

Intersection of the Orphan G Protein-Coupled Receptor, GPR19, with the Aging Process

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. GPCR Signaling
1.2. Aging and GPCR Functionality
1.3. GPCR-Based Control of Aging-Related Mechanisms
2. GPR19 and Aging-Related Activity
2.1. GPR19 and Energy Metabolism
2.2. GPR19 and Cell Cycle Regulation
2.3. GPR19 and Oxidative Stress
2.4. Circadian Rhythms
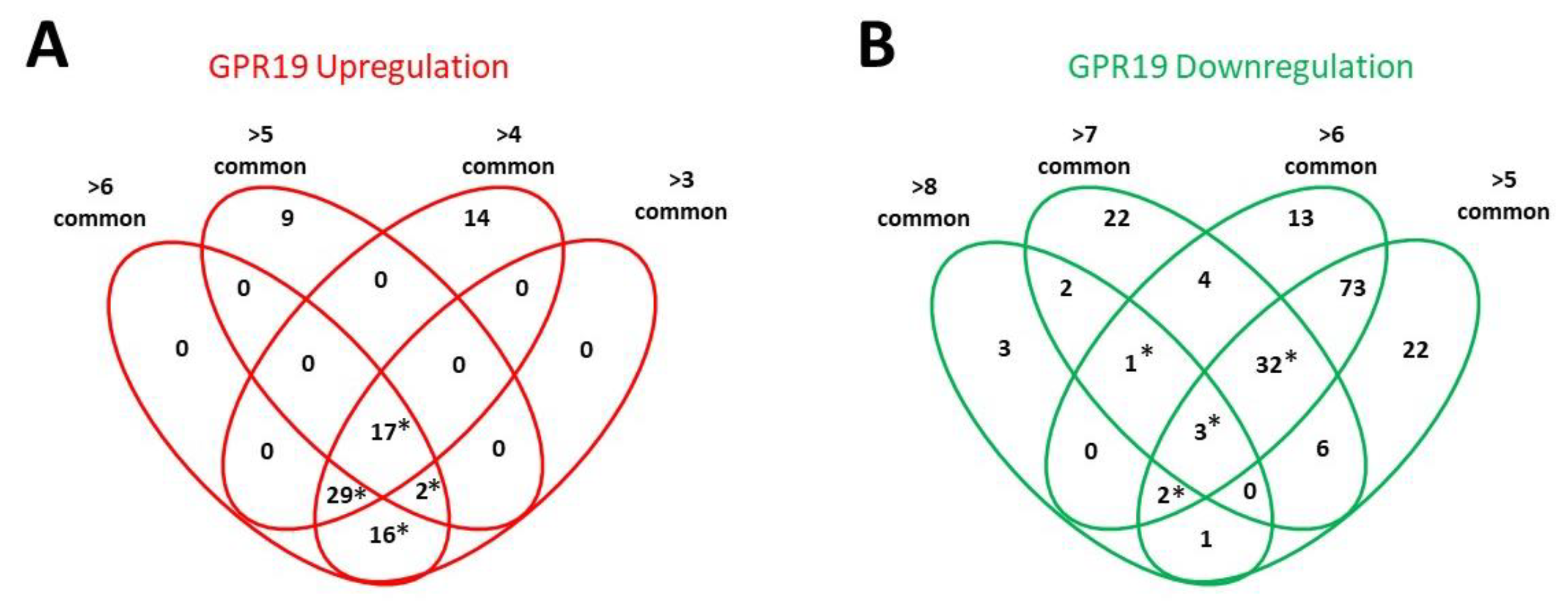
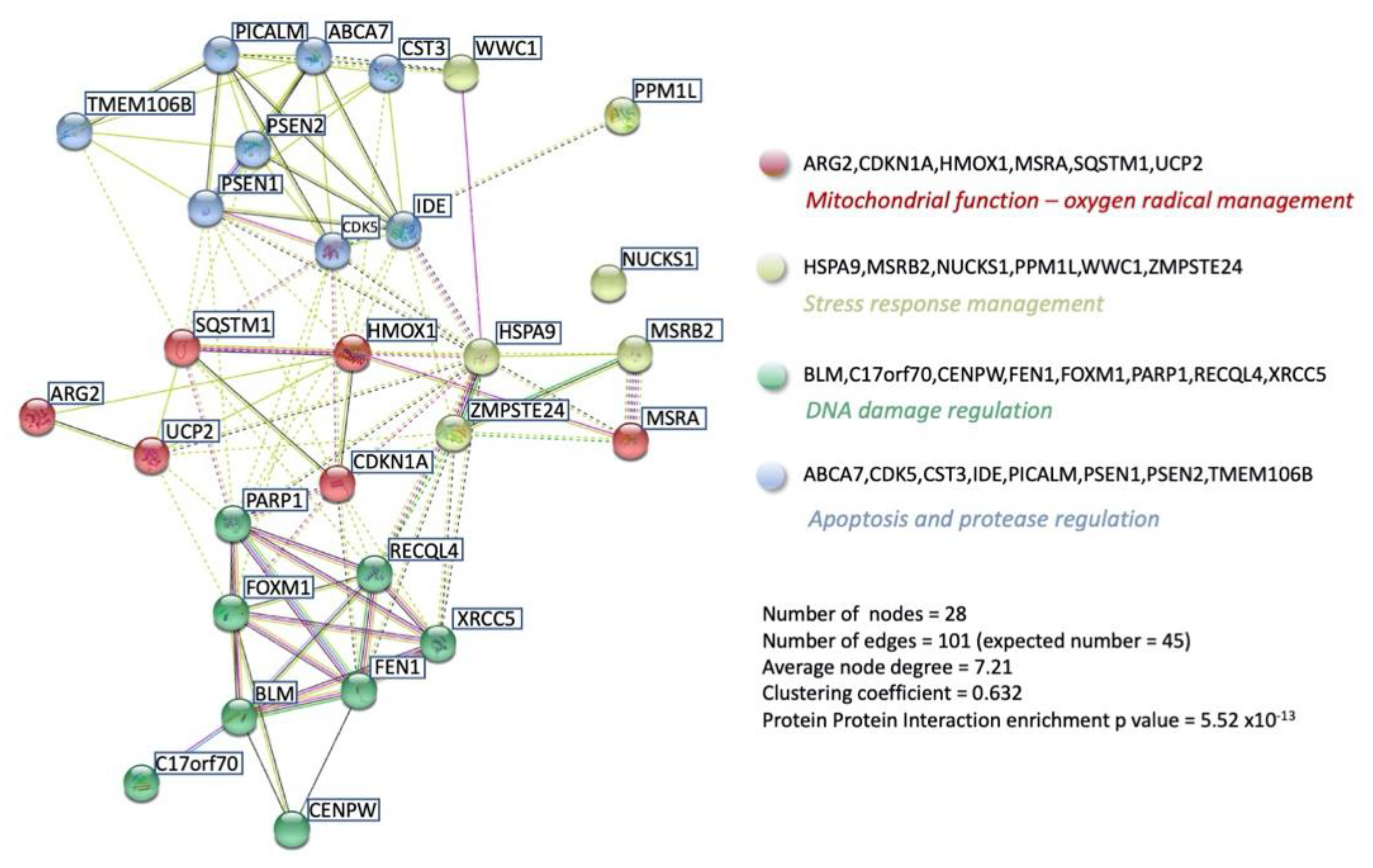
3. Functional GPR19 Molecular Signatures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Van Gastel, J.; Leysen, H.; Boddaert, J.; Vangenechten, L.; Luttrell, L.M.; Martin, B.; Maudsley, S. Aging-related modifications to G protein-coupled receptor signaling diversity. Pharmacol. Ther. 2021, 223, 107793. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Schiöth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Janssens, J.; Etienne, H.; Idriss, S.; Azmi, A.; Martin, B.; Maudsley, S. Systems-Level G Protein-Coupled Receptor Therapy Across a Neurodegenerative Continuum by the GLP-1 Receptor System. Front. Endocrinol. 2014, 5, 142. [Google Scholar] [CrossRef]
- Tse, L.H.; Wong, Y.H. GPCRs in Autocrine and Paracrine Regulations. Front. Endocrinol. 2019, 10, 428. [Google Scholar] [CrossRef]
- De Oliveira, P.G.; Ramos, M.L.S.; Amaro, A.J.; Dias, R.A.; Vieira, S.I. G(i/o)-Protein Coupled Receptors in the Aging Brain. Front. Aging Neurosci. 2019, 11, 89. [Google Scholar] [CrossRef]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef]
- Leysen, H.; Walter, D.; Christiaenssen, B.; Vandoren, R.; Harputluoğlu, İ.; van Loon, N.; Maudsley, S. GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease. Int. J. Mol. Sci. 2021, 22, 13387. [Google Scholar] [CrossRef]
- Del Castillo, J.; Katz, B. Interaction at end-plate receptors between different choline derivatives. Proc. R. Soc. Lond. B Biol. Sci. 1957, 146, 369–381. [Google Scholar]
- De Lean, A.; Stadel, J.M.; Lefkowitz, R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980, 255, 7108–7117. [Google Scholar] [CrossRef]
- Gardella, T.J.; Luck, M.D.; Jensen, G.S.; Schipani, E.; Potts, J.T., Jr.; Jüppner, H. Inverse agonism of amino-terminally truncated parathyroid hormone (PTH) and PTH-related peptide (PTHrP) analogs revealed with constitutively active mutant PTH/PTHrP receptors. Endocrinology 1996, 137, 3936–3941. [Google Scholar] [CrossRef] [PubMed]
- Gether, U.; Lin, S.; Kobilka, B.K. Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand-specific conformational changes. J. Biol. Chem. 1995, 270, 28268–28275. [Google Scholar] [CrossRef] [PubMed]
