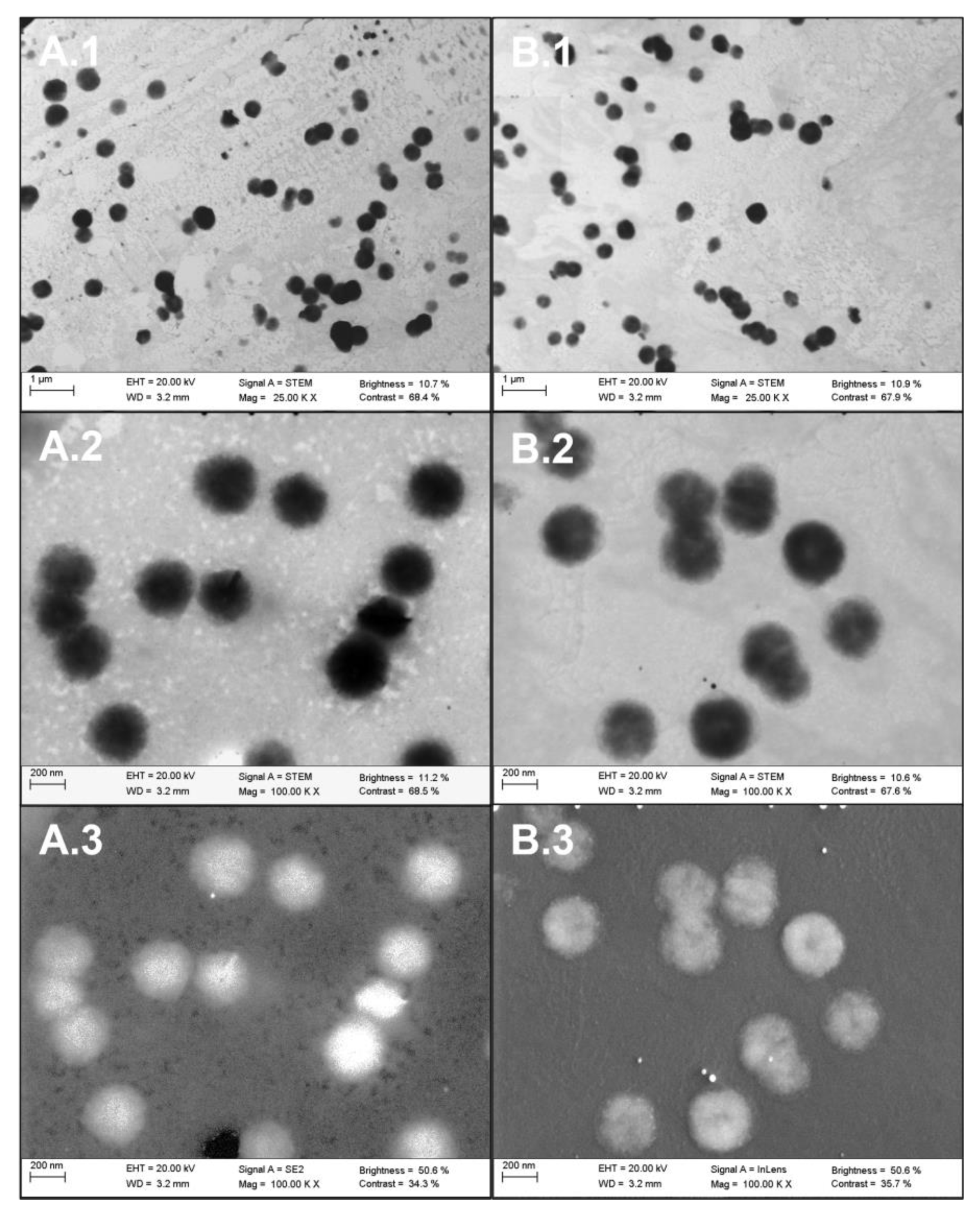
2.2.1. Visible Characterization of Extracellular Vesicles by Electron Microscopy
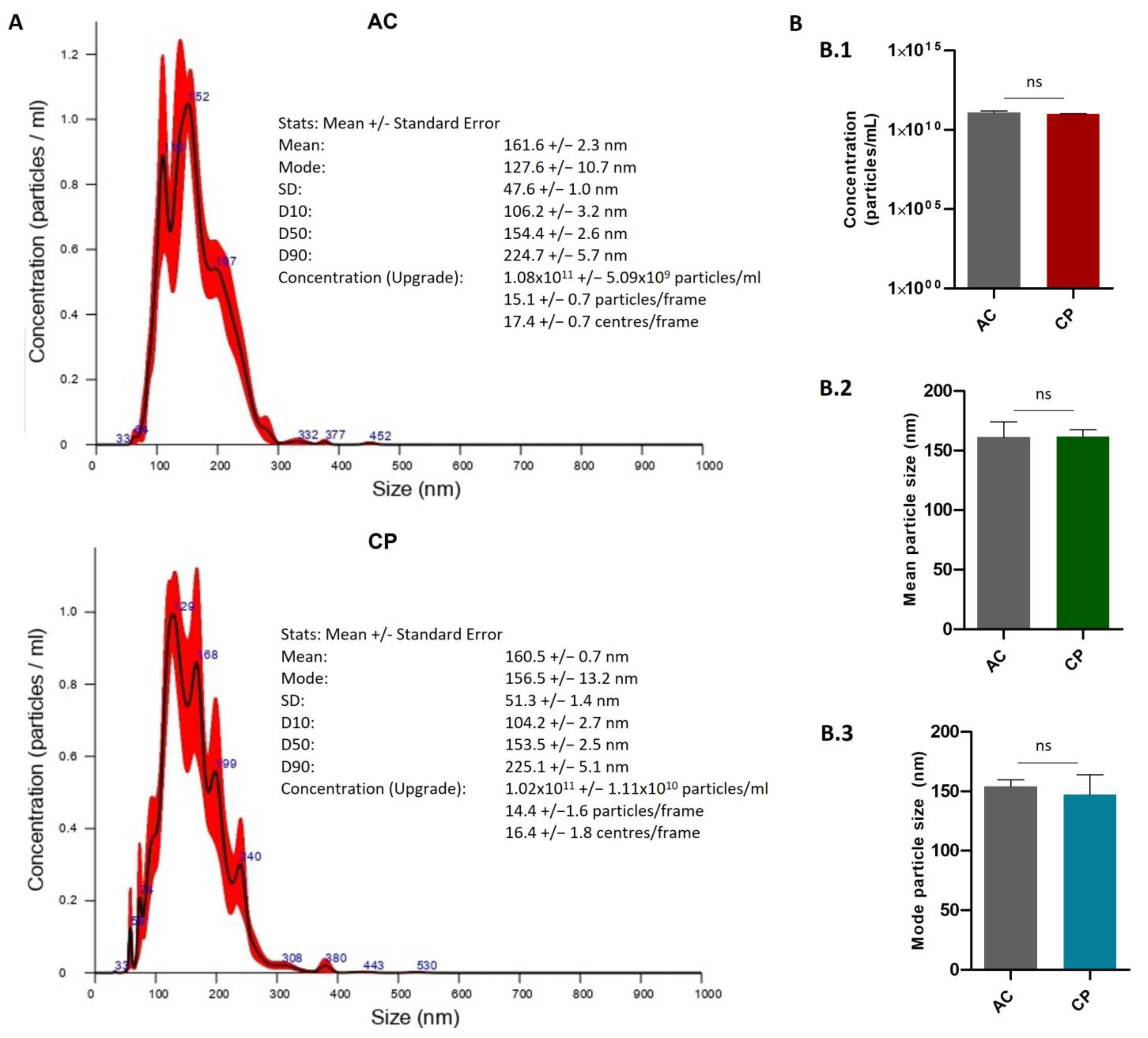
2.2.2. Quantitative Characterization of Extracellular Vesicles by Nanoparticle Tracking Analysis (NTA)
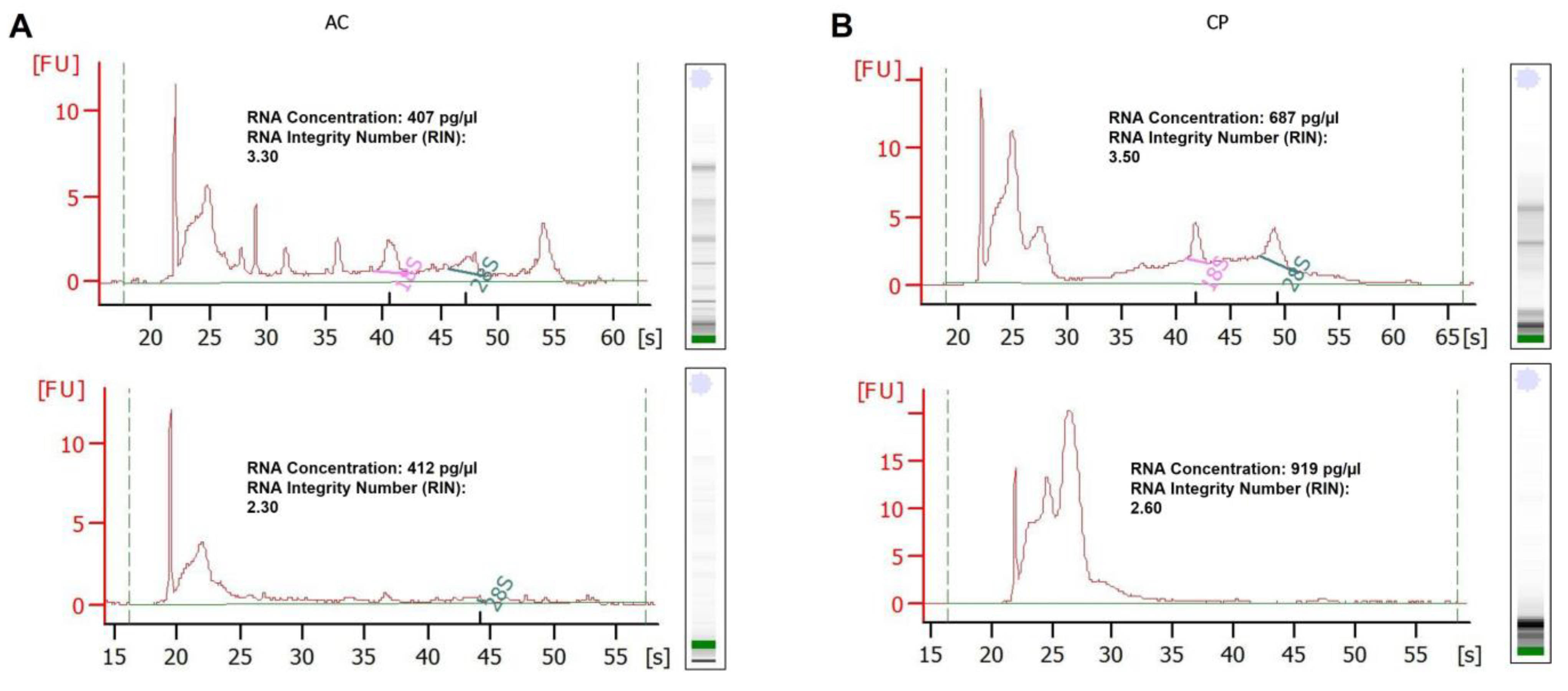
2.3. RNA Analysis of Integrity
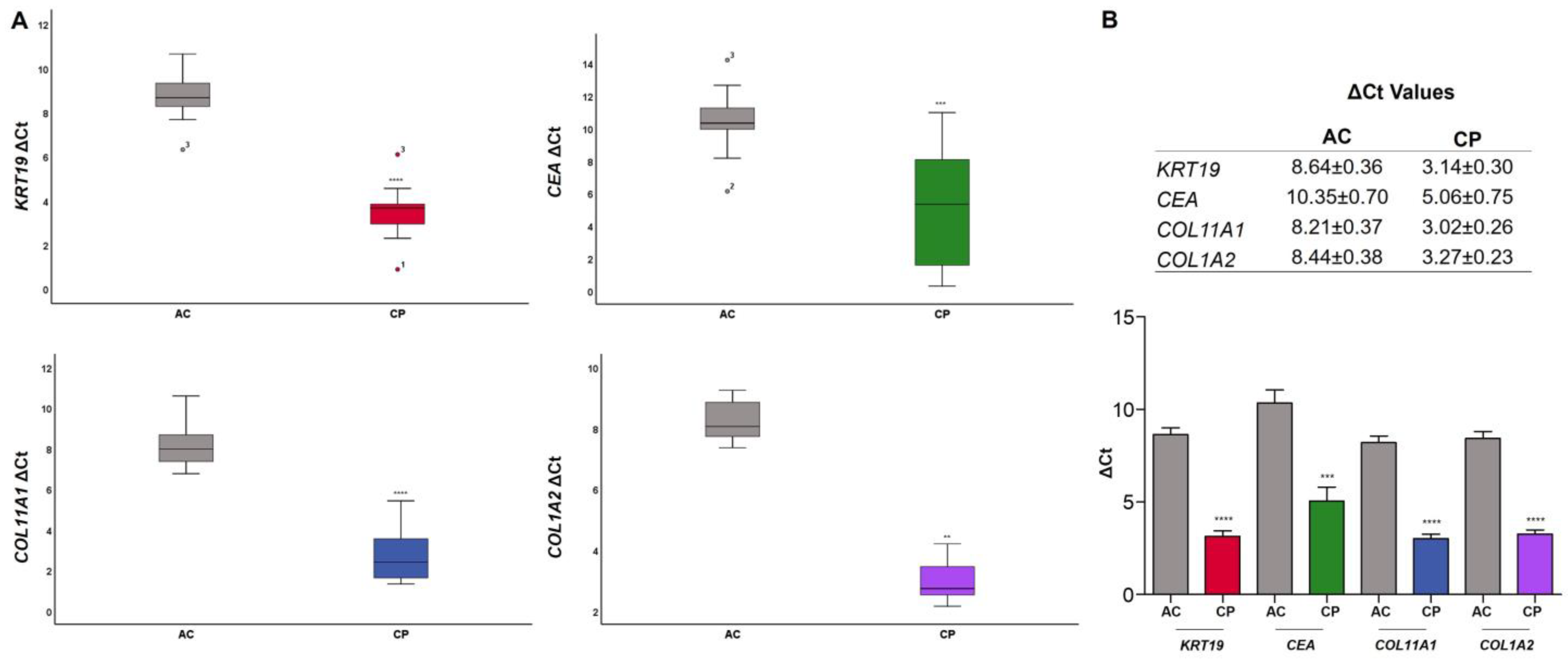
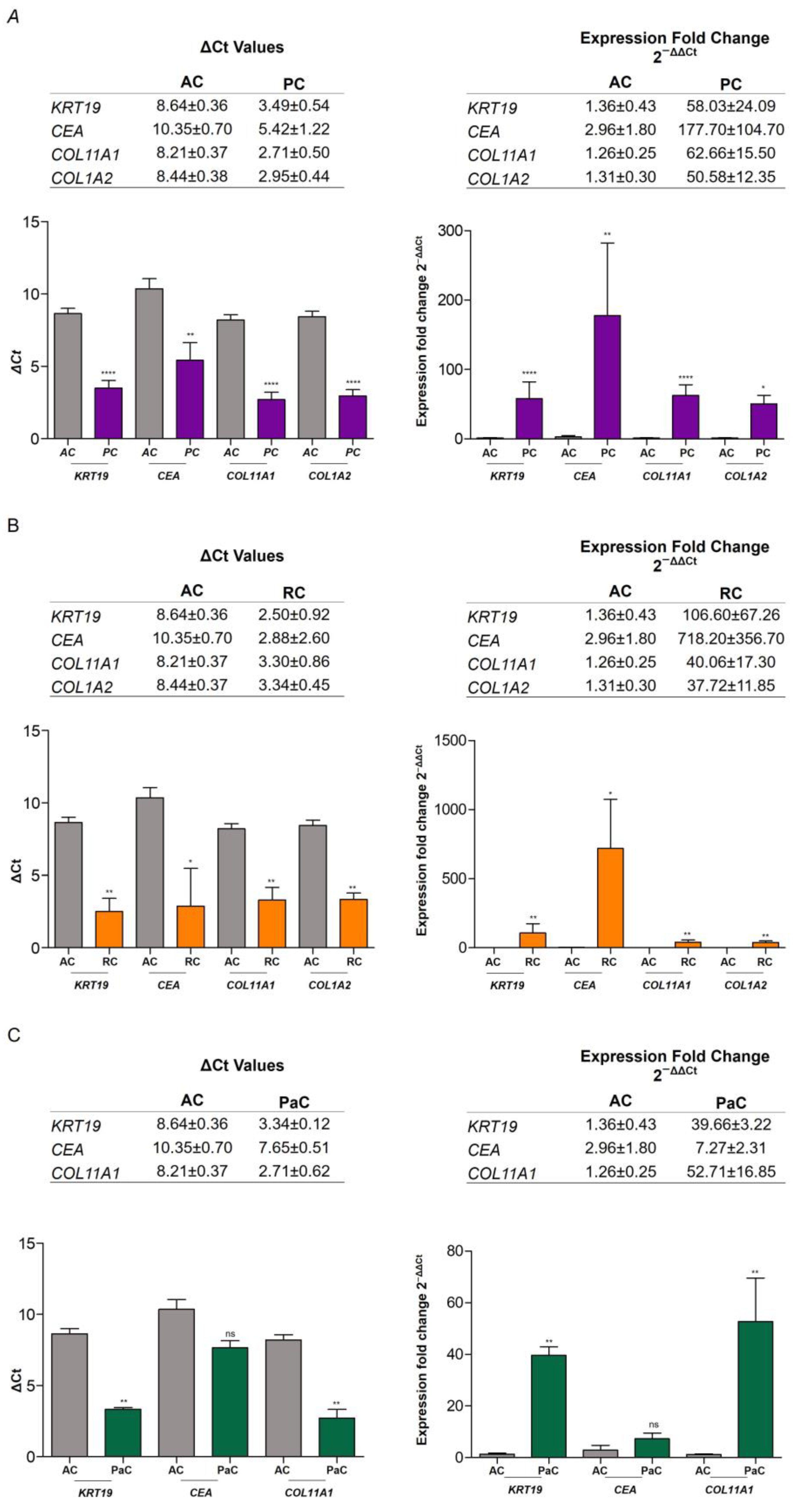
2.4. RNA Expression of Epithelial and Mesenchymal Markers
2.4.1. RNA Expression Level in Plasma of AC vs. CP
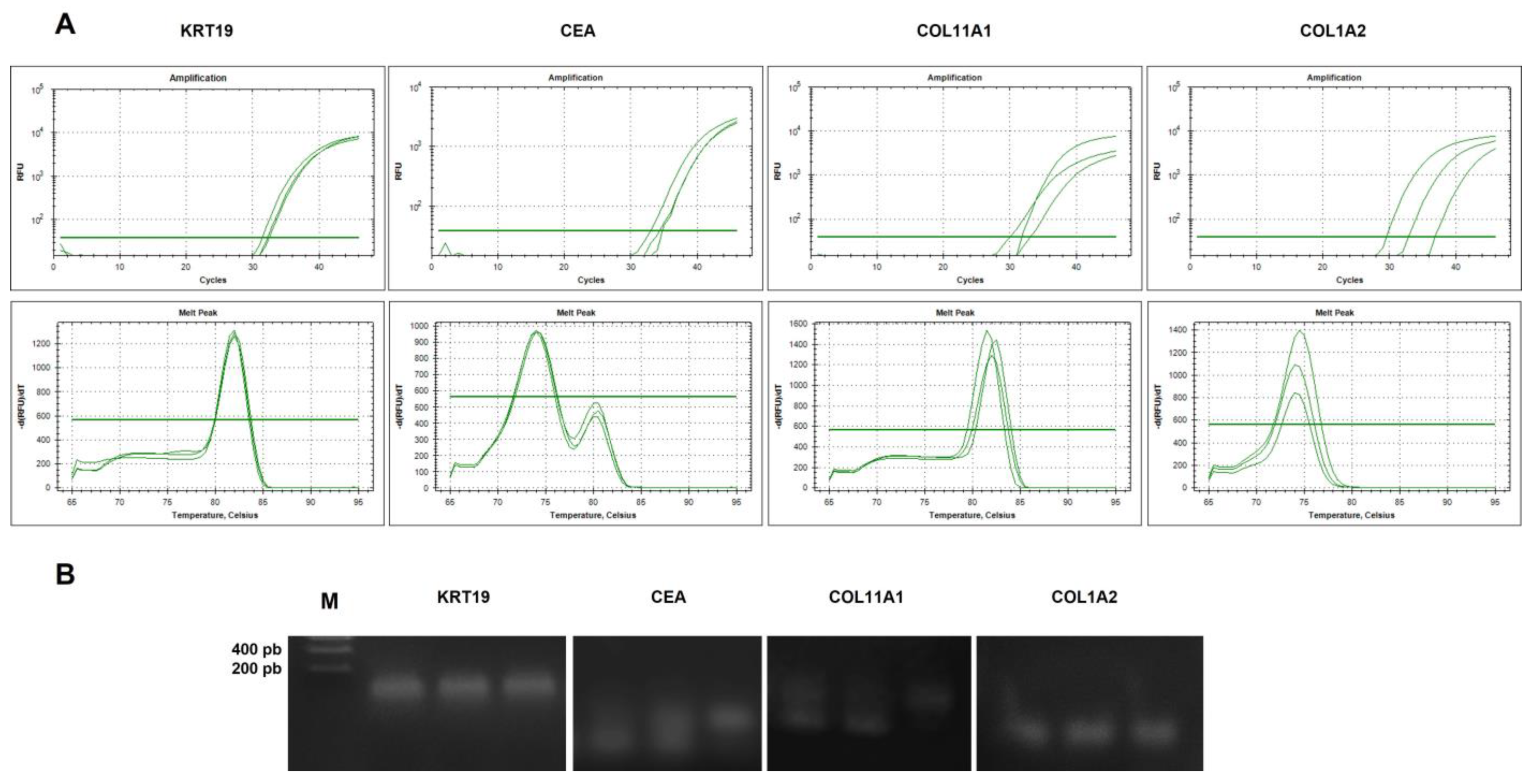
2.4.2. Analysis of Specificity of cDNA Amplicons
3. Discussion
4. Materials and Methods
4.1. Patients and Human Samples
4.2. Blood Sample Collection, Extracellular Vesicle Isolation, and RNA Purification
4.3. Extracellular Vesicle Characterization by Scanning Transmission Electron Microscopy
4.4. Extracellular Vesicle Characterization by Nanoparticle Tracking Analysis
4.5. Bioanalyzer Analysis of Total Purified RNA
4.6. Reverse Transcription and Real-Time PCR (qRT-PCR)
4.6.1. Reverse Transcription
4.6.2. qRT-PCR
4.7. Statistical Analysis
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esposito, A.; Criscitiello, C.; Locatelli, M.; Milano, M.; Curigliano, G. Liquid biopsies for solid tumors: Understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol. Ther. 2016, 157, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.M.; Lewitzky, M. Hunting for the ultimate liquid cancer biopsy—Let the TEP dance begin. Cell Commun. Signal. 2016, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Carozzo, S.; Pulliero, A.; Zhabayeva, D.; Ravetti, J.L.; Bersimbaev, R. Extracellular microRNA in liquid biopsy: Applicability in cancer diagnosis and prevention. Am. J. Cancer Res. 2016, 6, 1461–1493. [Google Scholar]
- Santarpia, M.; Liguori, A.; D’Aveni, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; Lazzari, C.; Altavilla, G.; Rosell, R. Liquid biopsy for lung cancer early detection. J. Thorac. Dis. 2018, 10, S882–S897. [Google Scholar] [CrossRef]
- Morán, T.; Felip, E.; Bosch-Barrera, J.; de Aguirre, I.; Ramirez, J.L.; Mesia, C.; Carcereny, E.; Roa, D.; Sais, E.; García, Y.; et al. Monitoring EGFR-T790M mutation in serum/plasma for prediction of response to third-generation EGFR inhibitors in patients with lung cancer. Oncotarget 2018, 9, 27074–27086. [Google Scholar] [CrossRef]
