
Aggregation and Gelation Behavior of Stereocomplexed Four-Arm PLA-PEG Copolymers Containing Neutral or Cationic Linkers

 , ,
, ,  and
and 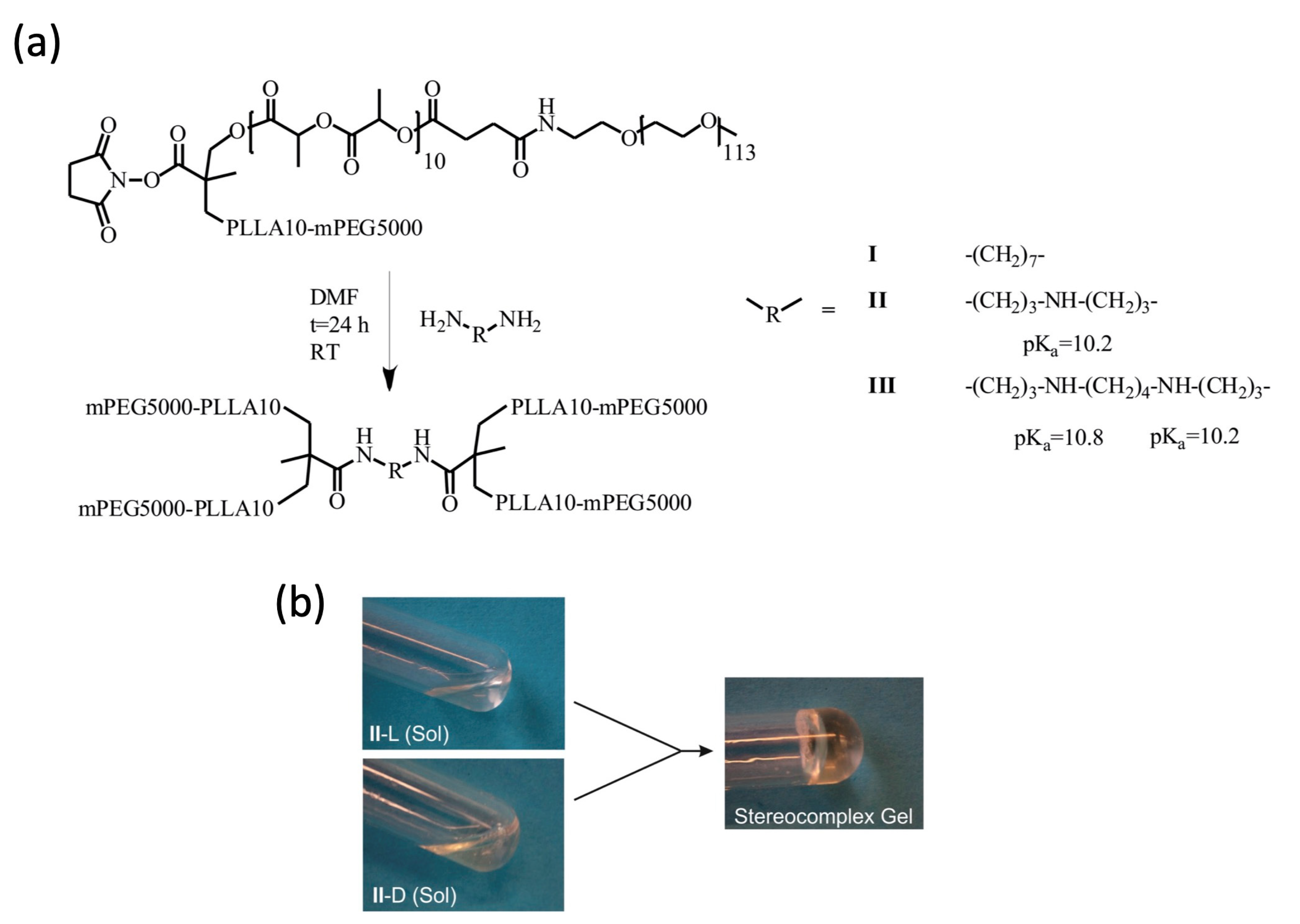{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
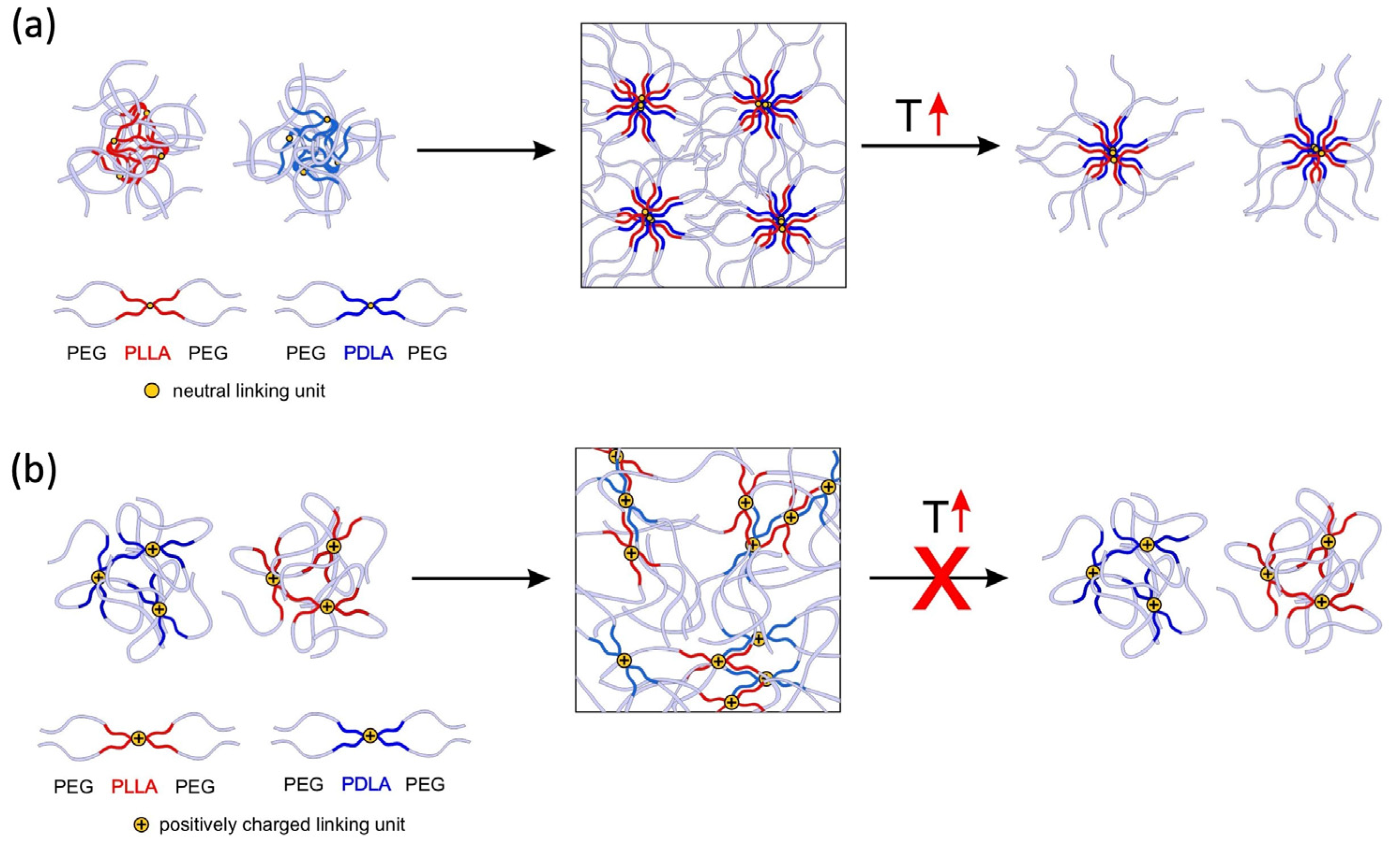
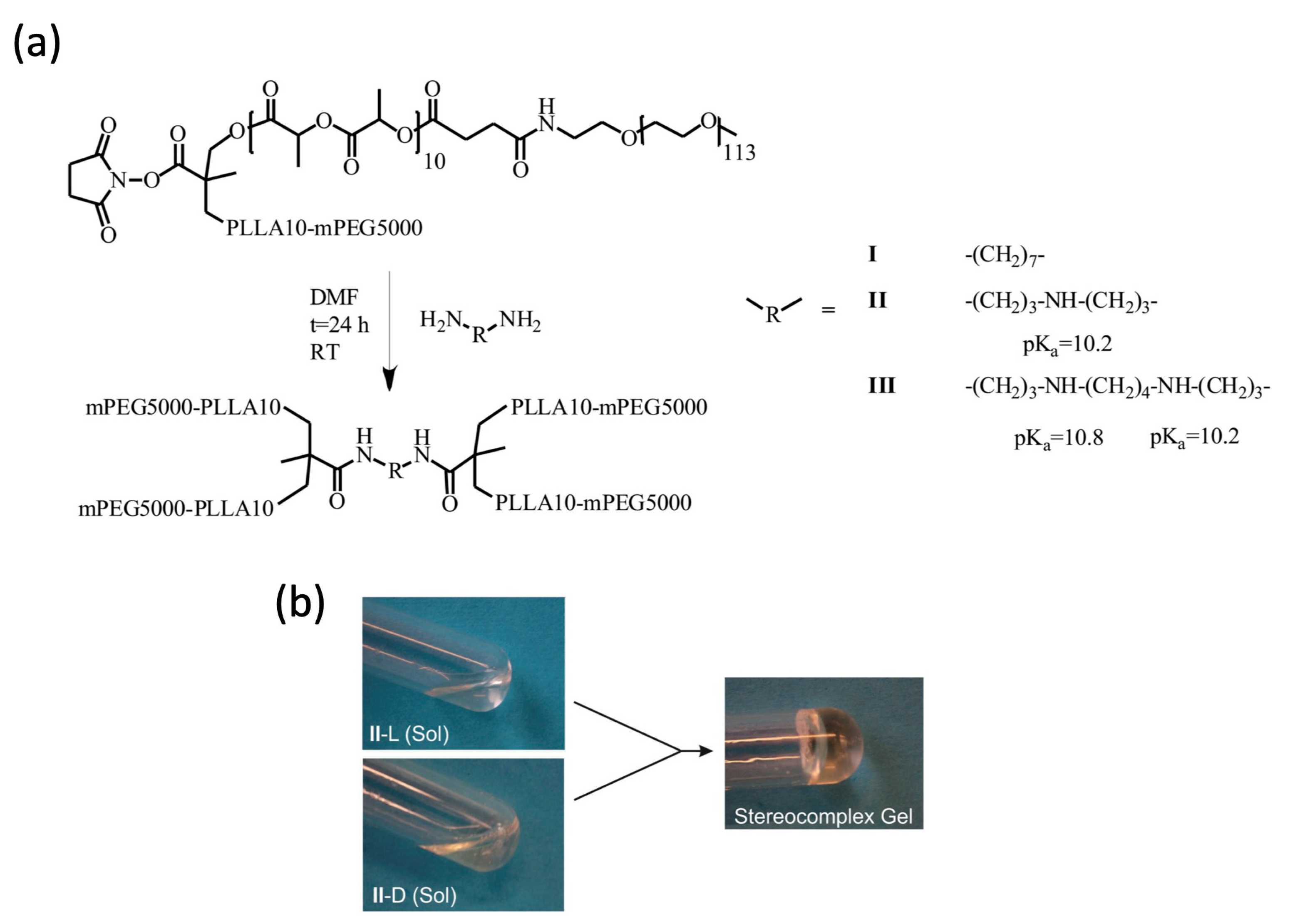
2.1. Gelation of Enantiomerically Pure Copolymers and Stereocomplexes Thereof
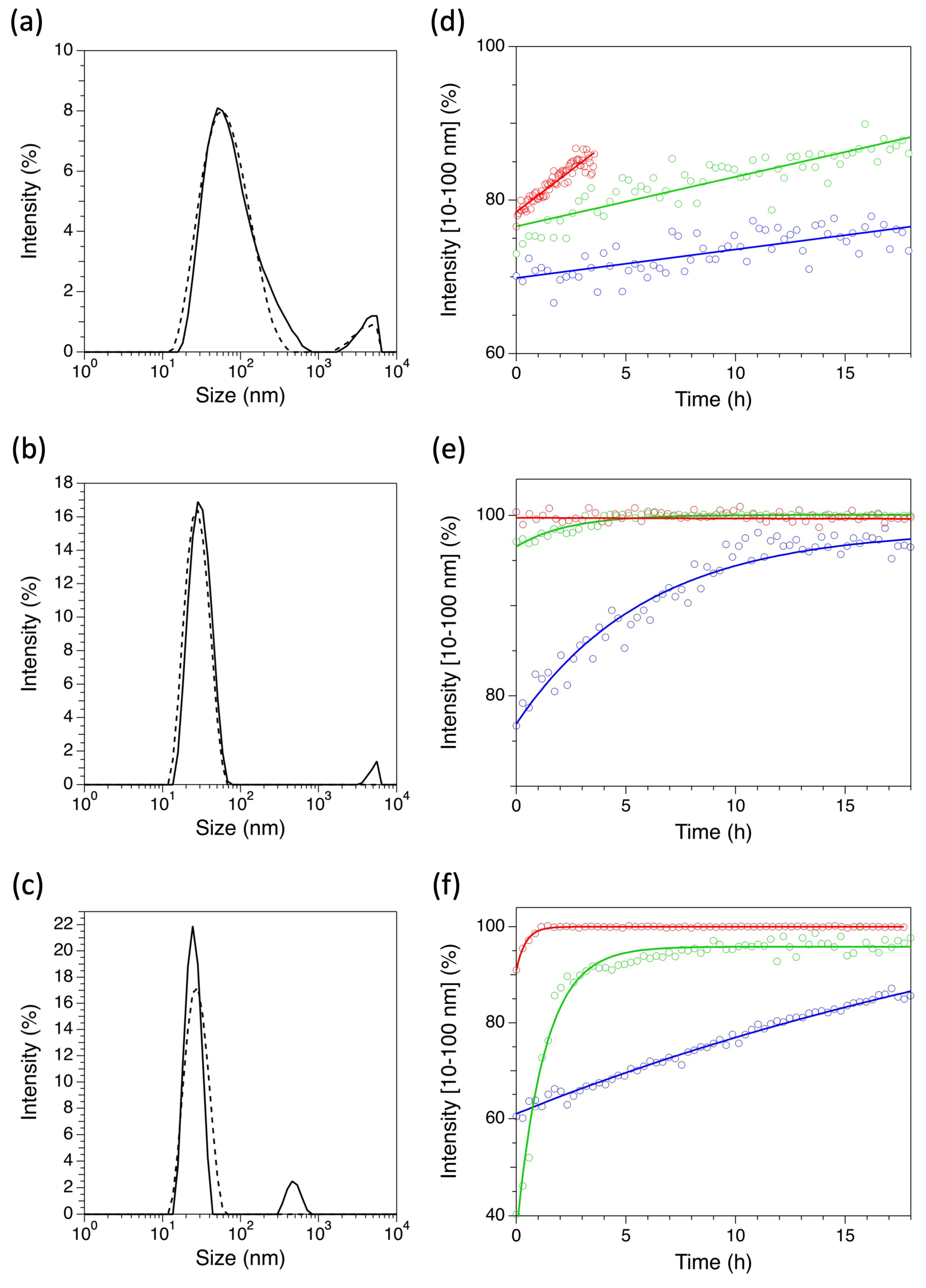
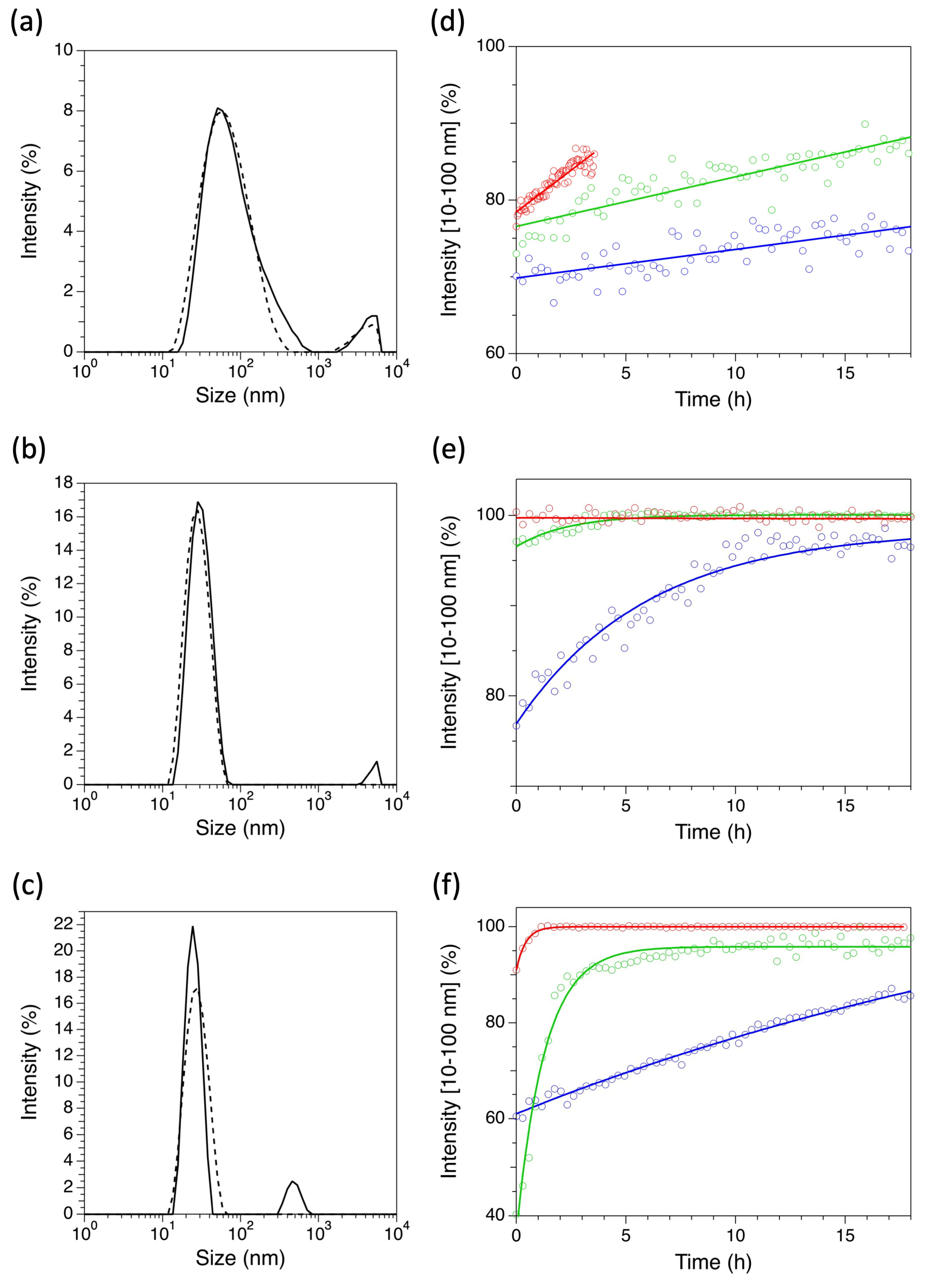
2.2. Dynamic Light Scattering Measurements
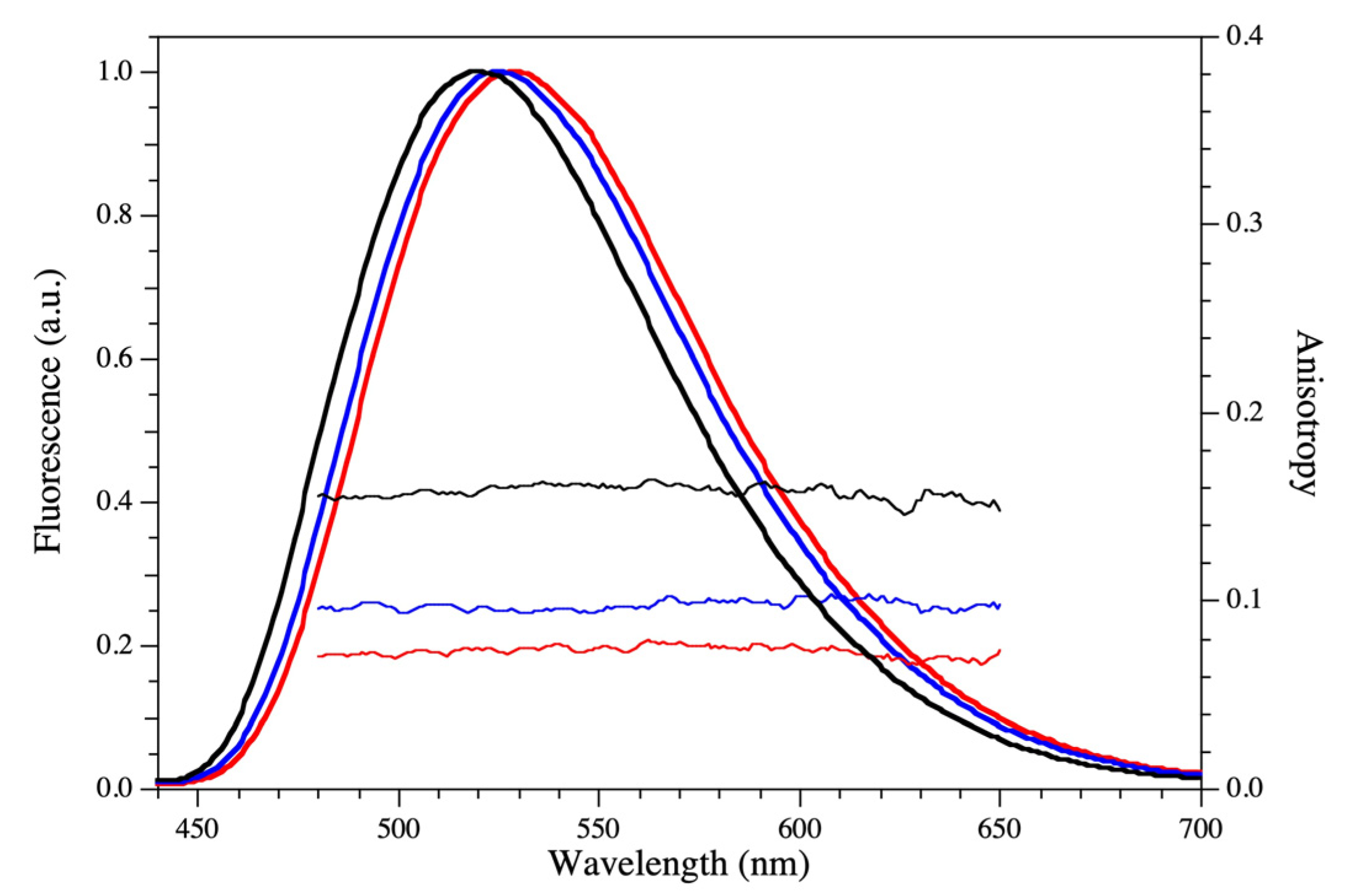
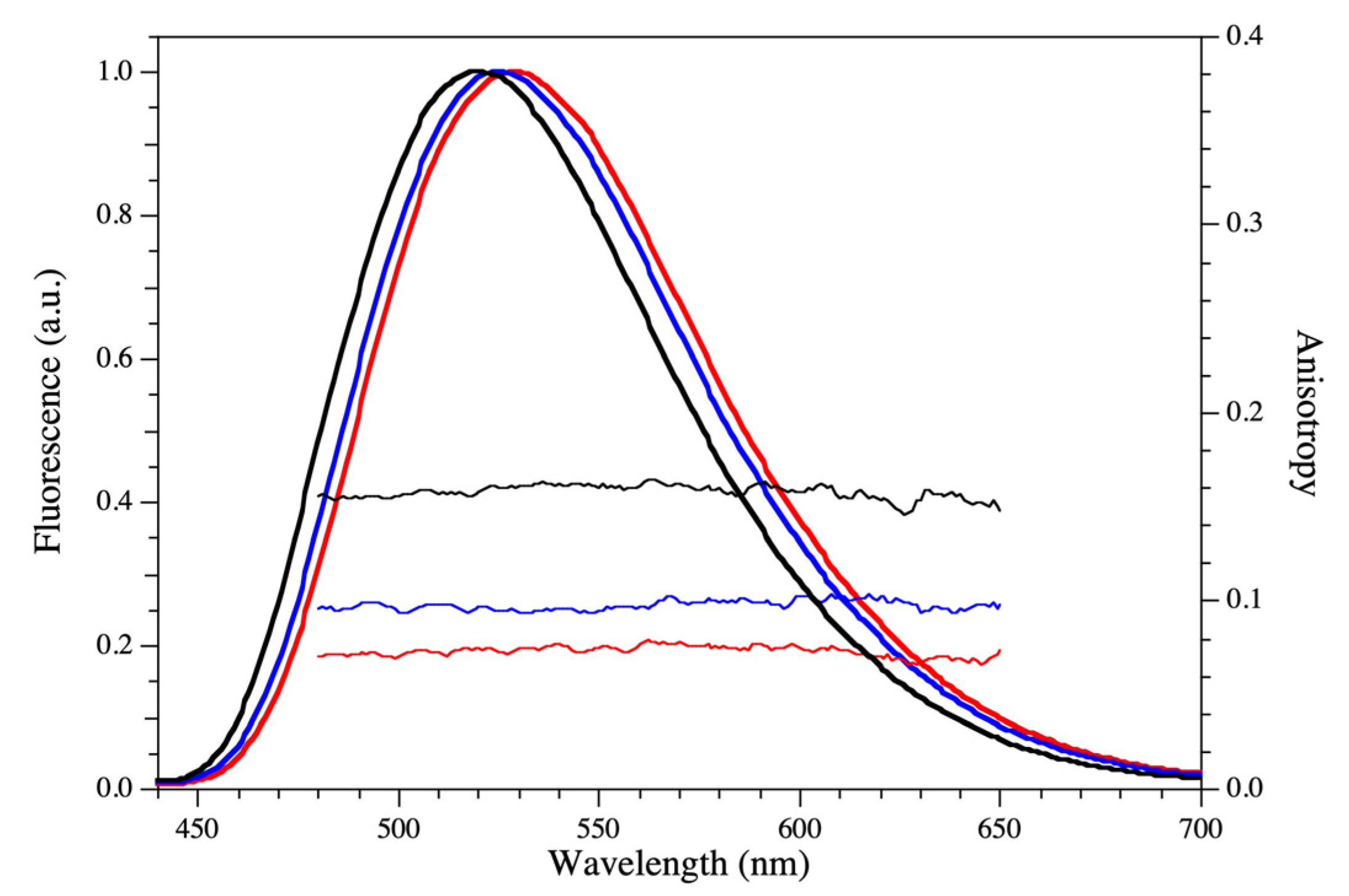
2.3. Fluorescence Analysis
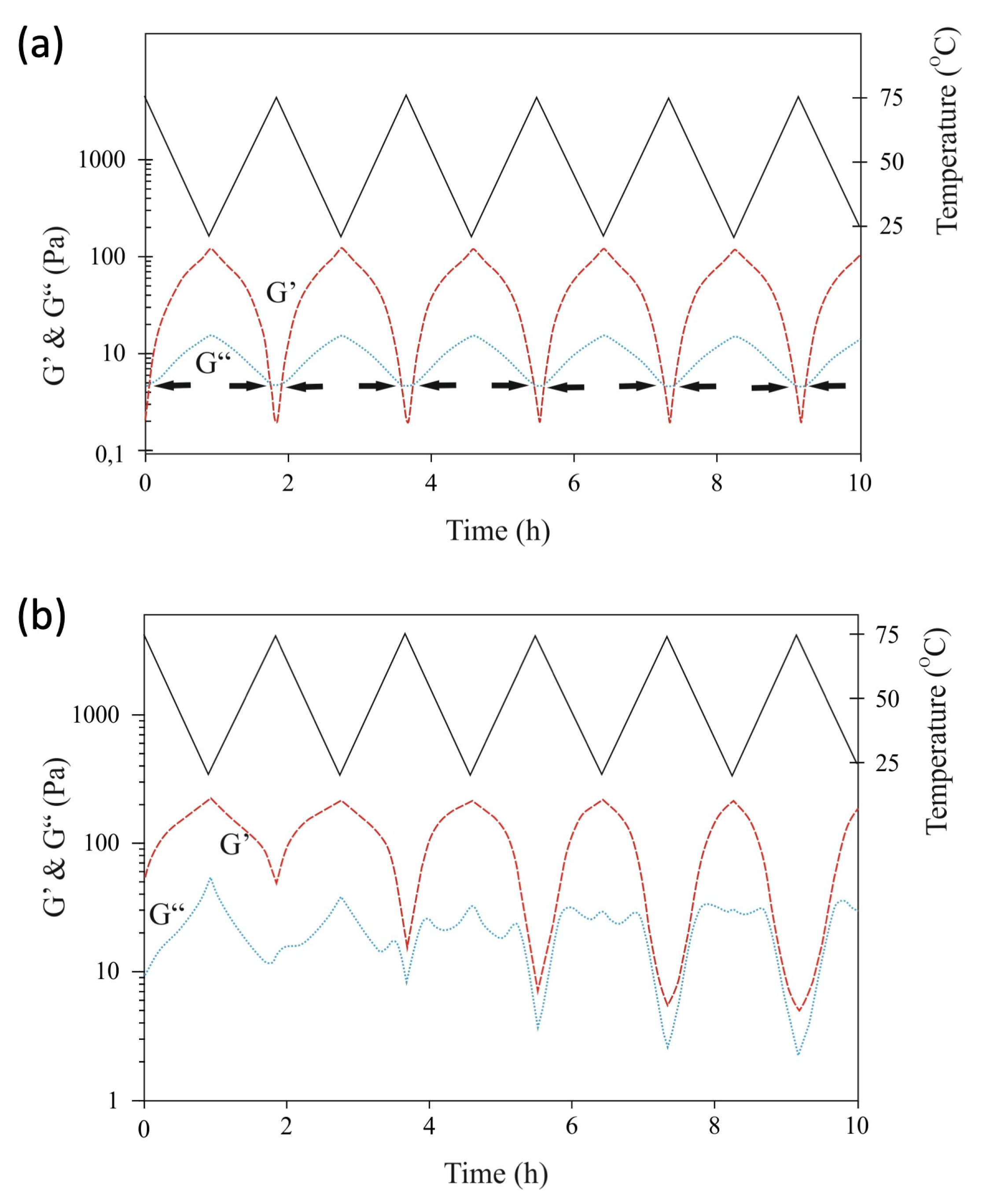
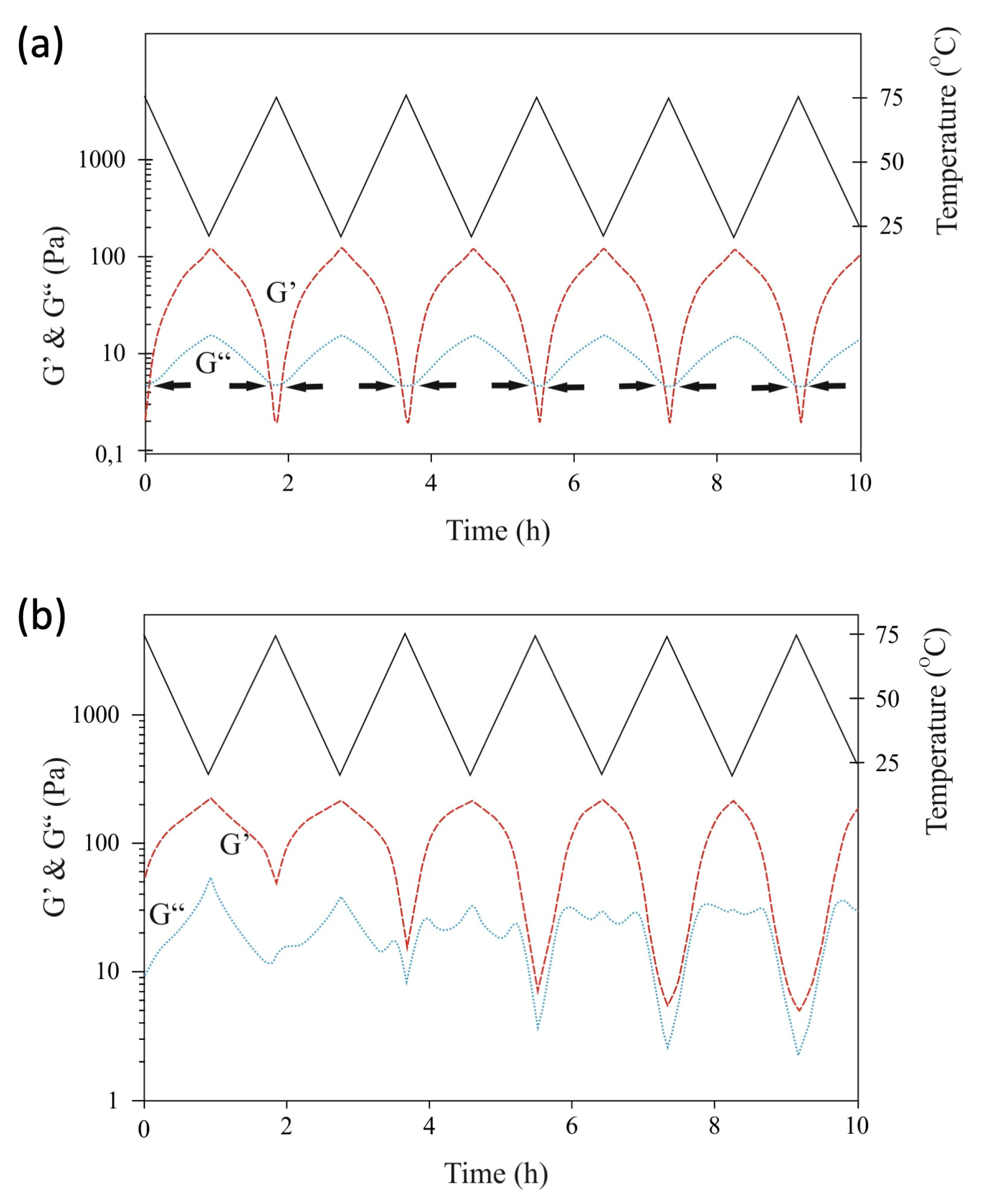
2.4. Rheology Measurements
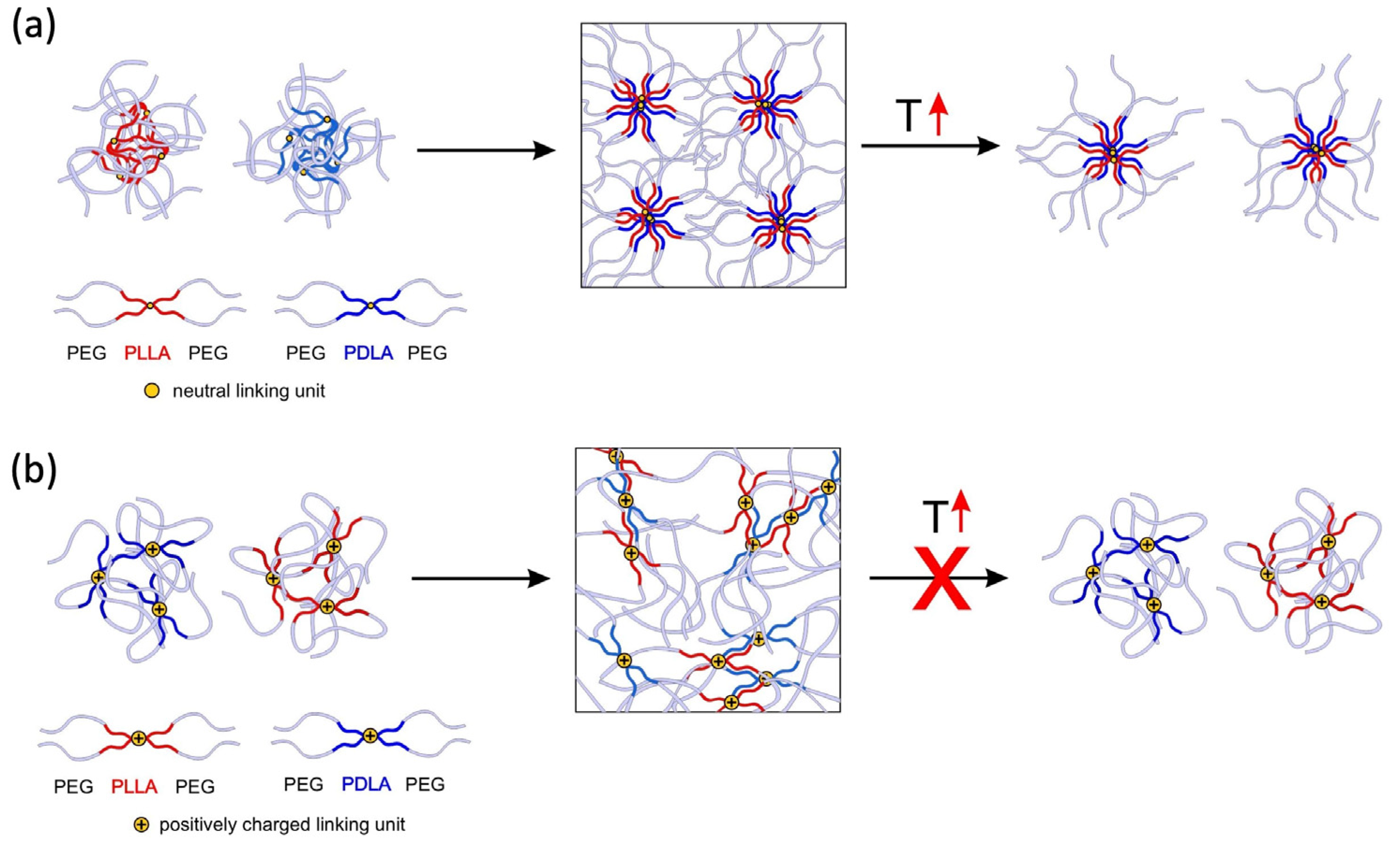
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock Polymers: Panacea or Pandora’s Box? Science 2012, 336, 434–440. [Google Scholar] [CrossRef]
- Huang, X.; Du, F.; Cheng, J.; Dong, Y.; Liang, D.; Ji, S.; Lin, S.-S.; Li, Z. Acid-Sensitive Polymeric Micelles Based on Thermoresponsive Block Copolymers with Pendent Cyclic Orthoester Groups. Macromolecules 2009, 42, 783–790. [Google Scholar] [CrossRef]
- Magnusson, J.P.; Khan, A.; Pasparakis, G.; Saeed, A.O.; Wang, W.; Alexander, C. Ion-Sensitive “Isothermal” Responsive Polymers Prepared in Water. J. Am. Chem. Soc. 2008, 130, 10852–10853. [Google Scholar] [CrossRef]
- Zhao, Y.; Bertrand, J.; Tong, X.; Zhao, Y. Photo-Cross-Linkable Polymer Micelles in Hydrogen-Bonding-Built Layer-By-Layer Films. Langmuir 2009, 25, 13151–13157. [Google Scholar] [CrossRef]
