

Evaluation of Epithelial–Mesenchymal Transition Markers in Autoimmune Thyroid Diseases

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
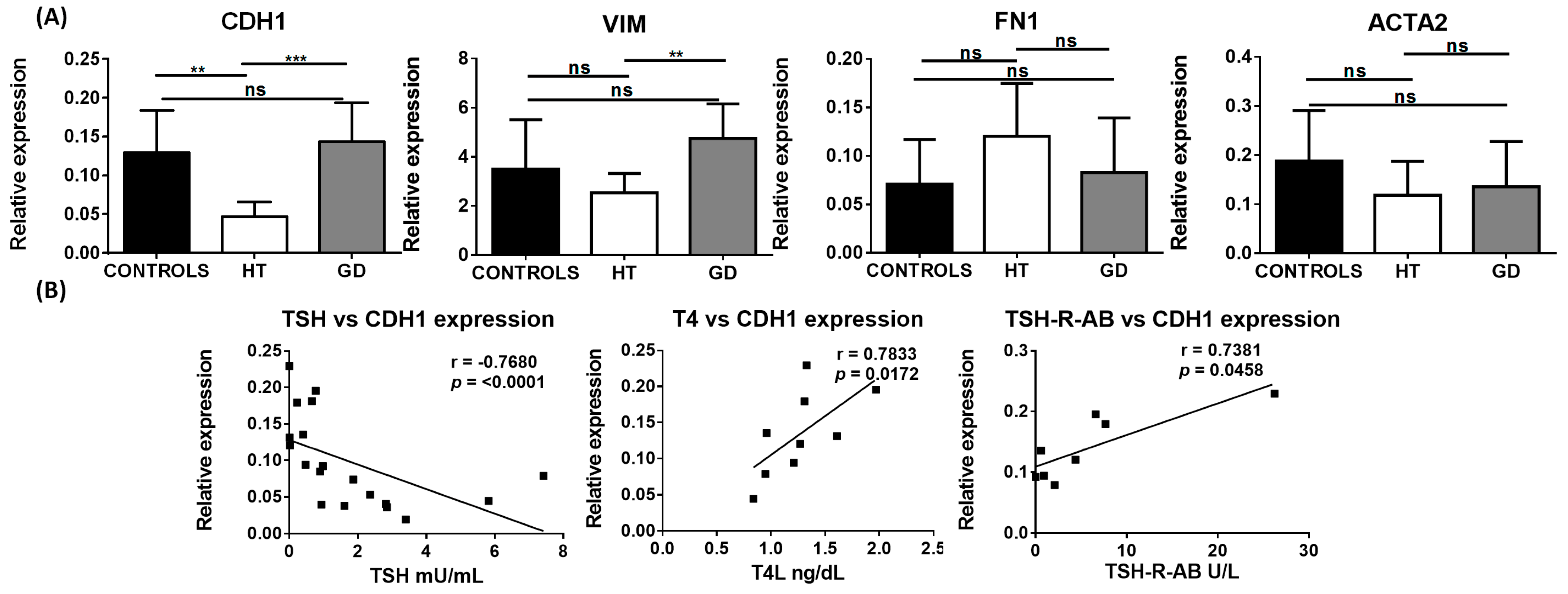
2.1. EMT Markers in AITD
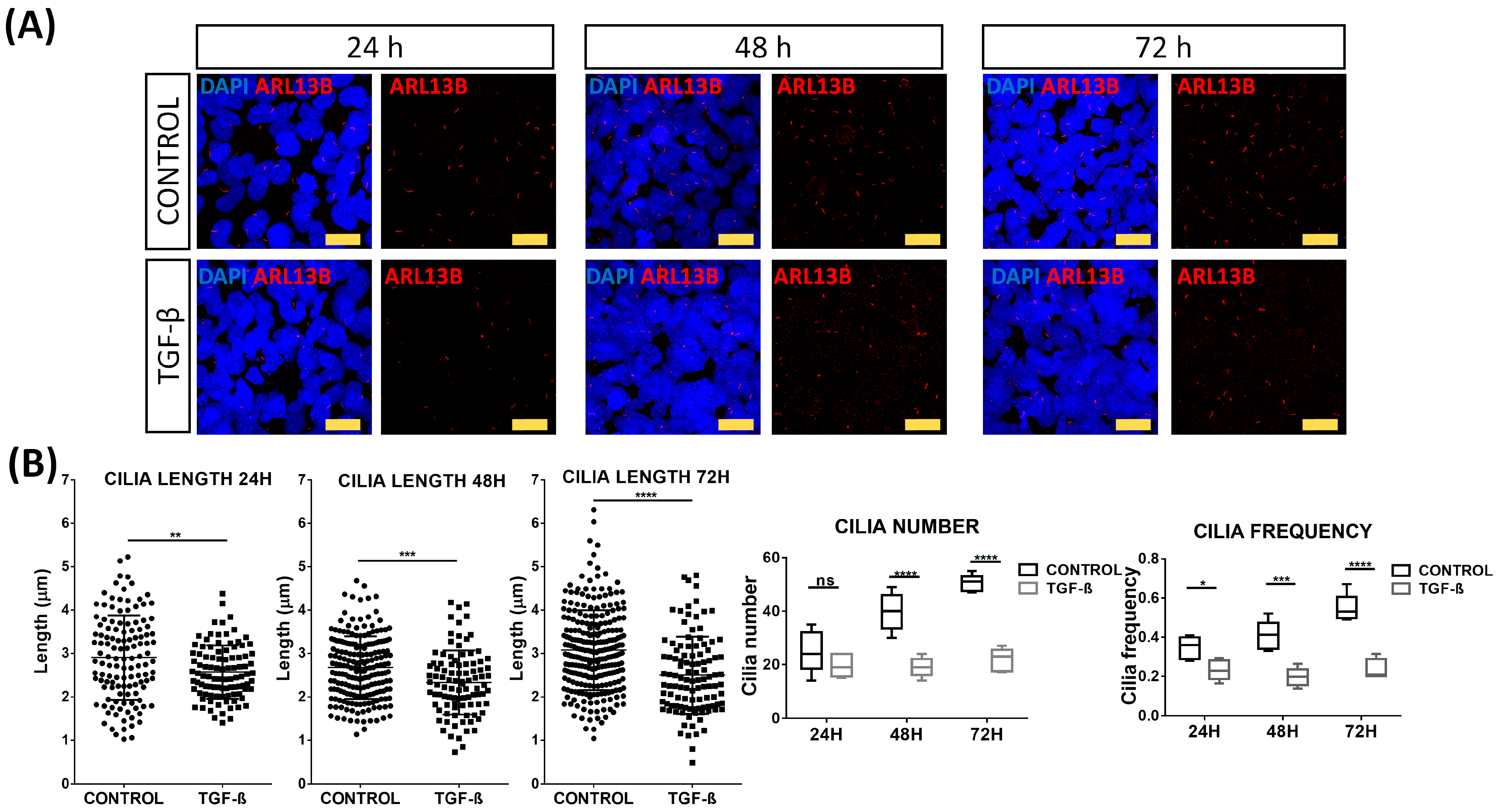
2.2. TGF-β Stimulation of Cultured Thyroid Cells
2.3. Primary Cilia Disruption after TGF-β Stimulation
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. RNA Isolation and RT-qPCR
4.3. Tissue Microarrays
4.4. Immunohistochemistry
4.5. Immunohistochemistry Score
4.6. Thyroid Cell Cultures
4.7. TGF-Β Stimulation Assays
4.8. Immunofluorescence Microscopy Analysis
4.9. Western Blot Analysis
4.10. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weetman, A.; DeGroot, L.J. Autoimmunity to the Thyroid Gland; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Mammen, J.S.R.; Cappola, A.R. Autoimmune Thyroid Disease in Women. JAMA 2021, 325, 2392–2393. [Google Scholar] [CrossRef]
- Bahn, R.S. Graves’ Ophthalmopathy. N. Engl. J. Med. 2010, 362, 726–738. [Google Scholar] [CrossRef]
- Burch, H.B.; Perros, P.; Bednarczuk, T.; Cooper, D.S.; Dolman, P.J.; Leung, A.M.; Mombaerts, I.; Salvi, M.; Stan, M.N. Management of Thyroid Eye Disease: A Consensus Statement by the American Thyroid Association and the European Thyroid Association. Eur. Thyroid J. 2022, 11, e220189. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Leví, A.M.; Marazuela, M. Pathogenesis of Thyroid Autoimmune Disease: The Role of Cellular Mechanisms. Endocrinol. Nutr. 2016, 63, 421–429. [Google Scholar] [CrossRef]
- Xiaoheng, C.; Yizhou, M.; Bei, H.; Huilong, L.; Xin, W.; Rui, H.; Lu, L.; Zhiguo, D. General and Specific Genetic Polymorphism of Cytokines-Related Gene in AITD. Mediat. Inflamm. 2017, 2017, 3916395. [Google Scholar] [CrossRef]
- Sacristán-Gómez, P.; Serrano-Somavilla, A.; González-Amaro, R.; Martínez-Hernández, R.; Marazuela, M. Analysis of Expression of Different Histone Deacetylases in Autoimmune Thyroid Disease. J. Clin. Endocrinol. Metab. 2021, 106, 3213–3227. [Google Scholar] [CrossRef]
- Prummel, M.F.; Wiersinga, W.M. Smoking and Risk of Graves’ Disease. JAMA 1993, 269, 479–482. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, A.; Vitales-Noyola, M.; Ramos-Levi, A.; Serrano-Somavilla, A.; González-Amaro, R.; Marazuela, M. Levels of Regulatory T Cells CD69(+)NKG2D(+)IL-10(+) Are Increased in Patients with Autoimmune Thyroid Disorders. Endocrine 2016, 51, 478–489. [Google Scholar] [CrossRef]