- Leff, P. The two-state model of receptor activation. Trends Pharmacol. Sci. 1995, 16, 89–97. [Google Scholar] [CrossRef]
- Parma, J.; Duprez, L.; van Sande, J.; Cochaux, P.; Gervy, C.; Mockel, J.; Dumont, J.; Vassart, G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 1993, 365, 649–651. [Google Scholar] [CrossRef]
- Pozvek, G.; Hilton, J.M.; Quiza, M.; Houssami, S.; Sexton, P.M. Structure/function relationships of calcitonin analogues as agonists, antagonists, or inverse agonists in a constitutively activated receptor cell system. Mol. Pharmacol. 1997, 51, 658–665. [Google Scholar] [CrossRef]
- Shenker, A.; Laue, L.; Kosugi, S.; Merendino, J.J., Jr.; Minegishi, T.; Cutler, G.B., Jr. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature 1993, 365, 652–654. [Google Scholar] [CrossRef]
- Samama, P.; Cotecchia, S.; Costa, T.; Lefkowitz, R.J. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993, 268, 4625–4636. [Google Scholar] [CrossRef]
- Ceresa, B.P.; Limbird, L.E. Mutation of an aspartate residue highly conserved among G-protein-coupled receptors results in nonreciprocal disruption of alpha 2-adrenergic receptor-G-protein interactions. A negative charge at amino acid residue 79 forecasts alpha 2A-adrenergic receptor sensitivity to allosteric modulation by monovalent cations and fully effective receptor/G-protein coupling. J. Biol. Chem. 1994, 269, 29557–29564. [Google Scholar]
- Morin, D.; Cotte, N.; Balestre, M.N.; Mouillac, B.; Manning, M.; Breton, C.; Barberis, C. The D136A mutation of the V2 vasopressin receptor induces a constitutive activity which permits discrimination between antagonists with partial agonist and inverse agonist activities. FEBS Lett. 1998, 441, 470–475. [Google Scholar] [CrossRef]
- Alewijnse, A.E.; Timmerman, H.; Jacobs, E.H.; Smit, M.J.; Roovers, E.; Cotecchia, S.; Leurs, R. The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol. Pharmacol. 2000, 57, 890–898. [Google Scholar] [PubMed]
- Pauwels, P.J.; Tardif, S.; Wurch, T.; Colpaert, F.C. Facilitation of constitutive alpha(2A)-adrenoceptor activity by both single amino acid mutation (Thr(373)Lys) and g(alphao) protein coexpression: Evidence for inverse agonism. J. Pharmacol. Exp. Ther. 2000, 292, 654–663. [Google Scholar] [PubMed]
- Maudsley, S.; Davidson, L.; Pawson, A.J.; Chan, R.; López de Maturana, R.; Millar, R.P. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res. 2004, 64, 7533–7544. [Google Scholar] [CrossRef] [PubMed]