- Moughton, A.O.; Hillmyer, M.A.; Lodge, T.P. Multicompartment Block Polymer Micelles. Macromolecules 2011, 45, 2–19. [Google Scholar] [CrossRef]
- Karayianni, M.; Pispas, S. Block copolymer solution self-assembly: Recent advances, emerging trends, and applications. J. Polym. Sci. 2021, 59, 1874–1898. [Google Scholar] [CrossRef]
- Kuperkar, K.; Patel, D.; Atanase, L.I.; Bahadur, P. Amphiphilic Block Copolymers: Their Structures, and Self-Assembly to Polymeric Micelles and Polymersomes as Drug Delivery Vehicles. Polymers 2022, 14, 4702. [Google Scholar] [CrossRef] [PubMed]
- Avsar, S.Y.; Kyropoulou, M.; Di Leone, S.; Schoenenberger, C.-A.; Meier, W.P.; Palivan, C.G. Biomolecules Turn Self-Assembling Amphiphilic Block Co-Polymer Platforms into Biomimetic Interfaces. Front. Chem. 2019, 6, 645. [Google Scholar] [CrossRef]
- Hennink, W.; van Nostrum, C. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Bustamante-Torres, M.; Romero-Fierro, D.; Arcentales-Vera, B.; Palomino, K.; Magaña, H.; Bucio, E. Hydrogels Classification According to the Physical or Chemical Interactions and as Stimuli-Sensitive Materials. Gels 2021, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Duan, L.; Zhang, Y.; Cao, J.; Zhang, K. Current hydrogel advances in physicochemical and biological response-driven biomedical application diversity. Signal Transduct. Target. Ther. 2021, 6, 426. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cid, P.; Jiménez-Rosado, M.; Romero, A.; Pérez-Puyana, V. Novel Trends in Hydrogel Development for Biomedical Applications: A Review. Polymers 2022, 14, 3023. [Google Scholar] [CrossRef]