- García-López, M.A.; Marazuela, M.; Sánchez-Madrid, F.; de la Fuente, H.; Monsiváis-Urenda, A.; Alvarado-Sánchez, B.; Figueroa-Vega, N.; González-Amaro, R. Regulatory T Cells in Human Autoimmune Thyroid Disease. J. Clin. Endocrinol. Metab. 2006, 91, 3639–3646. [Google Scholar] [CrossRef]
- Nanba, T.; Watanabe, M.; Inoue, N.; Iwatani, Y. Increases of the Th1/Th2 Cell Ratio in Severe Hashimoto’s Disease and in the Proportion of Th17 Cells in Intractable Graves’ Disease. Thyroid Off. J. Am. Thyroid Assoc. 2009, 19, 495–501. [Google Scholar] [CrossRef]
- ITO, C.; WATANABE, M.; OKUDA, N.; WATANABE, C.; IWATANI, Y. Association between the Severity of Hashimoto’s Disease and the Functional +874A/T Polymorphism in the Interferon-γ Gene. Endocr. J. 2006, 53, 473–478. [Google Scholar] [CrossRef]
- Widder, J.; Dorfinger, K.; Wilfing, A.; Trieb, K.; Pirich, K.; Loebenstein, R.; Niederle, B.; Gessl, A.; Spitzauer, S.; Grubeck-Loebenstein, B. The Immunoregulatory Influence of Transforming Growth Factor Beta in Thyroid Autoimmunity: TGF β Inhibits Autoreactivity in Graves’ Disease. J. Autoimmun. 1991, 4, 689–701. [Google Scholar] [CrossRef]
- Gianoukakis, A.G.; Khadavi, N.; Smith, T.J. Cytokines, Graves’ Disease, and Thyroid-Associated Ophthalmopathy. Thyroid 2008, 18, 953–958. [Google Scholar] [CrossRef]
- Ganesh, B.B.; Bhattacharya, P.; Gopisetty, A.; Prabhakar, B.S. Role of Cytokines in the Pathogenesis and Suppression of Thyroid Autoimmunity. J. Interferon Cytokine Res. 2011, 31, 721–731. [Google Scholar] [CrossRef]
- Vural, P.; Değirmencioğlu, S.; Doğru-Abbasoğlu, S.; Baki, M.; Özderya, A.; Karadağ, B.; Uysal, M. Arg25Pro (c.915G>C) Polymorphism of Transforming Growth Factor Β1 Gene Suggests an Association with Increased Risk for Hashimoto’s Thyroiditis. Int. Immunopharmacol. 2015, 28, 521–524. [Google Scholar] [CrossRef]
- Kutluturk, F.; Yarman, S.; Sarvan, F.O.; Kekik, C. Association of Cytokine Gene Polymorphisms (IL6, IL10, TNF-α, TGF-β and IFN-γ) and Graves’ Disease in Turkish Population. Endocr. Metab. Immune Disord. Drug Targets 2013, 13, 163–167. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, Y.; Qin, L.; Fang, X.; Zhang, C.; Yue, J.; Bai, W.; Wang, G.; Chen, Z.; Renz, H.; et al. IL-1β Augments TGF-β Inducing Epithelial-Mesenchymal Transition of Epithelial Cells and Associates with Poor Pulmonary Function Improvement in Neutrophilic Asthmatics. Respir. Res. 2021, 22, 216. [Google Scholar] [CrossRef]
