

Proximity Labeling to Identify β-Arrestin1 Binding Partners Downstream of Ligand-Activated G Protein-Coupled Receptors


Abstract
1. Introduction
2. Results
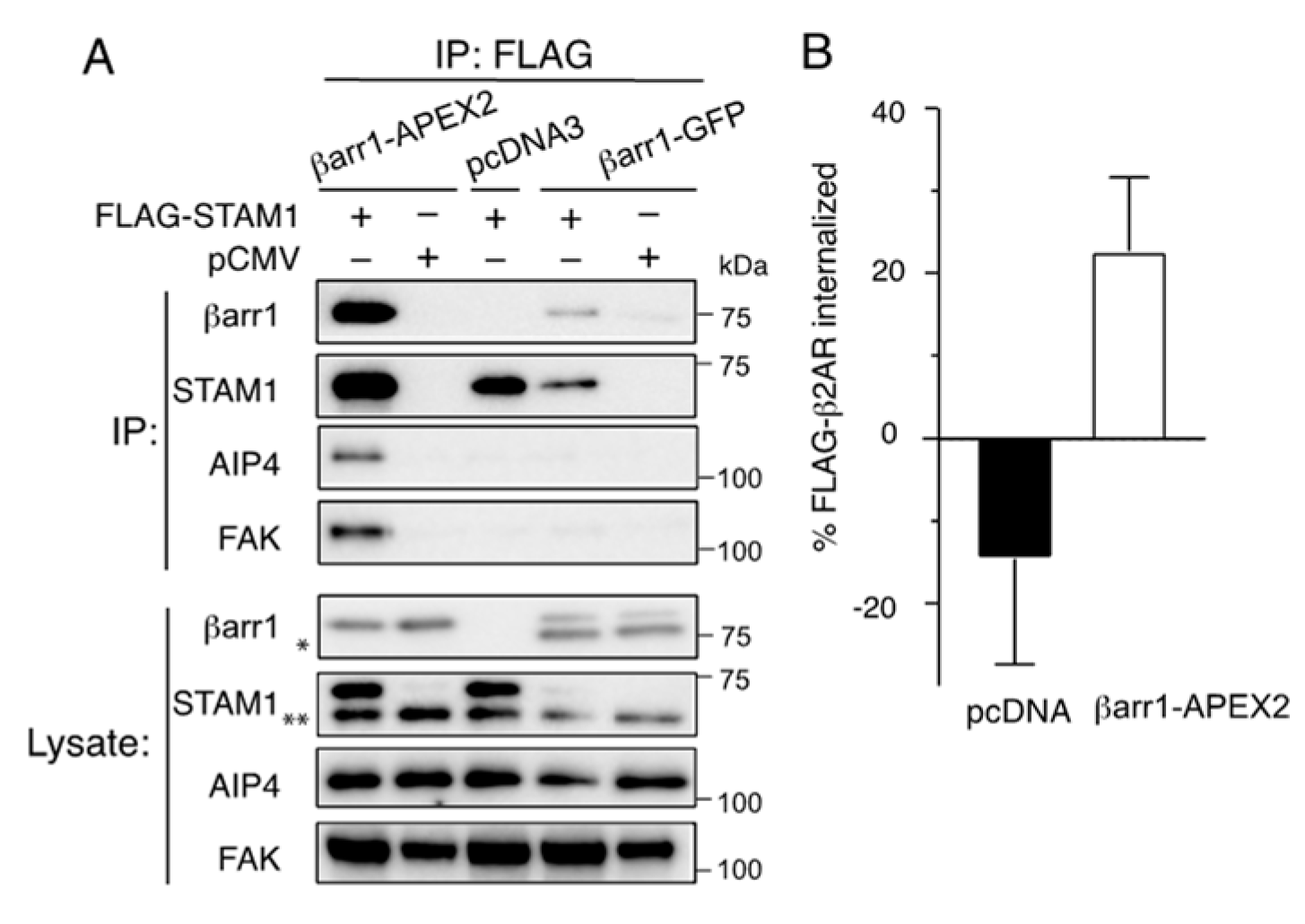
2.1. Characterization of β-Arrestin1-APEX2
2.2. Proximity Labeling by βarr1-APEX2

3. Discussion
4. Materials and Methods
4.1. Cell Culture, Antibodies, and Reagents
4.2. DNA Plasmids
4.3. Expression and Purification of Recombinant Proteins
4.4. Pulldown Assay
4.5. Coimmunoprecipitation
4.6. Biotin Labeling
4.7. Streptavidin Affinity Purification of Biotinylated Proteins
4.8. Sample Preparation for Mass Spectrometry Analysis
4.9. Receptor Surface Expression
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefkowitz, R.J. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998, 273, 18677–18680. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115 Pt 3, 455–465. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 2003, 375 Pt 3, 503–515. [Google Scholar] [CrossRef]
- Goodman, O.B., Jr.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Barlic, J.; Andrews, J.D.; Kelvin, A.A.; Bosinger, S.E.; DeVries, M.E.; Xu, L.; Dobransky, T.; Feldman, R.D.; Ferguson, S.S.; Kelvin, D.J. Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat. Immunol. 2000, 1, 227–233. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Dery, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Nat. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Modi, A.S.; Shukla, A.K.; Xiao, K.; Berthouze, M.; Ahn, S.; Wilkinson, K.D.; Miller, W.E.; Lefkowitz, R.J. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. USA 2009, 106, 6650–6655. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Marchese, A. Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 2010, 21, 2529–2541. [Google Scholar] [CrossRef]
- McDonald, P.H.; Cote, N.L.; Lin, F.T.; Premont, R.T.; Pitcher, J.A.; Lefkowitz, R.J. Identification of NSF as a beta-arrestin1-binding protein. Implications for beta2-adrenergic receptor regulation. J. Biol. Chem. 1999, 274, 10677–10680. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; McClatchy, D.B.; Shukla, A.K.; Zhao, Y.; Chen, M.; Shenoy, S.K.; Yates, J.R., 3rd; Lefkowitz, R.J. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12011–12016. [Google Scholar] [CrossRef]
- Lobingier, B.T.; Huttenhain, R.; Eichel, K.; Miller, K.B.; Ting, A.Y.; von Zastrow, M.; Krogan, N.J. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 2017, 169, 350–360.e12. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef]
- Paek, J.; Kalocsay, M.; Staus, D.P.; Wingler, L.; Pascolutti, R.; Paulo, J.A.; Gygi, S.P.; Kruse, A.C. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity Labeling. Cell 2017, 169, 338–349.e11. [Google Scholar] [CrossRef]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 2021, 18, 133–143. [Google Scholar] [CrossRef]