- Buwalda, S.J.; Boere, K.W.; Dijkstra, P.J.; Feijen, J.; Vermonden, T.; Hennink, W.E. Hydrogels in a historical perspective: From simple networks to smart materials. J. Control. Release 2014, 190, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.S.; Zhao, Y.; Forrest, M.L. Polyethylene Glycol Polyester Block Copolymers: Biocompatible Carriers for Nanoparticulate Drug Delivery. In Handbook of Harnessing Biomaterials in Nanomedicine: Preparation, Toxicity, and Applications; Jenny Stanford Publishing: Singapore, 2020; pp. 47–81. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef]
- Read, E.S.; Armes, S.P. Recent advances in shell cross-linked micelles. Chem. Commun. 2007, 38, 3021–3035. [Google Scholar] [CrossRef]
- Isoglu, I.; Ozsoy, Y.; Isoglu, S. Advances in Micelle-Based Drug Delivery: Cross-Linked Systems. Curr. Top. Med. Chem. 2017, 17, 1469–1489. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of Polyion Complex Micelles in an Aqueous Milieu from a Pair of Oppositely-Charged Block Copolymers with Poly(ethylene glycol) Segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar] [CrossRef]
- Gradzielski, M. Polyelectrolyte—Surfactant Complexes As a Formulation Tool for Drug Delivery. Langmuir ACS J. Surf. Colloids 2022, 38, 13330–13343. [Google Scholar] [CrossRef]
- Chen, L.; Xie, Z.; Hu, J.; Chen, X.; Jing, X. Enantiomeric PLA-PEG block copolymers and their stereocomplex micelles used as rifampin delivery. J. Nanoparticle Res. 2007, 9, 777–785. [Google Scholar] [CrossRef]