- Chen, K.; Wei, Y.; Sharp, G.C.; Braley-Mullen, H. Mechanisms of Spontaneous Resolution versus Fibrosis in Granulomatous Experimental Autoimmune Thyroiditis. J. Immunol. 2003, 171, 6236–6243. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Mu, Y.; Sa, N.; Wang, H.; Xu, W. Tumor Necrosis Factor α Induces Epithelial-Mesenchymal Transition and Promotes Metastasis via NF-ΚB Signaling Pathway-Mediated TWIST Expression in Hypopharyngeal Cancer. Oncol. Rep. 2014, 31, 321–327. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Steenvoorden, M.M.; Tolboom, T.C.; van der Pluijm, G.; Löwik, C.; Visser, C.P.; DeGroot, J.; Gittenberger-DeGroot, A.C.; DeRuiter, M.C.; Wisse, B.J.; Huizinga, T.W.; et al. Transition of Healthy to Diseased Synovial Tissue in Rheumatoid Arthritis Is Associated with Gain of Mesenchymalfibrotic Characteristics. Arthritis Res. Ther. 2006, 8, R165. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnäker, E.-M.; Tarner, I.H.; Robbins, P.D.; et al. Synovial Fibroblasts Spread Rheumatoid Arthritis to Unaffected Joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef]
- Bataille, F.; Rohrmeier, C.; Bates, R.; Weber, A.; Rieder, F.; Brenmoehl, J.; Strauch, U.; Farkas, S.; Fürst, A.; Hofstädter, F.; et al. Evidence for a Role of Epithelial Mesenchymal Transition during Pathogenesis of Fistulae in Crohn’s Disease. Inflamm. Bowel Dis. 2008, 14, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Weber, A.; Fürst, A.; Farkas, S.; Jehle, E.; Pesch, T.; Kellermeier, S.; Fried, M.; Rogler, G. Potential Role for SNAIL Family Transcription Factors in the Etiology of Crohnʼs Disease-Associated Fistulae. Inflamm. Bowel Dis. 2011, 17, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Ribatti, D.; Lisi, S. TGFβ1-Smad Canonical and -Erk Noncanonical Pathways Participate in Interleukin-17-Induced Epithelial-Mesenchymal Transition in Sjögren’s Syndrome. Lab. Investig. J. Tech. Methods Pathol. 2020, 100, 824–836. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Tamma, R.; Ribatti, D.; Lisi, S. The TGF-Β1 Signaling Pathway as an Attractive Target in the Fibrosis Pathogenesis of Sjögren’s Syndrome. Mediat. Inflamm. 2018, 2018, 1965935. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-Beta Induced Transdifferentiation of Mammary Epithelial Cells to Mesenchymal Cells: Involvement of Type I Receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-Beta1: Potential Role in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.-M.; Ng, Y.-Y.; Hill, P.A.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Transforming Growth Factor-β Regulates Tubular Epithelial-Myofibroblast Transdifferentiation in Vitro. Kidney Int. 1999, 56, 1455–1467. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Serrano-Somavilla, A.; Ramos-Leví, A.; Sampedro-Nuñez, M.; Lens-Pardo, A.; Muñoz De Nova, J.L.; Triviño, J.C.; González, M.U.; Torné, L.; Casares-Arias, J.; et al. Integrated MiRNA and MRNA Expression Profiling Identifies Novel Targets and Pathological Mechanisms in Autoimmune Thyroid Diseases. EBioMedicine 2019, 50, 329–342. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular Signalling by Primary Cilia in Development, Organ Function and Disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The Primary Cilium: A Signalling Centre during Vertebrate Development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Martin, A.; Hedinger, C.; Häberlin-Jakob, M.; Walt, H. Structure and Motility of Primary Cilia in the Follicular Epithelium of the Human Thyroid. Virchows Arch. B Cell Pathol. 1988, 55, 159–166. [Google Scholar] [CrossRef]
- Han, S.J.; Jung, J.K.; Im, S.-S.; Lee, S.-R.; Jang, B.-C.; Park, K.M.; Kim, J.I. Deficiency of Primary Cilia in Kidney Epithelial Cells Induces Epithelial to Mesenchymal Transition. Biochem. Biophys. Res. Commun. 2018, 496, 450–454. [Google Scholar] [CrossRef]
- Ehnert, S.; Sreekumar, V.; Aspera-Werz, R.H.; Sajadian, S.O.; Wintermeyer, E.; Sandmann, G.H.; Bahrs, C.; Hengstler, J.G.; Godoy, P.; Nussler, A.K. TGF-β(1) Impairs Mechanosensation of Human Osteoblasts via HDAC6-Mediated Shortening and Distortion of Primary Cilia. J. Mol. Med. Berl. Ger. 2017, 95, 653–663. [Google Scholar] [CrossRef]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.-H.; Moustakas, A. TGF-beta and the Smad Signaling Pathway Support Transcriptomic Reprogramming during Epithelial-Mesenchymal Cell Transition. Mol. Biol. Cell 2005, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Kardalas, E.; Maraka, S.; Papagianni, M.; Paltoglou, G.; Siristatidis, C.; Mastorakos, G. TGF-β Physiology as a Novel Therapeutic Target Regarding Autoimmune Thyroid Diseases: Where Do We Stand and What to Expect. Med. Kaunas Lith. 2021, 57, 621. [Google Scholar] [CrossRef] [PubMed]
- Tolboom, T.C.A.; van der Helm-Van Mil, A.H.M.; Nelissen, R.G.H.H.; Breedveld, F.C.; Toes, R.E.M.; Huizinga, T.W.J. Invasiveness of Fibroblast-like Synoviocytes Is an Individual Patient Characteristic Associated with the Rate of Joint Destruction in Patients with Rheumatoid Arthritis. Arthritis Rheum. 2005, 52, 1999–2002. [Google Scholar] [CrossRef]