- Conklin, B.R.; Bourne, H.R. Structural elements of G alpha subunits that interact with G beta gamma, receptors, and effectors. Cell 1993, 73, 631–641. [Google Scholar] [CrossRef]
- Ernst, O.P.; Hofmann, K.P.; Sakmar, T.P. Characterization of rhodopsin mutants that bind transducin but fail to induce GTP nucleotide uptake. Classification of mutant pigments by fluorescence, nucleotide release, and flash-induced light-scattering assays. J. Biol. Chem. 1995, 270, 10580–10586. [Google Scholar] [CrossRef] [PubMed]
- Farahbakhsh, Z.T.; Hideg, K.; Hubbell, W.L. Photoactivated conformational changes in rhodopsin: A time-resolved spin label study. Science 1993, 262, 1416–1419. [Google Scholar] [CrossRef]
- Franke, R.R.; Sakmar, T.P.; Graham, R.M.; Khorana, H.G. Structure and function in rhodopsin. Studies of the interaction between the rhodopsin cytoplasmic domain and transducin. J. Biol. Chem. 1992, 267, 14767–14774. [Google Scholar] [CrossRef]
- Donnelly, D.; Maudsley, S.; Gent, J.P.; Moser, R.N.; Hurrell, C.R.; Findlay, J.B. Conserved polar residues in the transmembrane domain of the human tachykinin NK2 receptor: Functional roles and structural implications. Biochem. J. 1999, 339 Pt 1, 55–61. [Google Scholar] [CrossRef]
- Van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Santos-Otte, P.; Luttrell, L.M.; Martin, B.; Maudsley, S. β-Arrestin Based Receptor Signaling Paradigms: Potential Therapeutic Targets for Complex Age-Related Disorders. Front. Pharmacol. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Wess, J. G-protein-coupled receptors: Molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J. 1997, 11, 346–354. [Google Scholar] [CrossRef]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Ward, R.; An, S.; Guo, X.X.; Li, W.; Xu, T.R. Biased signalling: The instinctive skill of the cell in the selection of appropriate signalling pathways. Biochem. J. 2015, 470, 155–167. [Google Scholar] [CrossRef]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Ferguson, S.S.G.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.T.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. β-arrestin-dependent formation of β2 adrenergic receptor-src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Van Gastel, J.; Etienne, H.; Azmi, A.; Maudsley, S. The synergistic GIT2-RXFP3 system in the brain and its importance in age-related disorders. Front. Aging Neurosci. 2016, 3, 8. [Google Scholar] [CrossRef]