- Im, S.H.; Im, D.H.; Park, S.J.; Chung, J.J.; Jung, Y.; Kim, S.H. Stereocomplex Polylactide for Drug Delivery and Biomedical Applications: A Review. Molecules 2021, 26, 2846. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Lee, H.J. pH-Controlled, Polymer-Mediated Assembly of Polymer Micelle Nanoparticles. Langmuir 2006, 23, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Buwalda, S.J.; Dijkstra, P.J.; Calucci, L.; Forte, C.; Feijen, J. Influence of Amide versus Ester Linkages on the Properties of Eight-Armed PEG-PLA Star Block Copolymer Hydrogels. Biomacromolecules 2009, 11, 224–232. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Stereocomplex formation between enantiomeric poly(lactic acid)s. XI. Mechanical properties and morphology of solution-cast films. Polymer 1999, 40, 6699–6708. [Google Scholar] [CrossRef]
- Jing, Y.; Quan, C.; Liu, B.; Jiang, Q.; Zhang, C. A Mini Review on the Functional Biomaterials Based on Poly(lactic acid) Stereocomplex. Polym. Rev. 2016, 56, 262–286. [Google Scholar] [CrossRef]
- de Jong, S.J.; De Smedt, S.C.; Wahls, M.W.C.; Demeester, J.; van den Bosch, J.J.K.; Hennink, W.E. Novel Self-Assembled Hydrogels by Stereocomplex Formation in Aqueous Solution of Enantiomeric Lactic Acid Oligomers Grafted To Dextran. Macromolecules 2000, 33, 3680–3686. [Google Scholar] [CrossRef]
- Hiemstra, C.; Zhong, Z.; Li, L.; Dijkstra, P.J.; Feijen, J. In-Situ Formation of Biodegradable Hydrogels by Stereocomplexation of PEG-(PLLA)8 and PEG-(PDLA)8 Star Block Copolymers. Biomacromolecules 2006, 7, 2790–2795. [Google Scholar] [CrossRef]
- Fujiwara, T.; Mukose, T.; Yamaoka, T.; Yamane, H.; Sakurai, S.; Kimura, Y. Novel thermo-responsive formation of a hydrogel by stereo-complexation between PLLA-PEG-PLLA and PDLA-PEG-PDLA block copolymers. Macromol. Biosci. 2001, 1, 204–208. [Google Scholar] [CrossRef]
