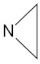
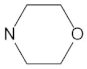
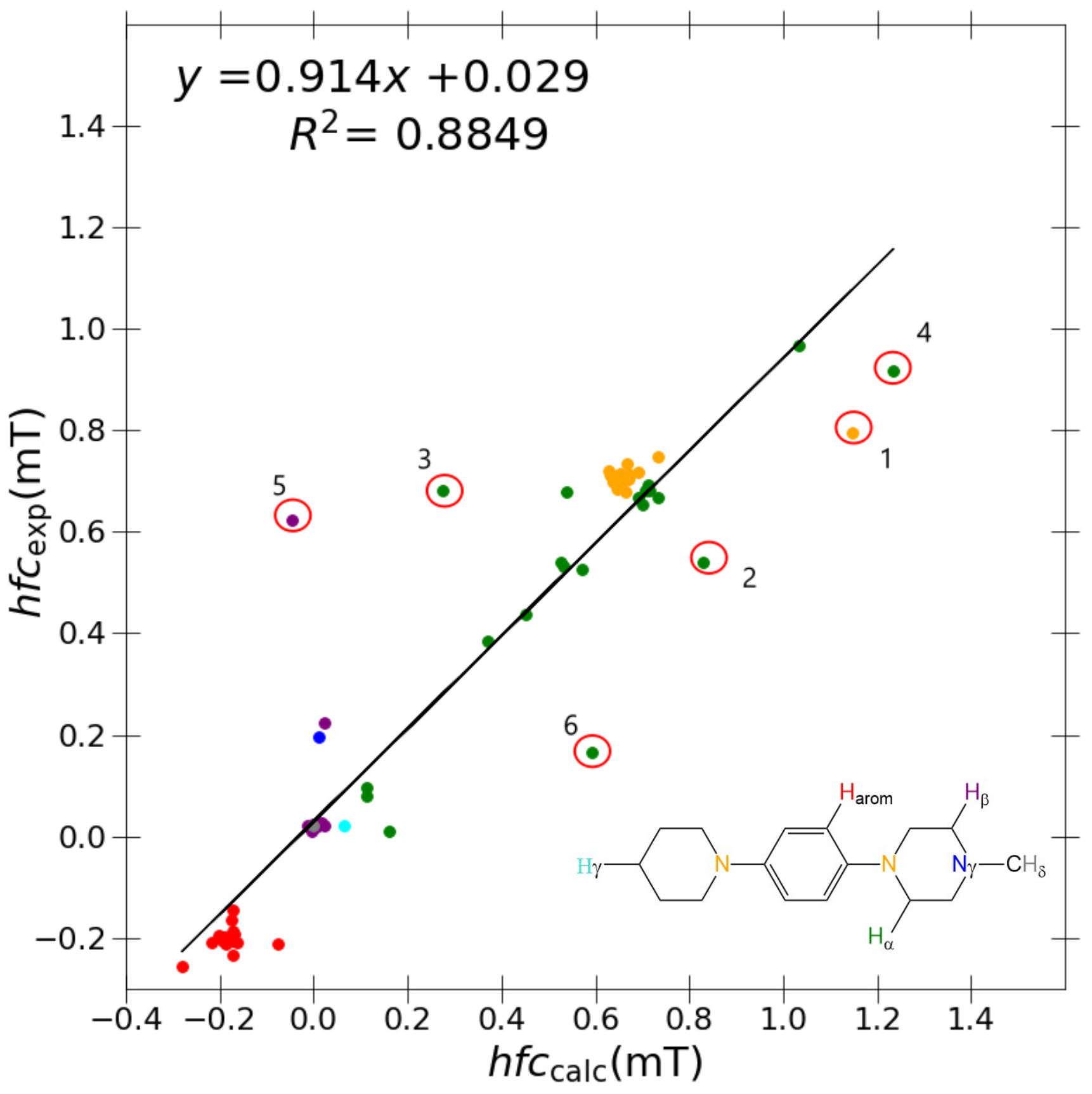
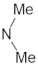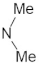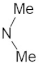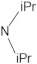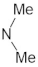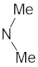A marginal improvement of ≈1% is seen in
Figure 3 using PBE0/6-31G(d,p)-J/PCM model for calculating a
and a
couplings in comparison to B3LYP/6-31G(d,p), hence promoting a greater
R value. However, outlier couplings degraded further, giving a marginally poorer correlation slope and
y-intercept as shown in
Table 4. It is difficult to conclude which results are better between each model, but based on the hierarchical importance of the
R value, the PBE0/6-31G(d,p)-J/PCM model is subjectively better.