- Maudsley, S.; Martin, B.; Janssens, J.; Etienne, H.; Jushaj, A.; van Gastel, J.; Willemsen, A.; Chen, H.; Gesty-Palmer, D.; Luttrell, L.M. Informatic deconvolution of biased GPCR signaling mechanisms from in vivo pharmacological experimentation. Methods 2016, 92, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, W.; Keselman, A.; Park, S.S.; Zhou, Y.; Wang, L.; Brenneman, R.; Martin, B.; Maudsley, S. Repetitive peroxide exposure reveals pleiotropic mitogen-activated protein kinase signaling mechanisms. J. Signal Transduct. 2011, 2011, 636951. [Google Scholar] [CrossRef]
- Davidson, L.; Pawson, A.J.; López de Maturana, R.; Freestone, S.H.; Barran, P.; Millar, R.P.; Maudsley, S. Gonadotropin-releasing hormone-induced activation of diacylglycerol kinase-zeta and its association with active c-src. J. Biol. Chem. 2004, 279, 11906–11916. [Google Scholar] [CrossRef]
- Maudsley, S.; Davidson, L.; Pawson, A.J.; Freestone, S.H.; López de Maturana, R.; Thomson, A.A.; Millar, R.P. Gonadotropin-releasing hormone functionally antagonizes testosterone activation of the human androgen receptor in prostate cells through focal adhesion complexes involving Hic-5. Neuroendocrinology 2006, 84, 285–300. [Google Scholar] [CrossRef]
- Van Gastel, J.; Leysen, H.; Santos-Otte, P.; Hendrickx, J.O.; Azmi, A.; Martin, B.; Maudsley, S. The RXFP3 receptor is functionally associated with cellular responses to oxidative stress and DNA damage. Aging 2019, 11, 11268–11313. [Google Scholar] [CrossRef]
- Van Gastel, J.; Cai, H.; Cong, W.N.; Chadwick, W.; Daimon, C.; Leysen, H.; Hendrickx, J.O.; de Schepper, R.; Vangenechten, L.; Van Turnhout, J.; et al. Multidimensional informatic deconvolution defines gender-specific roles of hypothalamic GIT2 in aging trajectories. Mech. Ageing Dev. 2019, 184, 111150. [Google Scholar] [CrossRef] [PubMed]
- Van Gastel, J.; Boddaert, J.; Jushaj, A.; Premont, R.T.; Luttrell, L.M.; Janssens, J.; Martin, B.; Maudsley, S. GIT2-A keystone in ageing and age-related disease. Ageing Res. Rev. 2018, 43, 46–63. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Gesty-Palmer, D.; Yuan, L.; Martin, B.; Wood, W.H., III; Lee, M.H.; Janech, M.G.; Tsoi, L.C.; Zheng, W.J.; Luttrell, L.M.; Maudsley, S. β-arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol. Endocrinol. 2013, 27, 296–314. [Google Scholar] [CrossRef]
- Maudsley, S.; Siddiqui, S.; Martin, B. Systems analysis of arrestin pathway functions. Prog. Mol. Biol. Transl. Sci. 2013, 118, 431–467. [Google Scholar]