- Li, S.; Vert, M. Synthesis, Characterization, and Stereocomplex-Induced Gelation of Block Copolymers Prepared by Ring-Opening Polymerization of l(d)-Lactide in the Presence of Poly(ethylene glycol). Macromolecules 2003, 36, 8008–8014. [Google Scholar] [CrossRef]
- Shi, X.; Wu, J.; Wang, Z.; Song, F.; Gao, W.; Liu, S. Synthesis and properties of a temperature-sensitive hydrogel based on physical crosslinking via stereocomplexation of PLLA-PDLA. RSC Adv. 2020, 10, 19759–19769. [Google Scholar] [CrossRef]
- Cao, H.; Chang, X.; Mao, H.; Zhou, J.; Wu, Z.L.; Shan, G.; Bao, Y.; Pan, P. Stereocomplexed physical hydrogels with high strength and tunable crystallizability. Soft Matter 2017, 13, 8502–8510. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Kunduru, K.R.; Doppalapudi, S.; Domb, A.J.; Khan, W. Poly(lactic acid) based hydrogels. Adv. Drug Deliv. Rev. 2016, 107, 192–205. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.; van Eerdenbrugh, B.; van Nostrum, C.; Bosch, J.K.-V.D.; Hennink, W. Physically crosslinked dextran hydrogels by stereocomplex formation of lactic acid oligomers: Degradation and protein release behavior. J. Control. Release 2001, 71, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, C.; Zhong, Z.; Van Tomme, S.R.; van Steenbergen, M.J.; Jacobs, J.J.L.; Otter, W.D.; Hennink, W.E.; Feijen, J. In vitro and in vivo protein delivery from in situ forming poly(ethylene glycol)—poly(lactide) hydrogels. J. Control. Release 2007, 119, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Hewitt, D.R.O.; Quah, S.P.; Zheng, B.; Mattei, G.S.; Khalifah, P.G.; Grubbs, R.B.; Bhatia, S.R. Impact of stereochemistry on rheology and nanostructure of PLA-PEO-PLA triblocks: Stiff gels at intermediate l/d-lactide ratios. Soft Matter 2018, 14, 7255–7263. [Google Scholar] [CrossRef]