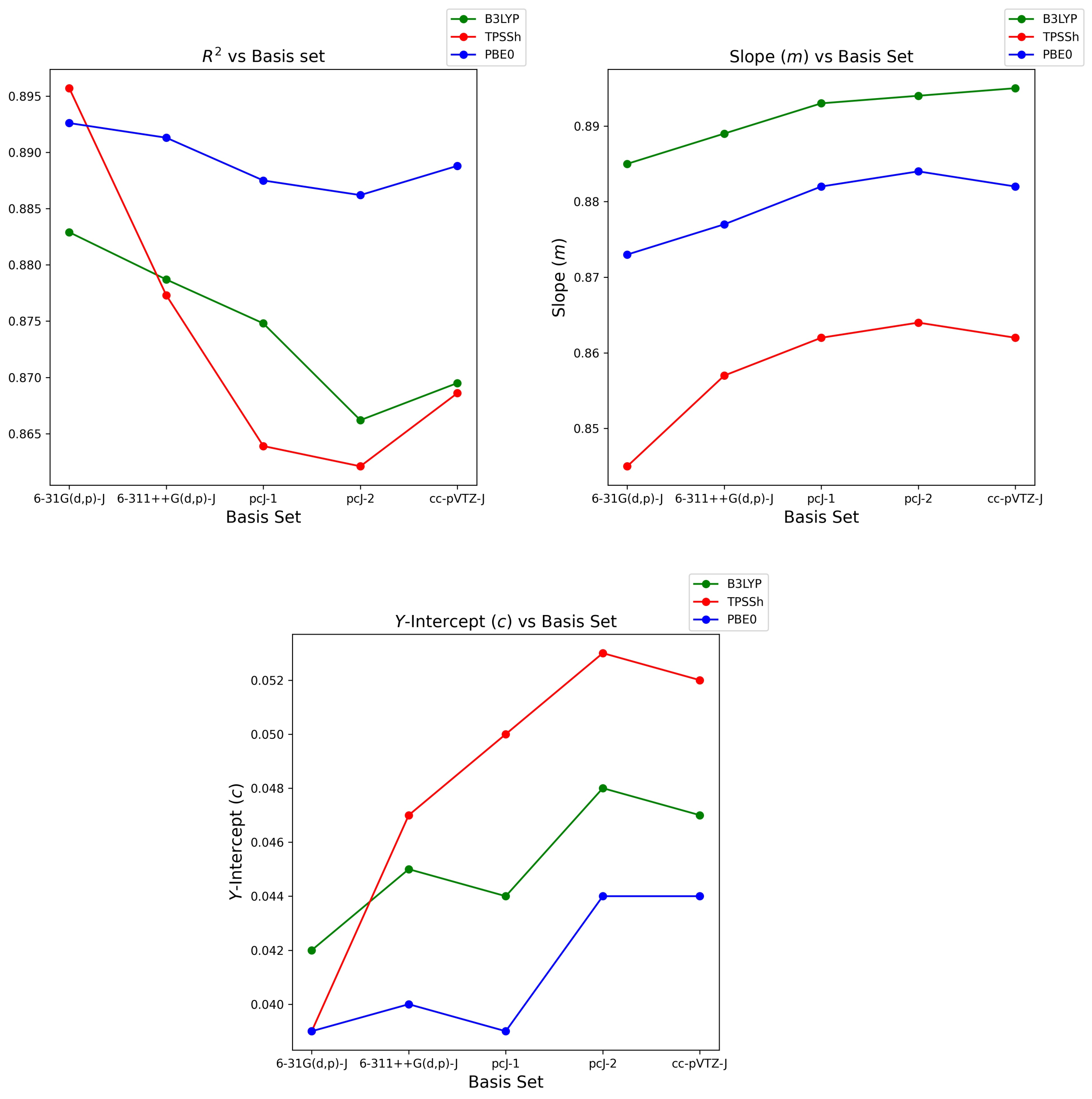
As seen in
Table 3, two opposing trends also appear here in
Table 4 as we correlate the change in basis set size with the linearly regressed parameters;
R, slope and
y-intercept across the three functionals. Trending the change in
R and
y-intercept, one observes improved values for each parameter as basis set size shrinks. Conversely, when examining the change in slope, a value closer to unity is calculated as the basis set size increases. The fact that underestimation of, namely
couplings, produces seemingly opposing trends eludes a causal explanation of these trends.
As shown, the results are not satisfactorily intuitive—and saying 6-31G(d,p) is a “better” basis set than cc-pVTZ-J is incorrect. One potential source of error within this works’ computational model lies with the implementation of an implicit solvation model, specifically the polarised continuum model (PCM). It is hard to judge how much this may effect the accuracy of the calculation but for some systems this may be considerable. Instead, one could implement an explicit solvation model to better replicate an experimental solvation environment. As shown in
Table 5, the improvement is considerable transitioning from a system absent of solvation to a system with implicit solvation, hence this more accurate model may bring potential improvements. In reality, due to the high computational cost associated with an explicit solvation model paired with this studies relatively large molecules, an explicit route may not be advised.
3.2. Examining Outliers
Common to all “J-style” basis set models tested is the constant set of detrimental outliers degrading calculated coupling quality significantly; hence, an analytic effort is applied hereafter for diagnosing the origin of such erroneous behaviour.