- Maudsley, S.; Martin, B.; Gesty-Palmer, D.; Cheung, H.; Johnson, C.; Patel, S.; Becker, K.G.; Wood, W.H., III; Zhang, Y.; Lehrmann, E.; et al. Delineation of a conserved arrestin-biased signaling repertoire in vivo. Mol. Pharmacol. 2015, 87, 706–717. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar]
- Pierce, K.L.; Maudsley, S.; Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 1489–1494. [Google Scholar] [CrossRef]
- Ferrand, A.; Kowalski-Chauvel, A.; Bertrand, C.; Escrieut, C.; Mathieu, A.; Portolan, G.; Pradayrol, L.; Fourmy, D.; Dufresne, M.; Seva, C. A novel mechanism for JAK2 activation by a G protein-coupled receptor, the CCK2R: Implication of this signaling pathway in pancreatic tumor models. J. Biol. Chem. 2005, 280, 10710–10715. [Google Scholar] [CrossRef]
- Li, H.; Eishingdrelo, A.; Kongsamut, S.; Eishingdrelo, H. G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal Transduct. Target. Ther. 2016, 1, 16018. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Guillin, O.; Diaz, J.; Griffon, N.; Sokoloff, P. GIPC recruits GAIP (RGS19) to attenuate dopamine D2 receptor signaling. Mol. Biol. Cell 2004, 15, 4926–4937. [Google Scholar] [CrossRef] [PubMed]
- Andreev, J.; Galisteo, M.L.; Kranenburg, O.; Logan, S.K.; Chiu, E.S.; Okigaki, M.; Cary, L.A.; Moolenaar, W.H.; Schlessinger, J. Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J. Biol. Chem. 2001, 276, 20130–20135. [Google Scholar] [CrossRef] [PubMed]
- Leysen, H.; Walter, D.; Clauwaert, L.; Hellemans, L.; van Gastel, J.; Vasudevan, L.; Martin, B.; Maudsley, S. The Relaxin-3 Receptor, RXFP3, Is a Modulator of Aging-Related Disease. Int. J. Mol. Sci. 2022, 23, 4387. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Zhang, Z.; Wu, Y.; Kang, J.; Pei, G. Beta-arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-kappaB activation. EMBO J. 2005, 24, 4237–4246. [Google Scholar] [CrossRef]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature 2011, 477, 349–353. [Google Scholar] [CrossRef]
- Premont, R.T.; Claing, A.; Vitale, N.; Freeman, J.L.; Pitcher, J.A.; Patton, W.A.; Moss, J.; Vaughan, M.; Lefkowitz, R.J. beta2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc. Natl. Acad. Sci. USA 1998, 95, 14082–14087. [Google Scholar] [CrossRef]
- Chadwick, W.; Martin, B.; Chapter, M.C.; Park, S.S.; Wang, L.; Daimon, C.M.; Brenneman, R.; Maudsley, S. GIT2 acts as a potential keystone protein in functional hypothalamic networks associated with age-related phenotypic changes in rats. PLoS ONE 2012, 7, e36975. [Google Scholar] [CrossRef]