- Loh, K.H.; Stawski, P.S.; Draycott, A.S.; Udeshi, N.D.; Lehrman, E.K.; Wilton, D.K.; Svinkina, T.; Deerinck, T.J.; Ellisman, M.H.; Stevens, B.; et al. Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell 2016, 166, 1295–1307.e21. [Google Scholar] [CrossRef]
- Martell, J.D.; Yamagata, M.; Deerinck, T.J.; Phan, S.; Kwa, C.G.; Ellisman, M.H.; Sanes, J.R.; Ting, A.Y. A split horseradish peroxidase for the detection of intercellular protein-protein interactions and sensitive visualization of synapses. Nature Biotechnol. 2016, 34, 774–780. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Chandan, N.R.; Abraham, S.; SenGupta, S.; Parent, C.A.; Smrcka, A.V. A network of Galphai signaling partners is revealed by proximity labeling proteomics analysis and includes PDZ-RhoGEF. Sci. Signal. 2022, 15, eabi9869. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.T.; Wang, J.; Paulo, J.A.; Jiang, X.; Gygi, S.P.; Rockman, H.A. Mapping Angiotensin II Type 1 Receptor-Biased Signaling Using Proximity Labeling and Proteomics Identifies Diverse Actions of Biased Agonists. J. Proteome Res. 2021, 20, 3256–3267. [Google Scholar] [CrossRef]
- Civciristov, S.; Huang, C.; Liu, B.; Marquez, E.A.; Gondin, A.B.; Schittenhelm, R.B.; Ellisdon, A.M.; Canals, M.; Halls, M.L. Ligand-dependent spatiotemporal signaling profiles of the mu-opioid receptor are controlled by distinct protein-interaction networks. J. Biol. Chem. 2019, 294, 16198–16213. [Google Scholar] [CrossRef]
- Barak, L.S.; Ferguson, S.S.; Zhang, J.; Caron, M.G. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 1997, 272, 27497–27500. [Google Scholar] [CrossRef]
- Alekhina, O.; Marchese, A. beta-Arrestin1 and Signal-transducing Adaptor Molecule 1 (STAM1) Cooperate to Promote Focal Adhesion Kinase Autophosphorylation and Chemotaxis via the Chemokine Receptor CXCR4. J. Biol. Chem. 2016, 291, 26083–26097. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Gurevich, V.V.; Vishnivetskiy, S.A.; Klug, C.S.; Marchese, A. A non-GPCR-binding partner interacts with a novel surface on beta-arrestin1 to mediate GPCR signaling. J. Biol. Chem. 2020, 295, 14111–14124. [Google Scholar] [CrossRef]
- Zhuo, Y.; Crecelius, J.M.; Marchese, A. G protein-coupled receptor kinase phosphorylation of distal C-tail sites specifies betaarrestin1-mediated signaling by chemokine receptor CXCR4. J. Biol. Chem. 2022, 298, 102351. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Curto, E.; Inoue, A.; Jenkins, L.; Raihan, S.Z.; Prihandoko, R.; Tobin, A.B.; Milligan, G. Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J. Biol. Chem. 2016, 291, 27147–27159. [Google Scholar] [CrossRef]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Kawakami, K.; Aoki, J.; Messer, K.; Sunahara, R.; et al. Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signaling to ERK. Sci. Signal. 2017, 10, eaal3395. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Marchese, A. The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4-promoted lysosomal degradation of the mammalian target of rapamycin antagonist DEPTOR. J. Biol. Chem. 2015, 290, 6810–6824. [Google Scholar] [CrossRef]