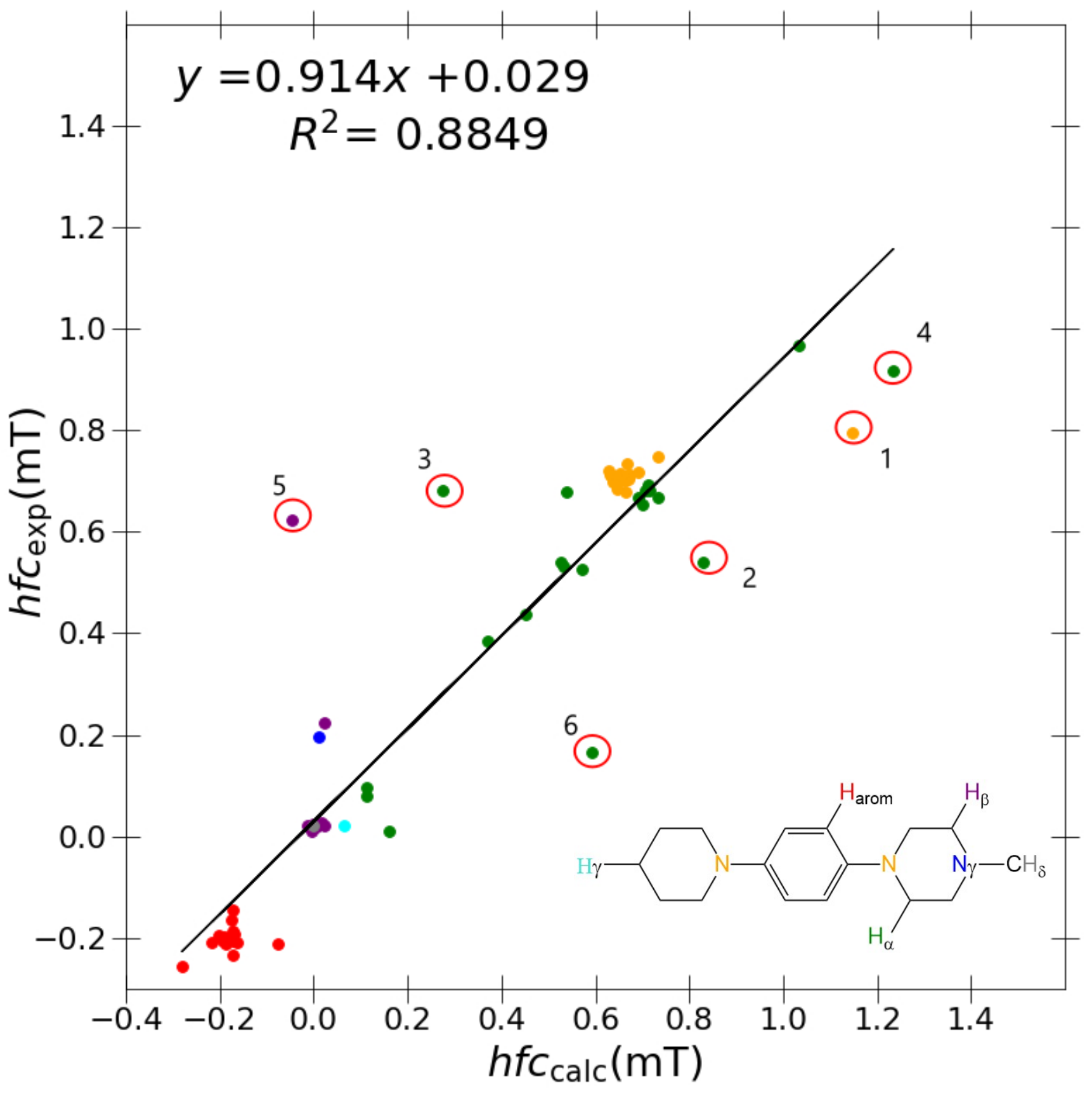
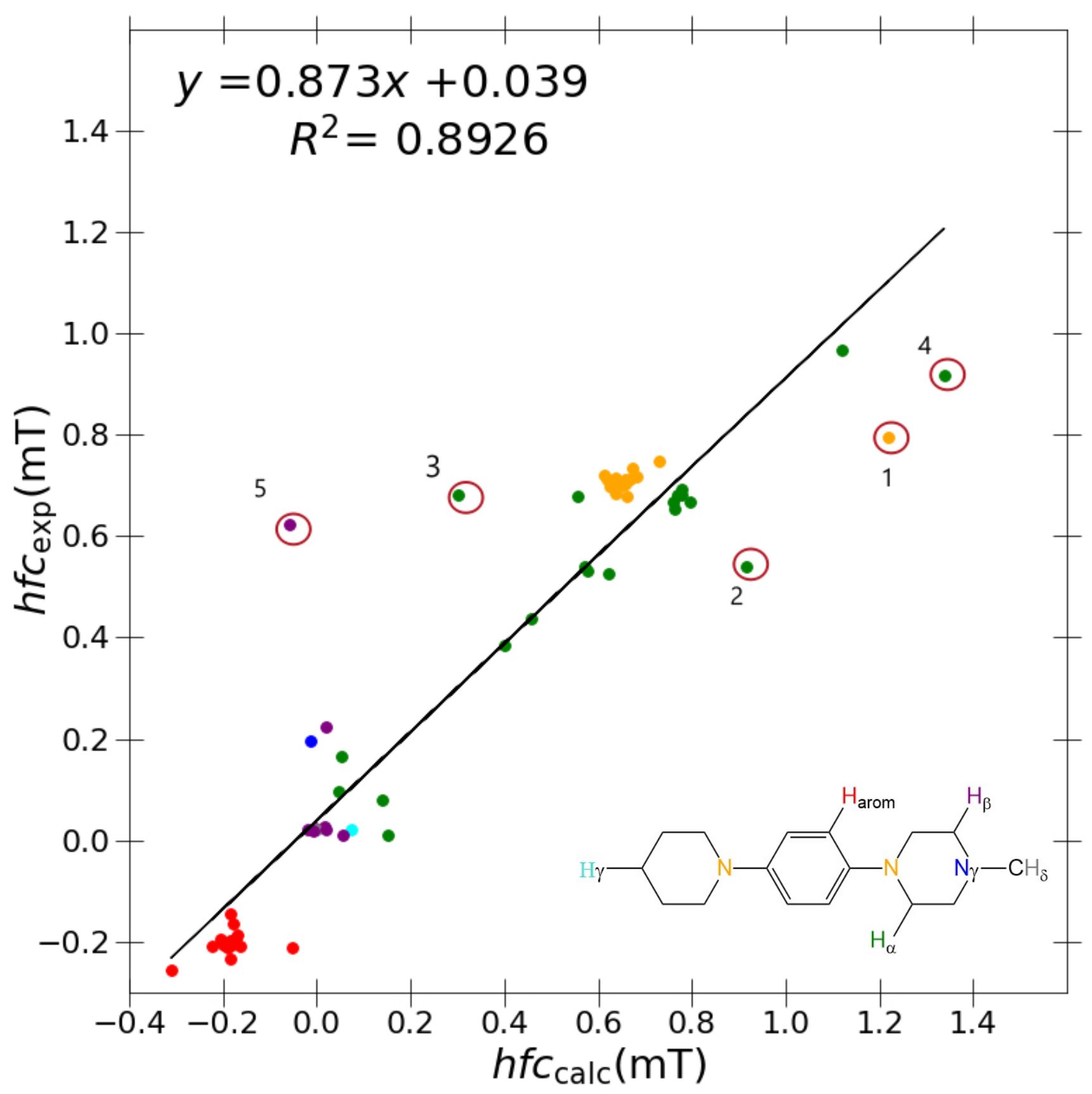
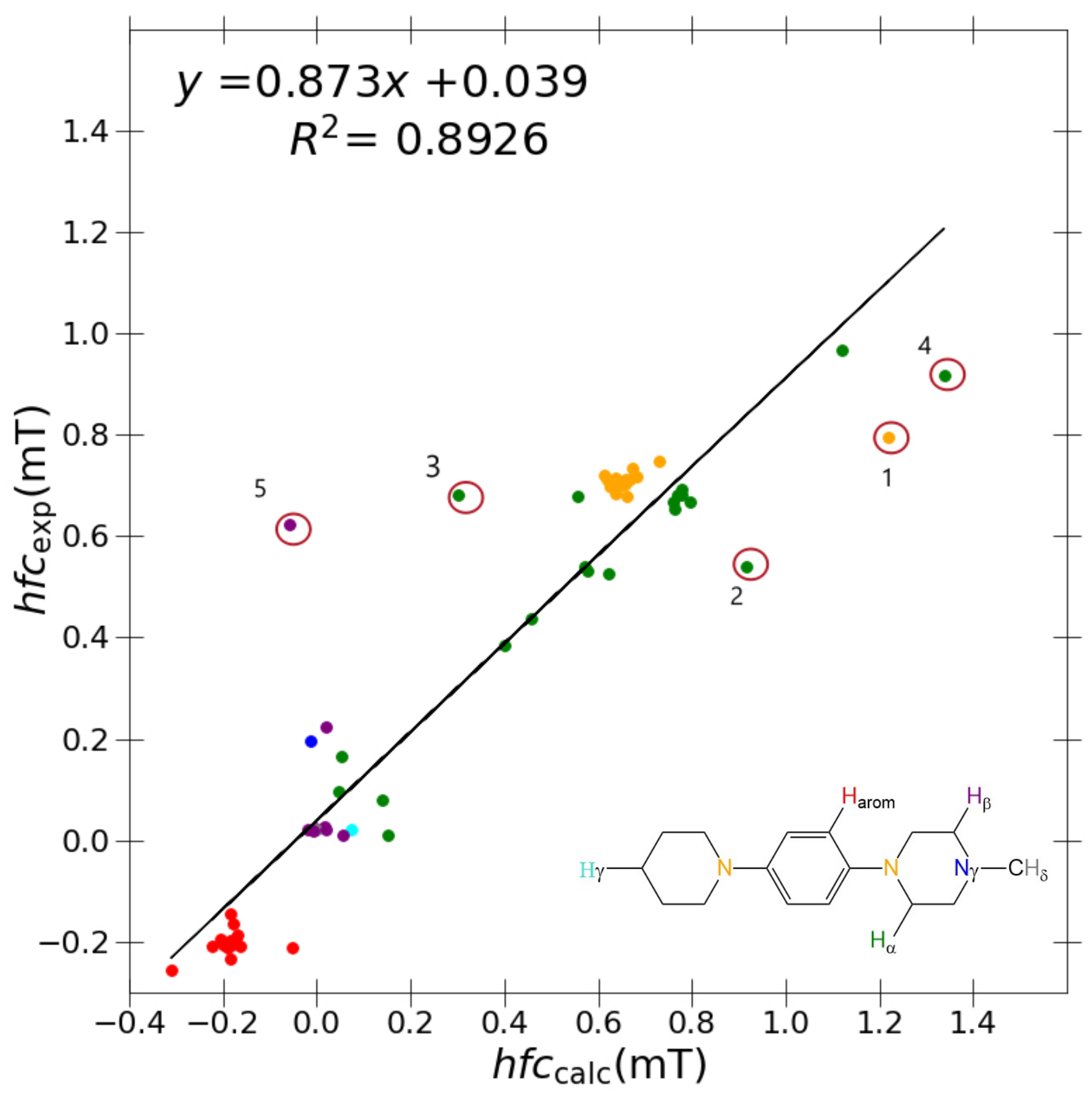
As shown in
Figure 3, problematic outliers remain, drastically depreciating the quality of the
R value (0.873), slope (0.8926), and
y-intercept (0.039) within the best-performing “J-style” system PBE0/6-31G(d,p)-J. Regardless of the basis set/functional deployed, these outliers still stem from two molecules exclusively for all “J-style” basis set calculations: DMeAzirA and DMeAzetA (shown in
Figure 5). From initial observations, this is an issue pertaining to strained fragments within each molecular structure that are not treated correctly by any hybrid-DFT functional and/or basis set tested.