- Martin, B.; Chadwick, W.; Janssens, J.; Premont, R.T.; Schmalzigaug, R.; Becker, K.G.; Lehrmann, E.; Wood, W.H.; Zhang, Y.; Siddiqui, S.; et al. GIT2 Acts as a Systems-Level Coordinator of Neurometabolic Activity and Pathophysiological Aging. Front. Endocrinol. 2016, 6, 191. [Google Scholar] [CrossRef]
- Siddiqui, S.; Lustig, A.; Carter, A.; Sankar, M.; Daimon, C.M.; Premont, R.T.; Etienne, H.; van Gastel, J.; Azmi, A.; Janssens, J.; et al. Genomic deletion of GIT2 induces a premature age-related thymic dysfunction and systemic immune system disruption. Aging 2017, 9, 706–740. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Cai, H.; Park, S.S.; Siddiqui, S.; Premont, R.T.; Schmalzigaug, R.; Paramasivam, M.; Seidman, M.; Bodogai, I.; Biragyn, A.; et al. Nuclear GIT2 is an ATM substrate and promotes DNA repair. Mol. Cell. Biol. 2015, 35, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Patel, S.A.; Park, S.S.; Luttrell, L.M.; Martin, B. Functional signaling biases in G protein-coupled receptors: Game Theory and receptor dynamics. Mini Rev. Med. Chem. 2012, 12, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; McDonald, P.H. Seeking Ligand Bias: Assessing GPCR Coupling to Beta-Arrestins for Drug Discovery. Drug Discov. Today Technol. 2010, 7, e37–e42. [Google Scholar] [CrossRef]
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Nieto, A.; Hara, M.R.; Quereda, V.; Grant, W.; Saunders, V.; Xiao, K.; McDonald, P.H.; Duckett, D.R. βarrestin-1 regulates DNA repair by acting as an E3-ubiquitin ligase adaptor for 53BP1. Cell Death Differ. 2020, 27, 1200–1213. [Google Scholar] [CrossRef]
- Bayer, P.; Gatenby, R.A.; McDonald, P.H.; Duckett, D.R.; Staňková, K.; Brown, J.S. Coordination games in cancer. PLoS ONE 2022, 17, e0261578. [Google Scholar] [CrossRef]
- McDonald, P.H.; Lefkowitz, R.J. Beta-Arrestins: New roles in regulating heptahelical receptors’ functions. Cell. Signal. 2001, 13, 683–689. [Google Scholar] [CrossRef]
- Dalle, S.; Ravier, M.A.; Bertrand, G. Emerging roles for β-arrestin-1 in the control of the pancreatic β-cell function and mass: New therapeutic strategies and consequences for drug screening. Cell. Signal. 2011, 23, 522–528. [Google Scholar] [CrossRef]
- Jiang, R.; Song, X.; Bali, P.; Smith, A.; Bayona, C.R.; Lin, L.; Cameron, M.D.; McDonald, P.H.; Kenny, P.J.; Kamenecka, T.M. Disubstituted piperidines as potent orexin (hypocretin) receptor antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 3890–3894. [Google Scholar] [CrossRef]