- Huang, W.-L.; Chen, Y.-L.; Yang, S.-C.; Ho, C.-L.; Wei, F.; Wong, D.T.; Su, W.C.; Lin, C.-C. Liquid biopsy genotyping in lung cancer: Ready for clinical utility? Oncotarget 2017, 8, 18590–18608. [Google Scholar] [CrossRef]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a new frontier of cancer liquid biopsy. Mol. Cancer 2022, 18, 21–56. [Google Scholar] [CrossRef]
- Cai, X.; Janku, F.; Zhan, Q.; Fan, J.-B. Accessing genetic information with liquid biopsies. Trends Genet. 2015, 31, 564–575. [Google Scholar] [CrossRef]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef]
- Möller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Joerger-Messerli, M.S.; Oppliger, B.; Spinelli, M.; Thomi, G.; di Salvo, I.; Schneider, P.; Schoeberlein, A. Extracellular Vesicles Derived from Wharton’s Jelly Mesenchymal Stem Cells Prevent and Resolve Programmed Cell Death Mediated by Perinatal Hypoxia-Ischemia in Neuronal Cells. Cell Transplant. 2018, 27, 168–180. [Google Scholar] [CrossRef]
- Raden, Y.; Shlomovitz, I.; Gerlic, M. Necroptotic extracellular vesicles—Present and future. Semin Cell Dev. Biol. 2021, 109, 106–113. [Google Scholar] [CrossRef]
- Sanwlani, R.; Gangoda, L. Role of Extracellular Vesicles in Cell Death and Inflammation. Cells 2021, 10, 2663. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Narang, P.; Shah, M.; Beljanski, V. Exosomal RNAs in diagnosis and therapies. Noncoding RNA Res. 2022, 7, 7–15. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Joshua Mitchell Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal. Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Qiao, F.; Pan, P.; Yan, J.; Sun, J.; Zong, Y.; Wu, Z.; Lu, X.; CheN, N.; Mi, R.; Ma, Y.; et al. Role of tumor-derived extracellular vesicles in cancer progression and their clinical applications. Int. J. Oncol. 2019, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Kok, V.C.; Yu, C.-C. Cancer-Derived Exosomes: Their Role in Cancer Biology and Biomarker Development. Int. J. Nanomed. 2020, 15, 8019–8036. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, X.F.; Zheng, S.; Jiang, W.-Z. Quantitative real-time RT-PCR detection for CEA, CK20 and CK19 mRNA in peripheral blood of colorectal cancer patients. J. Zhejiang Univ. Sci. B 2006, 7, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.G.; Weiss, L.M. Keratin expression in human tissues and neoplasms. Histopathology 2002, 40, 403–439. [Google Scholar] [CrossRef]