From a structural standpoint, each molecule shares a highly strained N-ring that projects significant problems onto the coupling calculation for each group present within the ring—with couplings being either drastically over/under-estimated. Four out of five outliers are present within each strained fragment, with the fifth outlier attached to the non-cyclic-nitrogen in DMeAzirA. On average, couplings appeared less correlated with experimental values in DMeAzirA compared to DMeAzetA, concluding that increased straining effects have a global impact on couplings within the molecule.
In comparison, couplings within the slightly less strained DMeAzetA system are better correlated with experimental results, yet still exhibit major outlier behaviour within the collated set of calculated couplings. Other couplings that are deemed “non-outliers” in the collated set reflect that they are independent of minor perturbations of other substituents in the instances that these perturbations do not originate from a strained ring fragment.
No other N-strained ring is present in this subset of N,N,N’,N’-tetrasubstituted p-phenylenediamines, concluding that this is an issue of ineptness within the computational model’s ability to treat couplings associated with highly strained N-ring systems. From molecular structure theory, the interior angle between atoms in three/four member rings is much less than ideal (compared to similar five or six member rings), forcing an increased overlap of electron density. In strained systems, some “wiggle” room is allowed (maybe forcing a group out of the plane) to alleviate this overlap, so a different conformation may be optimal for the DFT functional and/or basis set used.
Referencing
Table 6, a clear depreciating trend in correlation parameters is shown as one transitions from three-member towards six-member ring systems when assessing individual molecular performance with PBE0/6-31G(d,p)-J. DMeAzirA exhibits the largest ring strain within the three-member ring, imposing a poor coupling correlation, whereas the four-member DMeAzetA exhibits considerably better results, albeit still performing poorly within the set of the eleven molecules tested. In contrast, BPipB showcases the best correlation, with a marginal improvement observed transitioning from a five-member ring to a six-member ring. Such results reiterates the detrimental impact that ring strain imposes on smaller (three–four) ring systems.