- Gheldof, A.; Berx, G. Cadherins and Epithelial-to-Mesenchymal Transition. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2013; Volume 116, pp. 317–336. ISBN 978-0-12-394311-8. [Google Scholar]
- Loh, C.-Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Liu, X.; Huang, H.; Remmers, N.; Hollingsworth, M.A. Loss of E-Cadherin and Epithelial to Mesenchymal Transition Is Not Required for Cell Motility in Tissues or for Metastasis. Tissue Barriers 2014, 2, e969112. [Google Scholar] [CrossRef] [PubMed]
- Smyth, P.; Sheils, O.; Finn, S.; Martin, C.; O’Leary, J.; Sweeney, E.C. Real-Time Quantitative Analysis of E-Cadherin Expression in Ret/PTC-1-Activated Thyroid Neoplasms. Int. J. Surg. Pathol. 2001, 9, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin Contributes to Epithelial-Mesenchymal Transition Cancer Cell Mechanics by Mediating Cytoskeletal Organization and Focal Adhesion Maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Salmon, M.; Lin, X.; Zehner, Z.E. TGFβ1 Regulation of Vimentin Gene Expression during Differentiation of the C2C12 Skeletal Myogenic Cell Line Requires Smads, AP-1 and Sp1 Family Members. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2007, 1773, 427–439. [Google Scholar] [CrossRef]
- Byun, Y.; Chen, F.; Chang, R.; Trivedi, M.; Green, K.J.; Cryns, V.L. Caspase Cleavage of Vimentin Disrupts Intermediate Filaments and Promotes Apoptosis. Cell Death Differ. 2001, 8, 443–450. [Google Scholar] [CrossRef]
- Henzen-Logmans, S.C.; Mullink, H.; Ramaekers, F.C.; Tadema, T.; Meijer, C.J. Expression of Cytokeratins and Vimentin in Epithelial Cells of Normal and Pathologic Thyroid Tissue. Virchows Arch. A Pathol. Anat. Histopathol. 1987, 410, 347–354. [Google Scholar] [CrossRef]
- Viale, G.; Dell’Orto, P.; Coggi, G.; Gambacorta, M. Coexpression of Cytokeratins and Vimentin in Normal and Diseased Thyroid Glands. Lack of Diagnostic Utility of Vimentin Immunostaining. Am. J. Surg. Pathol. 1989, 13, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Lepreux, S.; Desmoulière, A. Human Liver Myofibroblasts during Development and Diseases with a Focus on Portal (Myo)Fibroblasts. Front. Physiol. 2015, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial Remodeling after Infarction: The Role of Myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The Role of α-Smooth Muscle Actin in Fibroblast-Mediated Matrix Contraction and Remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Smith, T.J.; Padovani-Claudio, D.A.; Lu, Y.; Raychaudhuri, N.; Fernando, R.; Atkins, S.; Gillespie, E.F.; Gianoukakis, A.G.; Miller, B.S.; Gauger, P.G.; et al. Fibroblasts Expressing the Thyrotropin Receptor Overarch Thyroid and Orbit in Graves’ Disease. J. Clin. Endocrinol. Metab. 