- Masai, K.; Nakagawa, K.; Yoshida, A.; Sakurai, H.; Watanabe, S.; Asamura, H.; Tsuta, K. Cytokeratin 19 expression in primary thoracic tumors and lymph node metastasis. Lung Cancer 2014, 86, 3. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, Y.; Yuan, Y.; Liu, B.; Pan, S.; Liu, Q.; Qi, X.; Zhou, H.; Dong, W.; Jia, L. The potential of exosomes derived from colorectal cancer as a biomarker. Clin. Chim. Acta 2019, 490, 186–193. [Google Scholar] [CrossRef]
- Bystricky, B.; Jurisova, S.; Karaba, M.; Minarik, G.; Benca, J.; Sedlácková, T.; Tothova, L.; Vlkova, B.; Cierna, Z.; Janega, P.; et al. Relationship Between Circulating Tumor Cells and Tissue Plasminogen Activator in Patients with Early Breast Cancer. Anticancer Res. 2017, 37, 1787–1791. [Google Scholar]
- Thomas, S.N.; Zhu, F.; Schnaar, R.L.; Alves, C.S.; Konstantopoulos, K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J. Biol. Chem. 2008, 283, 15647–15655. [Google Scholar] [CrossRef]
- Moertel, C.G.; Fleming, T.R.; Macdonald, J.S.; Haller, D.G.; Laurie, J.A.; Tangen, C. An evaluation of the carcinoembryonic antigen test for monitoring patients with resected colon cancer. JAMA 1993, 270, 943–947. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Gironella, M.; Castells, A.; Bujanda, L. Molecular markers in pancreatic cancer diagnosis. Clin. Chim. Acta 2013, 418, 22–29. [Google Scholar] [CrossRef]
- Shao, Y.; Sun, X.; He, Y.; Liu, C.; Liu, H. Elevated Levels of Serum Tumor Markers CEA and CA15-3 Are Prognostic Parameters for Different Molecular Subtypes of Breast Cancer. PLoS ONE 2015, 10, e0133830. [Google Scholar] [CrossRef]
- Grunnet, M.; Sorensen, J.B. Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer 2012, 76, 138–143. [Google Scholar] [CrossRef]
- Huang, M.-B.; Xia, M.; Gao, Z.; Zhou, H.; Liu, M.; Huang, S.; Zhen, R.; Wu, J.Y.; Roth, W.W.; Bond, V.C.; et al. Characterization of Exosomes in Plasma of Patients with Breast, Ovarian, Prostate, Hepatic, Gastric, Colon, and Pancreatic Cancers. J. Cancer Ther. 2019, 10, 382–399. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Korkaya, H.; Orsulic, S. Editorial: The tumor microenvironment: Recent advances and novel therapeutic approaches. Front. Cell Dev. Biol 2020, 8, 586176. [Google Scholar] [CrossRef]
- Kim, H.; Watkinson, J.; Varadan, V.; Anastassiou, D. Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC Med. Genom. 2010, 3, 51. [Google Scholar] [CrossRef]