Of interest also was examining the change in correlation values upon removal of both problematically strained systems, shown in
Table 7. As expected, all regression parameters improved significantly for all functional and basis set combinations, with similar trends to those observed in
Table 4. Most evident is the superior correlation trend attained by the smallest basis set-6-31G(d,p)-J, largely due to suspected basis set and functional error cancellation. As the basis set size increases towards cc-pVTZ-J, experimental and calculated values deviate due to calculated couplings becoming under-calculated, generally, with PBE0 functional retaining greatest robustness towards basis set size increase.
These strained fragments were also deemed problematic in the calculation of oxidation potentials in a related article [
2], yet it was observed that a higher-level electronic structure model treated such outliers appropriately, thus improving results significantly. In this article, a similar approach is trialled to improve DFT results by using a slightly more sophisticated model such as MP2. Due to the inordinate scaling of CCSD or CCSD(T), MP2 with N
(where N is one electron basis function) scaling possesses a satisfactory middle ground between computation time and accuracy.
3.3. Computational MP2 Analysis
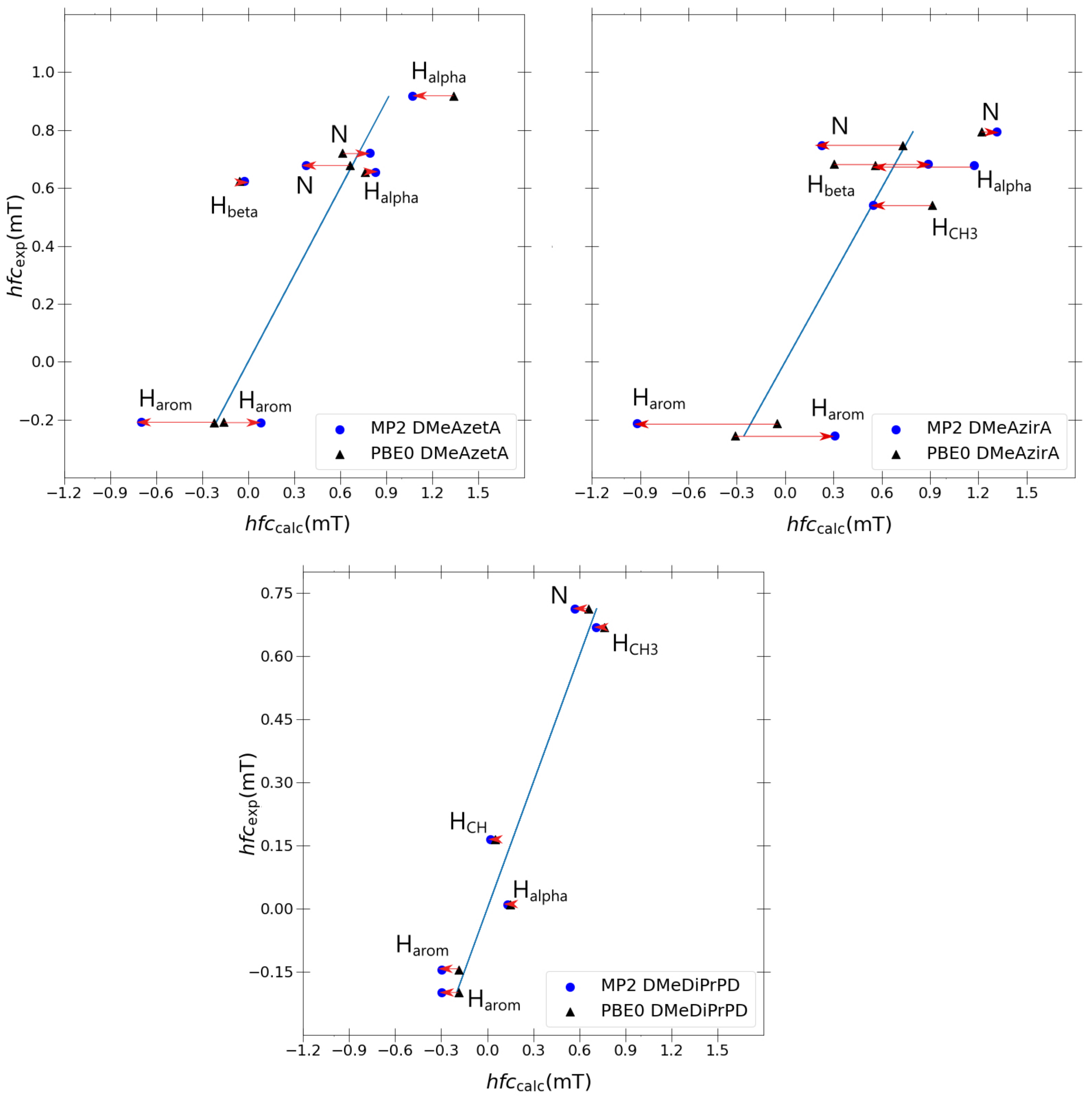
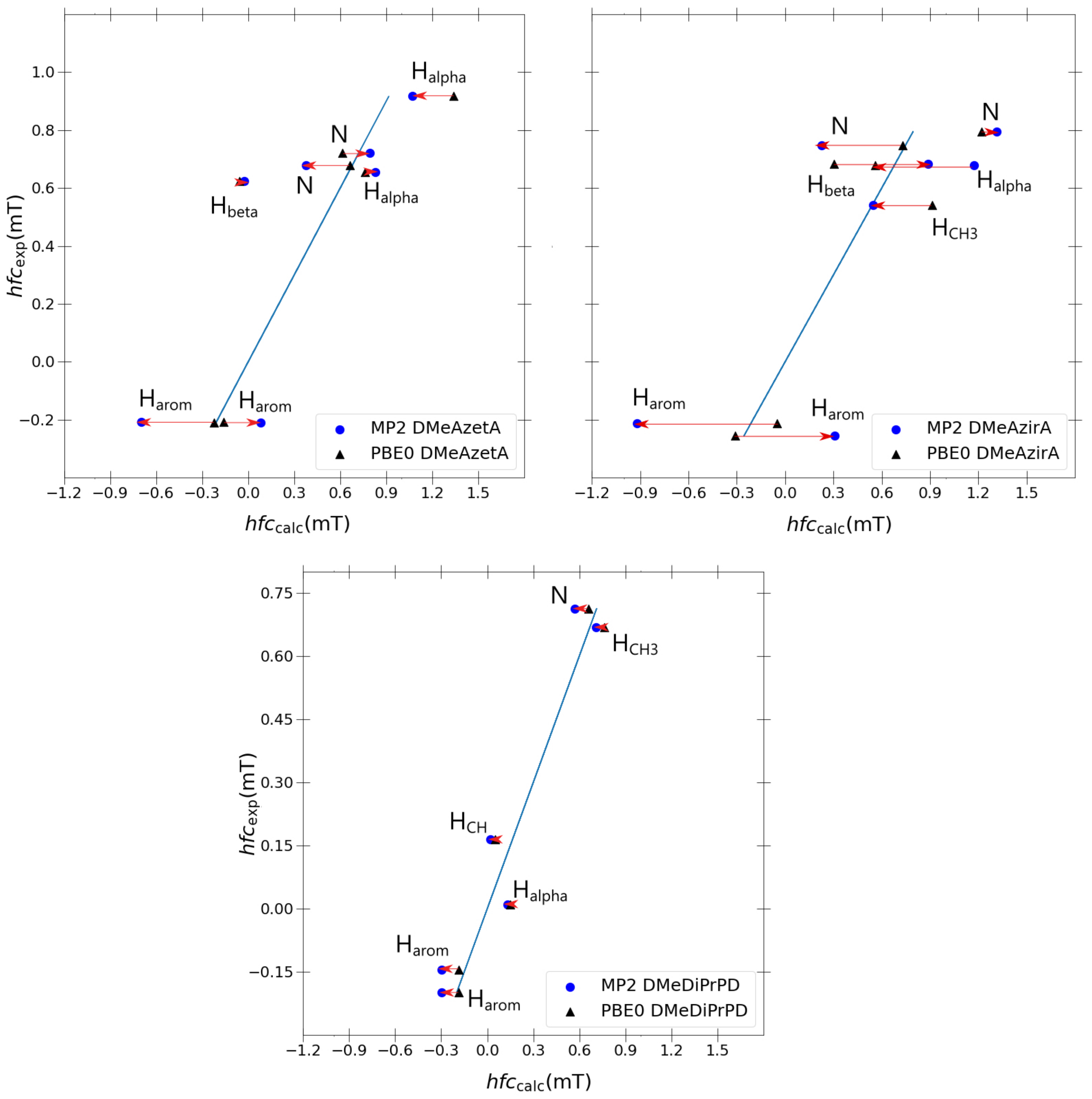
Taking inspiration from the success of the double-zeta Pople basis sets in the previous section, it was decided to re-optimise each structure using MP2/6-31G(d,p), utilising a J-augmented 6-31G(d,p)-J basis set for each MP2 single-point hfcc calculation. Three molecules were chosen for sampling the quality of MP2, namely the two outlier molecules (DMeAzetA and DMeAzirA) and one molecule that exhibited an excellent correlation with experimental results (DMeDiPrPD). A 1:1 comparison of each coupling with the blue ideal fitting line (
y =
x) is shown in
Figure 6 to observe the improvement, if any, that is gained transitioning from PBE0 to MP2.