2011, 96, 3827–3837. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Yamada, K.M. Fibronectins: Multifunctional Modular Glycoproteins. J. Cell Biol. 1982, 95, 369–377. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Gang, H.; Li, X.; Li, Z.; Wang, T.; Han, J.; Luo, T.; Wen, F.; Wu, X. Up-Regulation of Gastric Cancer Cell Invasion by Twist Is Accompanied by N-Cadherin and Fibronectin Expression. Biochem. Biophys. Res. Commun. 2007, 358, 925–930. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Strutz, F.; Müller, G.A. Renal Fibrosis: An Update. Curr. Opin. Nephrol. Hypertens. 2001, 10, 315–320. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, C.T.; Bhargava, M.; Torzilli, P.A. A Comparative Study of Fibronectin Cleavage by MMP-1, -3, -13, and -14. Cartilage 2012, 3, 267–277. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the Tumour Transition States Occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around Epithelial–Mesenchymal Plasticity in Cancer Metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-Mesenchymal Transitions: The Importance of Changing Cell State in Development and Disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.A.; Ajbro, K.D.; Koefoed, K.; Vestergaard, M.L.; Veland, I.R.; Henriques de Jesus, M.P.R.; Pedersen, L.B.; Benmerah, A.; Andersen, C.Y.; Larsen, L.A.; et al. TGF-β Signaling Is Associated with Endocytosis at the Pocket Region of the Primary Cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef]
- Kawasaki, M.; Ezura, Y.; Hayata, T.; Notomi, T.; Izu, Y.; Noda, M. TGF-β Suppresses Ift88 Expression in Chondrocytic ATDC5 Cells: TGF-β REGULATES IFT88. J. Cell. Physiol. 2015, 230, 2788–2795. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | HT | GD |
|---|---|---|
| N | 10 | 10 |
| Gender (F/M) | 10/0 | 9/1 |
| Age, years | 62 (57–70) | 47 (40–57) |
| Ophthalmopathy | 0 | 7 |
| TSH, mU/mL | 2.59 (1.68–3.26) | 0.44 (0.01–0.8) |
| T4, ng/dL | - | 1.29 (1.02–1.54) |
| TG-Ab, UI/mL | 626 (143.5–728.5) | 20 (20–2279) |
| TPO-Ab, UI/mL | 713.5 (434.75–1421.25) | 169 (20–578.5) |
| TSH-R-Ab, U/L | - | 5.63 (0.91–7.67) |
| Primer | Orientation | Sequence |
|---|---|---|
| CDH1 | FORWARD | GCCGAGAGCTACACGTTCAC |
| REVERSE | ACTTTGAATCGGGTGTCGAG | |
| VIM | FORWARD | CTCCCTCTGGTTGATACCCAC |
| REVERSE | GGTCATCGTGATGCTGAGAAG | |