- Galván, J.A.; García-Martínez, J.; Vázquez-Villa, F.; García-Ocaña, M.; García-Pravia, C.; Menéndez-Rodríguez, P.; González-del Rey, C.; Barneo-Serra, L.; de los Toyos, J.R. Validation of COL11A1/procollagen 11A1 expression in TGF-β1-activated immortalised human mesenchymal cells and in stromal cells of human colon adenocarcinoma. BMC Cancer 2014, 14, 867. [Google Scholar] [CrossRef]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Sanz-Pamplona, R.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Moreno, V. Excessive collagen turnover products are released during colorectal cancer progression and elevated in serum from metastatic colorectal cancer patients. Sci. Rep. 2016, 6, 30599. [Google Scholar] [CrossRef]
- Bager, C.L.; Willumsen, N.; Leeming, D.J.; Smith, V.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A.C. Collagen degradation products measured in serum can separate ovarian and breast cancer patients from healthy controls: A preliminary study. Cancer Biomark. 2015, 15, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Bager, C.L.; Leeming, D.J.; Smith, V.; Christiansen, C.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A.C. Serum biomarkers reflecting specific tumor tissue remodeling processes are valuable diagnostic tools for lung cancer. Cancer Med. 2014, 3, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Denève, E.; Nocca, D.; Coffy, A.; Vendrell, J.P.; Maudelonde, T.; Riethdorf, S.; Alix-Panabières, C. Circulating epithelial cells in patients with benign colon diseases. Clin. Chem. 2012, 58, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Abe, S.; Shibata, Y.; Yuki, H.; Suzuki, H.; Saito, H.; Sata, M.; Kato, S.; Tomoike, H. Elevated levels of cytokeratin 19 in the bronchoalveolar lavage fluid of patients with chronic airway inflammatory diseases—A specific marker for bronchial epithelial injury. Am. J. Respir. Crit. Care Med. 1997, 155, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Rule, A.H.; Goleski-Reilly, C.; Sachar, D.B.; Vandevoorde, J.; Janowitz, H.D. Circulating carcinoembryonic antigen (CEA): Relationship to clinical status of patients with inflammatory bowel disease. Gut 1973, 14, 880–884. [Google Scholar] [CrossRef]
- Delwiche, R.; Zamcheck, N.; Arcon, N. Carcinombryonic antigen in pancreatitis. Cancer 1973, 31, 328–330. [Google Scholar] [CrossRef]