- Mao, H.; Pan, P.; Shan, G.; Bao, Y. In Situ Formation and Gelation Mechanism of Thermoresponsive Stereocomplexed Hydrogels upon Mixing Diblock and Triblock Poly(Lactic Acid)/Poly(Ethylene Glycol) Copolymers. J. Phys. Chem. B 2015, 119, 6471–6480. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Wang, C.; Chang, X.; Cao, H.; Shan, G.; Bao, Y.; Pan, P. Poly(lactic acid)/poly(ethylene glycol) stereocomplexed physical hydrogels showing thermally-induced gel-sol-gel multiple phase transitions. Mater. Chem. Front. 2018, 2, 313–322. [Google Scholar] [CrossRef]
- Mao, H.; Shan, G.; Bao, Y.; Wu, Z.L.; Pan, P. Thermoresponsive physical hydrogels of poly(lactic acid)/poly(ethylene glycol) stereoblock copolymers tuned by stereostructure and hydrophobic block sequence. Soft Matter 2016, 12, 4628–4637. [Google Scholar] [CrossRef]
- Buwalda, S.J.; Calucci, L.; Forte, C.; Dijkstra, P.J.; Feijen, J. Stereocomplexed 8-armed poly(ethylene glycol)-poly(lactide) star block copolymer hydrogels: Gelation mechanism, mechanical properties and degradation behavior. Polymer 2012, 53, 2809–2817. [Google Scholar] [CrossRef]
- Kubisa, P.; Lapienis, G.; Biela, T. Star-shaped copolymers with PLA-PEG arms and their potential applications as biomedical materials. Polym. Adv. Technol. 2021, 32, 3857–3866. [Google Scholar] [CrossRef]