| FN1 | FORWARD | CCTCAATTGTTGTTCGCTGGAGCA |
| REVERSE | GGTGACGGAGTTTGCAGTTTC | |
| ACTA2 | FORWARD | TGGCTATCCAGGCGGTGCTGTCT |
| REVERSE | ATGGCATGGGGCAAGGCATAGC | |
| GAPDH | FORWARD | GCCCAATACGACCAAATCC |
| REVERSE | AGCCACATCGCTCAGACAC | |
| β-ACTIN | FORWARD | GCCGACAGGATGCAGAAGGA |
| REVERSE | CGGAGTACTTGCGCTCAGGA |
| Parameters | HT | GD | Controls |
|---|---|---|---|
| N | 17 | 16 | 16 |
| Gender (F/M) | 12/5 | 15/1 | 10/6 |
| Age, years | 62 (42–66) | 48 (43–59) | 57 (43–61) |
| Ophthalmopathy | 0 | 8 | 0 |
| Smoking | 1 | 6 | 2 |
| TSH, mU/mL | 3.48 (2.15–4.70) | 0.15 (0.01–5.04) | 1.63 (1.32–2.95) |
| T4, ng/dL | 1.34 (1.11–1.45) | 1.05 (0.95–1.51) | 1.34 (1.06–1.81) |
| TG-Ab, UI/mL | 80 (20.75–216.25) | 95 (23–117) | 12 (12–12) |
| TPO-Ab, UI/mL | 86 (22.5–297) | 175 (16–223) | 4 (4–10) |
| TSH-R-Ab, U/L | - | 5.22 (2.98–8.11) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacristán-Gómez, P.; Serrano-Somavilla, A.; Castro-Espadas, L.; Sánchez de la Blanca Carrero, N.; Sampedro-Núñez, M.; Muñoz-De-Nova, J.L.; Molina-Jiménez, F.; Rosell, A.; Marazuela, M.; Martínez-Hernández, R. Evaluation of Epithelial–Mesenchymal Transition Markers in Autoimmune Thyroid Diseases. Int. J. Mol. Sci. 2023, 24, 3359. https://doi.org/10.3390/ijms24043359
Sacristán-Gómez P, Serrano-Somavilla A, Castro-Espadas L, Sánchez de la Blanca Carrero N, Sampedro-Núñez M, Muñoz-De-Nova JL, Molina-Jiménez F, Rosell A, Marazuela M, Martínez-Hernández R. Evaluation of Epithelial–Mesenchymal Transition Markers in Autoimmune Thyroid Diseases. International Journal of Molecular Sciences. 2023; 24(4):3359. https://doi.org/10.3390/ijms24043359
Chicago/Turabian StyleSacristán-Gómez, Pablo, Ana Serrano-Somavilla, Lía Castro-Espadas, Nuria Sánchez de la Blanca Carrero, Miguel Sampedro-Núñez, José Luis Muñoz-De-Nova, Francisca Molina-Jiménez, Alejandra Rosell, Mónica Marazuela, and Rebeca Martínez-Hernández. 2023. "Evaluation of Epithelial–Mesenchymal Transition Markers in Autoimmune Thyroid Diseases" International Journal of Molecular Sciences 24, no. 4: 3359. https://doi.org/10.3390/ijms24043359
APA StyleSacristán-Gómez, P., Serrano-Somavilla, A., Castro-Espadas, L., Sánchez de la Blanca Carrero, N., Sampedro-Núñez, M., Muñoz-De-Nova, J. L., Molina-Jiménez, F., Rosell, A., Marazuela, M., & Martínez-Hernández, R. (2023). Evaluation of Epithelial–Mesenchymal Transition Markers in Autoimmune Thyroid Diseases. International Journal of Molecular Sciences, 24(4), 3359. https://doi.org/10.3390/ijms24043359