- Maestranzi, S.; Przemioslo, R.; Mitchell, H.; Sherwood, R.A. The effect of benign and malignant liver disease on the tumour markers CA19-9 and CEA. Ann. Clin. Biochem. 1998, 35, 99–103. [Google Scholar] [CrossRef]
- Bulut, I.; Arbak, P.; Coskun, A.; Balbay, O.; Annakkaya, A.N.; Yavuz, O.; Gülcan, E. Comparison of serum CA 19.9, CA 125 and CEA levels with severity of chronic obstructive pulmonary disease. Med. Princ Pract. 2009, 18, 289–293. [Google Scholar] [CrossRef]
- Takahashi, N.; Shimada, T.; Ishibashi, Y.; Oyake, N.; Murakami, Y. Transient elevation of serum tumor markers in a patient with hypothyroidism. Am. J. Med. Sci. 2007, 333, 387–389. [Google Scholar] [CrossRef]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours, 8th ed.; Wiley Blackwell: Oxford, UK, 2017. [Google Scholar]
- Soo, C.Y.; Song, Y.; Zheng, Y.; Campbell, E.C.; Riches, A.C.; Gunn-Moore, F.; Powis, S.J. Nanoparticle tracking analysis monitors microvesicle and exosome secretion from immune cells. Immunology 2012, 136, 192–197. [Google Scholar] [CrossRef]
- Sorop, A.; Iacob, R.; Iacob, S.; Constantinescu, D.; Chitoiu, L.; Fertig, T.E.; Dinischiotu, A.; Chivu-Economescu, M.; Bacalbasa, N.; Savu, L.; et al. Plasma Small Extracellular Vesicles Derived miR-21-5p and miR-92a-3p as Potential Biomarkers for Hepatocellular Carcinoma Screening. Front. Genet. 2020, 11, 712. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Sokolenko, A.P.; Imyanitov, E.N. Molecular Diagnostics in Clinical Oncology. Front. Mol. Biosci. 2018, 5, 76. [Google Scholar] [CrossRef]
- Lassere, M.N. The Biomarker-Surrogacy Evaluation Schema: A review of the biomarker-surrogate literature and a proposal for a criterion-based, quantitative, multidimensional hierarchical levels of evidence schema for evaluating the status of biomarkers as surrogate endpoints. Stat. Methods Med. Res. 2008, 17, 303–340. [Google Scholar]
- WHO. Biomarkers in Risk Assessment: Validity and Validation; WHO: Geneva, Switzerland, 2001. [Google Scholar]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar]
- LeBleu, V.S.; Kalluri, R. Exosomes as a Multicomponent Biomarker Platform in Cancer. Trends Cancer 2020, 6, 767–774. [Google Scholar] [CrossRef]
- Morasso, C.; Ricciardi, A.; Sproviero, D.; Truffi, M.; Albasini, S.; Piccotti, F.; Sottotetti, F.; Mollica, L.; Cereda, C.; Sorrentino, L.; et al. Fast quantification of extracellular vesicles levels in early breast cancer patients by Single Molecule Detection Array (SiMoA). Breast Cancer Res. Treat. 2022, 192, 65–74. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Welton, J.; Staffurth, J.; Court, J.; Mason, M.D.; Tabi, Z.; Clayton, A. Can urinary exosomes act as treatment response markers in prostate cancer? J. Transl. Med. 2009, 7, 4. [Google Scholar] [CrossRef]