- English, E.J.; Mahn, S.A.; Marchese, A. Endocytosis is required for C-X-C chemokine receptor type 4 (CXCR4)-mediated Akt activation and anti-apoptotic signaling. J. Biol. Chem. 2018, 293, 11470–11480. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Trejo, J.; Benovic, J.L.; Marchese, A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 2007, 282, 36971–36979. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic. Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Cullen, P.J. Endosomal sorting and signalling: An emerging role for sorting nexins. Nat. Rev. Mol. Cell Biol. 2008, 9, 574–582. [Google Scholar] [CrossRef]
- Bendris, N.; Schmid, S.L. Endocytosis, Metastasis and Beyond: Multiple Facets of SNX9. Trends Cell Biol. 2017, 27, 189–200. [Google Scholar] [CrossRef]
- Yarar, D.; Waterman-Storer, C.M.; Schmid, S.L. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev. Cell 2007, 13, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schoneberg, J.; Ullrich, A.; Lampe, A.; Muller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Kovoor, A.; Celver, J.; Abdryashitov, R.I.; Chavkin, C.; Gurevich, V.V. Targeted construction of phosphorylation-independent beta-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999, 274, 6831–6834. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Benovic, J.L. Visual arrestin binding to rhodopsin. Diverse functional roles of positively charged residues within the phosphorylation-recognition region of arrestin. J. Biol. Chem. 1995, 270, 6010–6016. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.M.; Gurevich, V.V.; Prossnitz, E.R.; Engen, J.R. Conformational Differences Between Arrestin2 and Pre-activated Mutants as Revealed by Hydrogen Exchange Mass Spectrometry. J. Mol. Biol. 2005, 351, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Chereau, D.; Kerff, F.; Graceffa, P.; Grabarek, Z.; Langsetmo, K.; Dominguez, R. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 16644–16649. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, M.; Berman, K.S.; Cobb, M.H. Isolation of TAO1, a protein kinase that activates MEKs in stress-activated protein kinase cascades. J. Biol. Chem. 1998, 273, 28625–28632. [Google Scholar] [CrossRef]
- Chen, Z.; Raman, M.; Chen, L.; Lee, S.F.; Gilman, A.G.; Cobb, M.H. TAO (thousand-and-one amino acid) protein kinases mediate signaling from carbachol to p38 mitogen-activated protein kinase and ternary complex factors. J. Biol. Chem. 2003, 278, 22278–22283. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Heid, H.W.; Mundel, T.M.; Kruger, M.; Reiser, J.; Kriz, W. Synaptopodin: An actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol. 1997, 139, 193–204. [Google Scholar] [CrossRef]