- Chen, L.; Ci, T.; Yu, L.; Ding, J. Effects of Molecular Weight and Its Distribution of PEG Block on Micellization and Thermogellability of PLGA-PEG-PLGA Copolymer Aqueous Solutions. Macromolecules 2015, 48, 3662–3671. [Google Scholar] [CrossRef]
- Abebe, D.G.; Fujiwara, T. Controlled Thermoresponsive Hydrogels by Stereocomplexed PLA-PEG-PLA Prepared via Hybrid Micelles of Pre-Mixed Copolymers with Different PEG Lengths. Biomacromolecules 2012, 13, 1828–1836. [Google Scholar] [CrossRef]
- Mukose, T.; Fujiwara, T.; Nakano, J.; Taniguchi, I.; Miyamoto, M.; Kimura, Y.; Teraoka, I.; Lee, C.W. Hydrogel Formation between Enantiomeric B-A-B-Type Block Copolymers of Polylactides(PLLA or PDLA: A) and Polyoxyethylene(PEG: B); PEG-PLLA-PEG and PEG-PDLA-PEG. Macromol. Biosci. 2004, 4, 361–367. [Google Scholar] [CrossRef]
- Wennink, J.; Signori, F.; Karperien, M.; Bronco, S.; Feijen, J.; Dijkstra, P. Introducing small cationic groups into 4-armed PLLA-PEG copolymers leads to preferred micellization over thermo-reversible gelation. Polymer 2013, 54, 6894–6901. [Google Scholar] [CrossRef]
- Signore, G.; Abbandonato, G.; Storti, B.; Stöckl, M.; Subramaniam, V.; Bizzarri, R. Imaging the static dielectric constant in vitro and in living cells by a bioconjugable GFP chromophore analog. Chem. Commun. 2013, 49, 1723–1725. [Google Scholar] [CrossRef]
- Abbandonato, G.; Polli, D.; Viola, D.; Cerullo, G.; Storti, B.; Cardarelli, F.; Salomone, F.; Nifosì, R.; Signore, G.; Bizzarri, R. Simultaneous Detection of Local Polarizability and Viscosity by a Single Fluorescent Probe in Cells. Biophys. J. 2018, 114, 2212–2220. [Google Scholar] [CrossRef]