- Vitale, S.R.; Helmijr, J.A.; Gerritsen, M.; Coban, H.; van Dessel, L.F.; Beije, N.; van der Vlugt-Daane, M.; Vigneri, P.; Sieuwerts, A.M.; Dits, N.; et al. Detection of tumor-derived extracellular vesicles in plasma from patients with solid cancer. BMC Cancer 2021, 21, 315. [Google Scholar] [CrossRef]
- Eitan, E.; Green, J.; Bodogai, M.; Mode, N.A.; Bæk, R.; Jørgensen, M.M.; Freeman, D.W.; Witwer, K.W.; Zonderman, A.B.; Biragyn, A.; et al. Age-Related Changes in Plasma Extracellular Vesicle Characteristics and Internalization by Leukocytes. Sci. Rep. 2017, 7, 1342. [Google Scholar] [CrossRef]
- Hotten, N.; Byappanahalli, A.M.; Vannoy, M.; Omoniyi, V.; Evan, M.K. Influences of age, race, and sex on extracellular vesicle characteristics. Theranostics 2022, 12, 4459–4476. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Li, T.; Huang, Y.; Sun, J.; Zhu, Y.; Yang, Y.; Lu, Z.J. RNA biomarkers: Frontier of precision medicine for cancer. Noncoding RNA 2017, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Zaporozhchenko, I.A.; Ponomaryova, A.A.; Rykova, E.Y.; Laktionov, P.P. The potential of circulating cell-free RNA as a cancer biomarker: Challenges and opportunities. Expert Rev. Mol. Diagn. 2018, 18, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.P.; Diamandis, E.P.; Blasutig, I.M. The long journey of cancer biomarkers from the bench to the clinic. Clin. Chem. 2013, 59, 147–157. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with amulti-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Klein, E.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clemen, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Kogure, A.; Yoshioka, Y.; Ochiya, T. Extracellular Vesicles in Cancer Metastasis: Potential as Therapeutic Targets and Materials. Int. J. Mol. Sci. 2020, 21, 4463. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical and Pathological Features of Patients Included in This Study (n = 30) | |||
| Clinic pathological features | AC (n = 10) | CP (n = 20) | |
| Age median (range) | 40.30 (25–55) | 67.50 (41–91) | |
| Gender | |||
| Female | 7 (70.00%) | 3 (15.00%) | |