- Pestonjamasp, K.N.; Pope, R.K.; Wulfkuhle, J.D.; Luna, E.J. Supervillin (p205): A novel membrane-associated, F-actin-binding protein in the villin/gelsolin superfamily. J. Cell Biol. 1997, 139, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Jacques, K.M.; Stauffer, S.; Kubosaki, A.; Zhu, K.; Hirsch, D.S.; Resau, J.; Zheng, Y.; Randazzo, P.A. ARAP1: A point of convergence for Arf and Rho signaling. Mol. Cell 2002, 9, 109–119. [Google Scholar] [CrossRef]
- Bhandari, D.; Robia, S.L.; Marchese, A. The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol. Biol. Cell 2009, 20, 1324–1339. [Google Scholar] [CrossRef]
- St-Germain, J.R.; Samavarchi Tehrani, P.; Wong, C.; Larsen, B.; Gingras, A.C.; Raught, B. Variability in Streptavidin-Sepharose Matrix Quality Can Significantly Affect Proximity-Dependent Biotinylation (BioID) Data. J. Proteome Res. 2020, 19, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Udeshi, N.D.; Lam, S.S.; Loh, K.H.; Cox, K.J.; Pedram, K.; Carr, S.A.; Ting, A.Y. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef]
- D’Agostino, G.; Artinger, M.; Locati, M.; Perez, L.; Legler, D.F.; Bianchi, M.E.; Ruegg, C.; Thelen, M.; Marchese, A.; Rocchi, M.B.L.; et al. beta-Arrestin1 and beta-Arrestin2 Are Required to Support the Activity of the CXCL12/HMGB1 Heterocomplex on CXCR4. Front. Immunol. 2020, 11, 550824. [Google Scholar] [CrossRef]
- Zhuo, Y.; Vishnivetskiy, S.A.; Zhan, X.; Gurevich, V.V.; Klug, C.S. Identification of receptor binding-induced conformational changes in non-visual arrestins. J. Biol. Chem. 2014, 289, 20991–21002. [Google Scholar] [CrossRef]
- Hanson, S.M.; Gurevich, V.V. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J. Biol. Chem. 2006, 281, 3458–3462. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Benovic, J.L. Arrestin: Mutagenesis, expression, purification, and functional characterization. In Meth Enzymol; Krzystof, P., Ed.; Academic Press: New York, NY, USA, 2000; Volume 315, pp. 422–437. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Annotation | Symbol | Name | Abundance Ratio (CXCL12/Basal) | UniProt Accession Number |
|---|---|---|---|---|
| Actin Cytoskeleton | SYNPO | SYNPO protein | 1.33 ± 0.42 | A7MD96 |
| Signal Transduction | TAOK1 | Serine/threonine-protein kinase TAO1 (TAO—thousand and one amino acid) | 1.24 ± 0.24 | Q7L7X3 |
| Membrane Trafficking | SNX9 | Sorting nexin 9 | 1.23 ± 0.19 | Q9Y5X1 |
| Cholesterol Biosynthesis | DHCR24 | Delta(24)-sterol reductase | 1.24 ± 0.08 | Q15392 |
| Translation Regulation | BZW2 | eIF5-mimic protein 1 | 1.22 ± 0.17 | Q9Y6E2 |
| Functional Annotation | Symbol | Name | Abundance Ratio (CXCL12/Basal) | UniProt Accession Number | |