- Kumbhakar, M.; Nath, S.; Mukherjee, T.; Pal, H. Solvation dynamics in triton-X-100 and triton-X-165 micelles: Effect of micellar size and hydration. J. Chem. Phys. 2004, 121, 6026–6033. [Google Scholar] [CrossRef] [PubMed]
- Chassenieux, C.; Nicolai, T.; Benyahia, L. Rheology of associative polymer solutions. Curr. Opin. Colloid Interface Sci. 2011, 16, 18–26. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signori, F.; Wennink, J.W.H.; Bronco, S.; Feijen, J.; Karperien, M.; Bizzarri, R.; Dijkstra, P.J. Aggregation and Gelation Behavior of Stereocomplexed Four-Arm PLA-PEG Copolymers Containing Neutral or Cationic Linkers. Int. J. Mol. Sci. 2023, 24, 3327. https://doi.org/10.3390/ijms24043327
Signori F, Wennink JWH, Bronco S, Feijen J, Karperien M, Bizzarri R, Dijkstra PJ. Aggregation and Gelation Behavior of Stereocomplexed Four-Arm PLA-PEG Copolymers Containing Neutral or Cationic Linkers. International Journal of Molecular Sciences. 2023; 24(4):3327. https://doi.org/10.3390/ijms24043327
Chicago/Turabian StyleSignori, Francesca, Jos W. H. Wennink, Simona Bronco, Jan Feijen, Marcel Karperien, Ranieri Bizzarri, and Pieter J. Dijkstra. 2023. "Aggregation and Gelation Behavior of Stereocomplexed Four-Arm PLA-PEG Copolymers Containing Neutral or Cationic Linkers" International Journal of Molecular Sciences 24, no. 4: 3327. https://doi.org/10.3390/ijms24043327
APA StyleSignori, F., Wennink, J. W. H., Bronco, S., Feijen, J., Karperien, M., Bizzarri, R., & Dijkstra, P. J. (2023). Aggregation and Gelation Behavior of Stereocomplexed Four-Arm PLA-PEG Copolymers Containing Neutral or Cationic Linkers. International Journal of Molecular Sciences, 24(4), 3327. https://doi.org/10.3390/ijms24043327