| Male | 3 (30.00%) | 17 (85.00%) | |
| Clinical and Pathological Features of Patients with Tumoral Process (n = 20) | |||
| Prostate cancer (PC) (n = 8) | Renal cancer (RC) (n = 3) | ||
| Age mean (range) | 73.25 (64–91) | Age mean (range) | 63.67 (63–65) |
| Histological type | Histological type | ||
| Adenocarcinoma | 8 | CCRC | 3 |
| Clinical stage | Clinical stage | ||
| Tis/I/II | 1 | Tis/I/II | 0 |
| III/IV | 7 | III/IV | 3 |
| Pancreatic Cancer (PaC) (n = 3) | Melanoma (Me) (n = 2) | ||
| Age mean (range) | 50.33 (41–65) | Age | 57–83 |
| Histological type | Histological type | ||
| Neuroendocrine carcinoma | 3 | Melanoma | 2 |
| Clinical stage | Clinical stage | ||
| Tis/I/II | 0 | Tis/I/II | 1 |
| III/IV | 3 | III/IV | 1 |
| Others (n = 4) | |||
| Age | Histological Type | Clinical stage | |
| Bladder carcinoma | 73 | Urotelial carcinoma | IV |
| Colorectal carcinoma | 63 | Adenocarcinoma/neuroendocrine carcinoma | I/IV |
| Ovarian cancer | 70 | Papillarly serous carcinoma | III |
| Lung cancer | 76 | Small-cell carcinoma | IV |
| Patient | Sex | RNA Expression | |||
|---|---|---|---|---|---|
| ΔCt KRT19 | ΔCt CEA | ΔCt COL11A1 | ΔCt COL1A2 | ||
| AC1 | Female | 8.27 | 8.16 | 10.59 | 9.25 |
| AC2 | Female | 8.58 | 6.12 | 9.5 | 7.36 |
| AC3 | Female | 6.31 | 14.21 | 8.27 | 8.85 |
| AC4 | Male | 7.68 | 10.32 | 6.76 | 9.35 |
| AC5 | Female | 8.67 | 10.56 | 7.54 | 10.81 |
| AC6 | Male | 9.35 | 9.95 | 8.67 | 7.73 |
| AC7 | Male | 9.32 | 11.25 | 7.36 | 6.64 |
| AC8 | Female | 8.65 | 10.32 | 8.45 | 8.27 |
| AC9 | Female | 10.65 | 9.99 | 7.25 | 7.79 |
| AC10 | Female | 8.95 | 12.65 | 7.68 | 8.32 |
| Mean | 8.64 | 10.35 | 8.21 | 8.44 | |
| SEM | 0.36 | 0.70 | 0.37 | 0.37 | |
| Patient | Stage | Sex | RNA Expression | |||
|---|---|---|---|---|---|---|
| ΔCt KRT19 | ΔCt CEA | ΔCt COL11A1 | ΔCt COL1A2 | |||
| Prostate cancer | IV | Male | 0.88 | 5.41 | 1.33 | 2.59 |
| Prostate cancer | IV | Male | 3.84 | 6.7 | 2.51 | 2.87 |
| Prostate cancer | IV | Male | 6.09 | 10.97 | 3.66 | 4.2 |
| Prostate cancer | IV | Male | 2.29 | 0.68 | 2.28 | |
| Prostate cancer | IV | Male | 3.61 | 3.92 | 5.42 | |
| Prostate cancer | IIB | Male | 3.71 | 1.57 | 1.62 | 2.15 |
| Prostate cancer | IV | Male | 2.94 | 5.24 | 1.32 | |
| Prostate cancer | IV | Male | 4.55 | 8.87 | 3.55 | |
| Renal cancer | IV | Male | 3.81 | 8.07 | 2.81 | 3.45 |
| Renal cancer | IV | Male | 2.97 | 0.28 | 2.11 | 2.52 |