Comparing changes transitioning from PBE0 to MP2 for each molecule in
Figure 6, it is apparent that the discrepancies between experimental and calculated values increase significantly as we compare the well-behaved DMeDiPrPD system with the ill-behaved DMeAzetA and DMeAzirA systems. This behaviour is quite possibly a direct effect of the strained rings—most evidently seen in DMeAzirA and reducing in effect as we transition to DMeAzetA—with DMeDiPrPD behaving appropriately due to an absence of such straining effects. Such large changes suggest that strained compounds are more sensitive to how electron correlation is treated compared to non-strained compounds, as one might have expected. However, the large changes attained from going to MP2 do not improve on the quality of correlation as was the case in the previous study of redox potentials related to such
N,N,N’,N’-tetrasubstituted
p-phenylenediamines [
2].
Regarding individual coupling group comparisons, MP2 always gives poorer couplings in comparison to PBE0, with a similar trend observed for a couplings (except for one a coupling in DMeAzetA). On average, couplings are split 50:50 between each model, with and couplings actually improving from PBE0 to MP2. One coupling was present in favour of PBE0. From this, one can conclude that particular coupling groups are degraded when transitioning from PBE0 to MP2, but others may or may not improve.
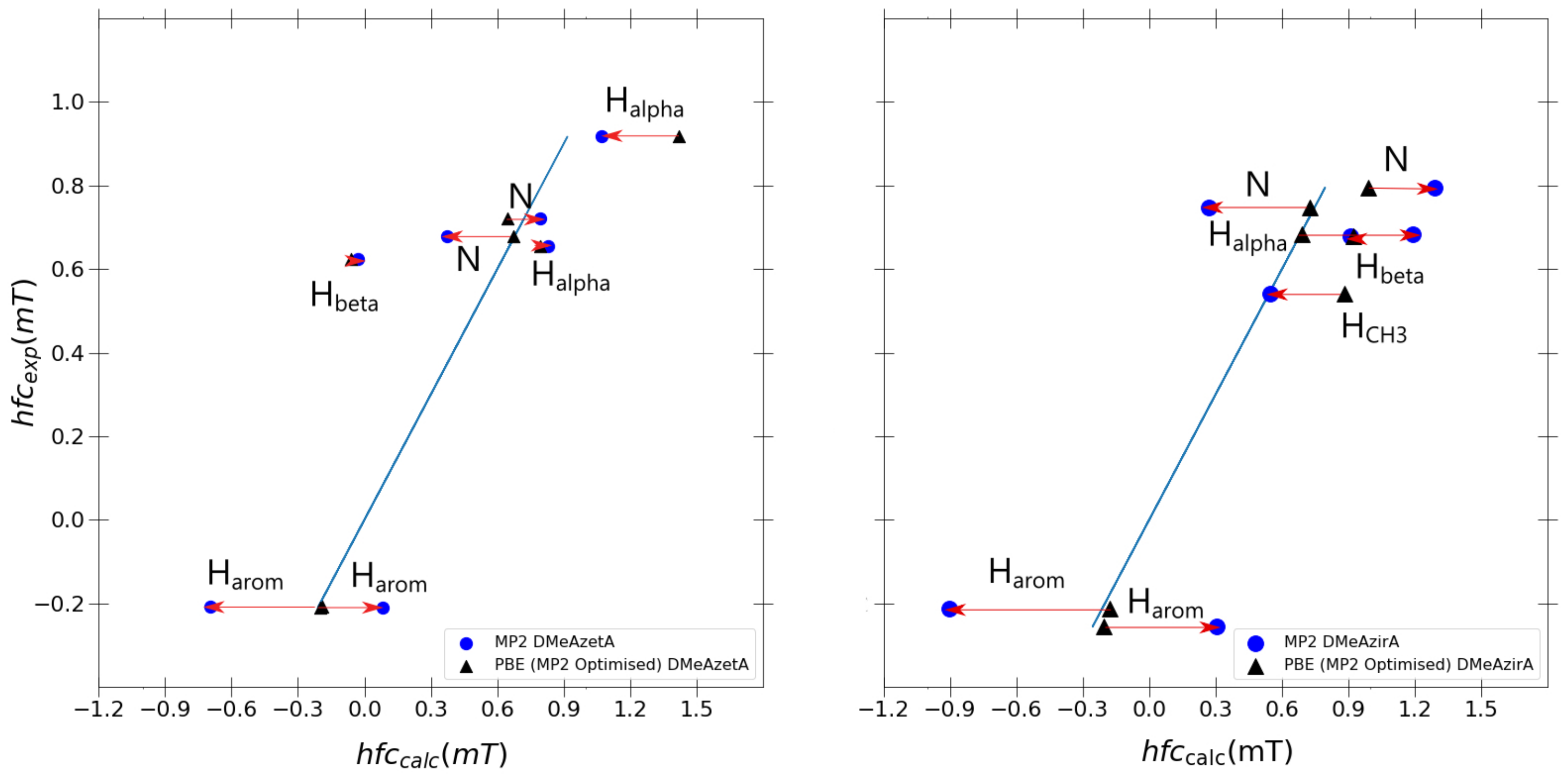
The question arises now whether the changes going from PBE0 to MP2 calculations are due to changes in geometry upon performing an MP2 geometry optimization or else due to MP2 calculation of spin density at the nuclei. Hence, it is of interest to compare the hfcc results of a single-point PBE0/6-31G(d,p)-J and MP2/6-31G(d,p)-J calculation with both retaining MP2/6-31G(d,p) optimised geometries for each outlier molecule, DMeAzetA and DMeAzirA.