|---|---|---|---|---|---|
| Actin Cytoskeleton and Cell Motility | SVIL | Supervillin | 1.23 ± 0.23 | O95425 | |
| ARAP1 | Arf-GAP with Rho-GAP domain, ANK repeat and PH domain-containing protein 1 | 1.24 ± 0.08 | Q96P48 | *** | |
| SPAG1 | Sperm associated antigen 1, isoform CRA_b | 1.42 ± 0.40 | A0A024R9D8 | ||
| DSC1 | Desmocollin-1 | 1.42 ± 0.68 | Q08554 | ||
| NEXN | Nexilin | 1.30 | Q0ZGT2 | ** | |
| FMNL1 | Formin-like protein 1 | 1.24 ± 0.40 | O95466 | ||
| CD44 | CD44 antigen | 1.22 ± 0.30 | P16070 | ||
| CAP2 | Adenylyl cyclase-associated protein 2 | 1.23 | P40123 | ** | |
| Membrane Trafficking | VAC14 | Protein VAC14 homolog | 1.22 | Q08AM6 | *** |
| HPCAL1 | Hippocalcin-like protein 1 | 1.26 | P37235 | * | |
| Protein Quality Control | AGAP3 | Arf-GAP with GTPase, ANK repeat and PH domain-containing protein 3 | 1.21 ± 0.15 | Q96P47 | |
| Translation Regulation | SF4 | Splicing factor 4 | 1.26 ± 0.01 | Q08170 | |
| CUX1 | Homeobox protein cut-like 1 | 1.26 ± 0.11 | P39880 | ||
| LCMT2 | cDNA FLJ76457, highly similar to Homo sapiens leucine carboxyl methyltransferase 2 (LCMT2), mRNA | 1.31 ± 0.11 | A8K972/O60294 | ||
| ZNF622 | Zinc finger protein 622 | 1.32 ± 0.58 | Q969S3 | ||
| WDR33 | pre-mRNA 3′ end processing protein WDR33 | 1.21 | Q9C0J8 | ** | |
| RPL37 | 60S ribosomal protein L37 | 1.36 | P61927 | ** | |
| Epigenetic Regulation | PPHLN1 | Periphilin-1 | 1.27 ± 0.01 | Q8NEY8/F8W0Q9 | |
| ANP32E | Acidic leucine-rich nuclear phosphoprotein 32 family member E | 3.97 ± 4.26 | Q9BTT0 | ||
| Cytoskeleton Organization | CSKI2 | Caskin-2 | 1.25 ± 0.35 | Q8WXE0 | |
| FLJ61294 | cDNA FLJ61294, highly similar to Keratin, type I cytoskeletal 17 | 1.23 ± 0.19 | B4DJM5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuo, Y.; Robleto, V.L.; Marchese, A. Proximity Labeling to Identify β-Arrestin1 Binding Partners Downstream of Ligand-Activated G Protein-Coupled Receptors. Int. J. Mol. Sci. 2023, 24, 3285. https://doi.org/10.3390/ijms24043285
Zhuo Y, Robleto VL, Marchese A. Proximity Labeling to Identify β-Arrestin1 Binding Partners Downstream of Ligand-Activated G Protein-Coupled Receptors. International Journal of Molecular Sciences. 2023; 24(4):3285. https://doi.org/10.3390/ijms24043285
Chicago/Turabian StyleZhuo, Ya, Valeria L. Robleto, and Adriano Marchese. 2023. "Proximity Labeling to Identify β-Arrestin1 Binding Partners Downstream of Ligand-Activated G Protein-Coupled Receptors" International Journal of Molecular Sciences 24, no. 4: 3285. https://doi.org/10.3390/ijms24043285
APA StyleZhuo, Y., Robleto, V. L., & Marchese, A. (2023). Proximity Labeling to Identify β-Arrestin1 Binding Partners Downstream of Ligand-Activated G Protein-Coupled Receptors. International Journal of Molecular Sciences, 24(4), 3285. https://doi.org/10.3390/ijms24043285