| Renal cancer | III | Male | 0.73 | 0.28 | 4.98 | 4.06 |
| Pancreatic cancer | IV | Male | 3.15 | 6.84 | 2.11 | |
| Pancreatic cancer | IV | Male | 3.56 | 8.58 | 3.96 | |
| Pancreatic cancer | IV | Male | 3.31 | 7.54 | 2.07 | |
| Melanoma | 0 | Male | 1.51 | 6 | 2.99 | 3.25 |
| Melanoma | IV | Female | 3.02 | 9.42 | 4.58 | 2.78 |
| Bladder carcinoma | IV | Female | 3.18 | 1.93 | 2.98 | 3.61 |
| Colorectal carcinoma | IV | Male | 5.34 | 4.61 | 3.65 | 4.44 |
| Ovarian cancer | III | Female | 2.1 | 3.53 | 3.25 | |
| Lung cancer | IV | Male | 2.29 | 0.73 | 3.1 | |
| Mean | 3.14 | 5.06 | 3.01 | 3.27 | ||
| SEM | 0.30 | 0.75 | 0.26 | 0.23 | ||
| Primer Name | Forward Primers | Reverse Primers |
|---|---|---|
| ACTB (Housekeeping) | 5′AGCCTCGCCTTTGCCGA 3′ | 5′CTGGTGCCTGGGGCG 3′ |
| KRT19 | 5′CAGCCACTACTACACGACCATC 3′ | 5′CAAACTTGGTTCGGAAGTCATC 3′ |
| CEA | 5′AATGGGATACCGCAGCAAC 3′ | 5′GAGAGACCAGGAGAAGTTCCAGAT 3′ |
| COL11A1 | 5′AATGGAGCTGATGGACCACA 3′ | 5’TCCTTTGGGACCGCCTAC 3′ |
| COL1A2 | 5′TCAAACTGGCTGCCAGCAT 3′ | 5’CAAGAAACACGTCTGGCTAGG 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alen, B.O.; Estévez-Pérez, L.S.; Otero Alén, M.; Domínguez Hormaetxe, S.; Simón, L.; Concha, Á. Expression of Epithelial and Mesenchymal Markers in Plasmatic Extracellular Vesicles as a Diagnostic Tool for Neoplastic Processes. Int. J. Mol. Sci. 2023, 24, 3578. https://doi.org/10.3390/ijms24043578
Alen BO, Estévez-Pérez LS, Otero Alén M, Domínguez Hormaetxe S, Simón L, Concha Á. Expression of Epithelial and Mesenchymal Markers in Plasmatic Extracellular Vesicles as a Diagnostic Tool for Neoplastic Processes. International Journal of Molecular Sciences. 2023; 24(4):3578. https://doi.org/10.3390/ijms24043578
Chicago/Turabian StyleAlen, Begoña O., Lara Sofía Estévez-Pérez, María Otero Alén, Saioa Domínguez Hormaetxe, Laureano Simón, and Ángel Concha. 2023. "Expression of Epithelial and Mesenchymal Markers in Plasmatic Extracellular Vesicles as a Diagnostic Tool for Neoplastic Processes" International Journal of Molecular Sciences 24, no. 4: 3578. https://doi.org/10.3390/ijms24043578
APA StyleAlen, B. O., Estévez-Pérez, L. S., Otero Alén, M., Domínguez Hormaetxe, S., Simón, L., & Concha, Á. (2023). Expression of Epithelial and Mesenchymal Markers in Plasmatic Extracellular Vesicles as a Diagnostic Tool for Neoplastic Processes. International Journal of Molecular Sciences, 24(4), 3578. https://doi.org/10.3390/ijms24043578