As shown in
Figure 7, one observes that the PBE0 hyperfine couplings remain stagnant compared to
Figure 6 differences between PBE0 and MP2 calculated values originating solely from the differences in the spin-density in the hfcc MP2 single-point calculation. Strained effects are still in total effect in both MP2 and PBE0 optimised systems for DMeAzetA and DMeAzirA, with the geometry change between PBE0 and MP2 contributing little to no effect towards improved hfccs (when comparing each molecules performance in
Figure 6 and
Figure 7). Therefore, the following regression analysis will be based with
Figure 6 in mind.
The usual linear regression model is used in
Table 8 to compare
R, slope and
y-intercept values retained by MP2 and PBE0. For ease of comparison, both problematic systems and a model DMeDiPrPD system (that achieved satisfactory results) are used to statistically determine if any improvement is gained transitioning from the best performing DFT functional PBE0 to a higher-level method such as MP2.
Evidently, PBE0 remains superior to MP2 for DMeAzetA and DMeDiPrPD as the
R, slope, and
y-intercept retain better values. With respect to DMeAzirA, one actually sees an improved
R value, whereas a quite drastic degradation in slope and
y-intercept is concurrently observed when transitioning from PBE0 to MP2. These opposing correlation trends for DMeAzirA are explained simply by observing the holistic data point distribution with respect to the fitted line in each separate plot of MP2 and PBE0 in
Figure 6. A closer to linear relationship is seen with a superior
R for MP2. Yet, factoring a depreciated slope and
y-intercept, one concludes that there is a large error from the true fitted line. Moreover, this error originates from the overestimation of calculated couplings at the MP2 level of theory, which is clearly seen upon comparison with corresponding absolute experimental values.
A reference calculation was also performed (
Table 8) using
B97XD optimised using MP2/6-31G(d,p) and a single-point calculation ran with a 6-31G(d,p)-J basis set and PCM solvation model. Surprisingly, DMeAzirA improved significantly over MP2 and PBE0 showing signs for improved correlation upon realisation of the
B97XD functional. Meanwhile, DMeAzetA and DMeDiPrPD correlation values remain stationary in comparison to MP2 and PBE0. In order to compare this result when functionals previously tested, it was necessary to perform a similar single-point calculation with an
B97XD/6-31G(d,p) optimised geometry for each of the three test molecules. It is immediately apparent that correlation parameter performance degrades significantly for DMeAzirA in comparison to the MP2/6-31G(d,p) optimised equivalent calculation. Thus, this result falls in line with similar calculations using B3LYP, TPSSh and PBE0 functionals.
As a means to further consolidate discussion of the MP2 results, it was of interest to run a simple single-point calculation using the CFOUR [
25] quantum chemistry program. An MP2 calculation was run with an MP2/6-31G(d,p) optimised geometry without solvation parameters and compared to an equivalent calculation run in the Gaussian program. CFOUR by implementation only prints the spin density of each nuclei in the output, hence an extra conversion is needed to calculate the isotropic hfcc a
of each nuclei N:
where
is the total spin expectation value, g
and g
are the electron and nuclear g-factors, and
and
are the electron and nuclear magnetrons.
is the nuclear spin density. As shown in
Table 5, the results are significantly similar to conclude the MP2 results presented in
Table 8 are consistent. Shown also are the DMeAzetA experimental results, which are in much better agreement with MP2 Gaussian results in a solvated environment, further exemplifying the critical improving effect a solvation model provides.
Therefore, to calculate N,N,N’,N’-tetrasubstituted p-phenylenediamines hfccs, using MP2 would be irrational as the benefit is only marginal for a subset of couplings taking into consideration also the extra cost with respect to hybrid DFT methods. The final synopsis would be to therefore continue with PBE0/6-31G(d,p)-J/PCM for calculating hfccs of N,N,N’,N’-tetrasubstituted p-phenylenediamines, but a case can be made for using B97XD with an MP2-optimised geometry, especially for improving couplings in the strained DMeAzirA molecule.


 ,
, 
 ): ; blue (
): ; blue ( ): ; red (
): ; red ( ): ; green (
): ; green ( ): ; purple (
): ; purple ( ): ; cyan (
): ; cyan ( ): ; grey (
): ; grey ( ): .
): .
 ): ; blue (
): ; blue ( ): ; red (
): ; red ( ): ; green (
): ; green ( ): ; purple (
): ; purple ( ): ; cyan (
): ; cyan ( ): ; grey (
): ; grey ( ): .
): .



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}