- Pydi, S.P.; Barella, L.F.; Zhu, L.; Meister, J.; Rossi, M.; Wess, J. β-Arrestins as Important Regulators of Glucose and Energy Homeostasis. Annu. Rev. Physiol. 2022, 84, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Wess, J. The Two β-Arrestins Regulate Distinct Metabolic Processes: Studies with Novel Mutant Mouse Models. Int. J. Mol. Sci. 2022, 23, 495. [Google Scholar] [CrossRef] [PubMed]
- Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Santos-Otte, P.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. Int. J. Mol. Sci. 2018, 19, 2919. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wang, L.; Zhang, J.; Dong, W.; Zhang, T.; Ni, Y.; Cao, H.; Wang, K.; Li, Y.; Wang, Y.; et al. ARRB1 enhances the chemosensitivity of lung cancer through the mediation of DNA damage response. Oncol. Rep. 2017, 37, 761–767. [Google Scholar] [CrossRef]
- Sood, R.; Ritov, G.; Richter-Levin, G.; Barki-Harrington, L. Selective increase in the association of the β2 adrenergic receptor, β Arrestin-1 and p53 with Mdm2 in the ventral hippocampus one month after underwater trauma. Behav. Brain Res. 2013, 240, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Cong, W.N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Fang, M.; Ni, B.; Lu, D.; Martin, B.; Maudsley, S. Central role of the EGF receptor in neurometabolic aging. Int. J. Endocrinol. 2012, 2012, 739428. [Google Scholar] [CrossRef]
- Mezhnina, V.; Ebeigbe, O.P.; Poe, A.; Kondratov, R.V. Circadian Control of Mitochondria in Reactive Oxygen Species Homeostasis. Antioxid. Redox Signal. 2022, 37, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Epel, E. Stress and telomere shortening: Insights from cellular mechanisms. Ageing Res. Rev. 2022, 73, 101507. [Google Scholar] [CrossRef]
- Ayala, J.C.; Grismaldo, A.; Sequeda-Castañeda, L.G.; Aristizábal-Pachón, A.F.; Morales, L. Oxidative Stress in ICU Patients: ROS as Mortality Long-Term Predictor. Antioxidants 2021, 10, 1912. [Google Scholar] [CrossRef]
- Wang, R.; Ross, C.A.; Cai, H.; Cong, W.N.; Daimon, C.M.; Carlson, O.D.; Egan, J.M.; Siddiqui, S.; Maudsley, S.; Martin, B. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front. Physiol. 2014, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; D’Souza, G.X.; Shi, J.; Ritter, A.; Suazo, J.; Sabbagh, M.N. The Cause of Alzheimer’s Disease: The Theory of Multipathology Convergence to Chronic Neuronal Stress. Aging Dis. 2022, 13, 37–60. [Google Scholar] [CrossRef]
- Son, J.M.; Lee, C. Aging: All roads lead to mitochondria. Semin. Cell Dev. Biol. 2021, 116, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Munro, D.; Pamenter, M.E. Comparative studies of mitochondrial reactive oxygen species in animal longevity: Technical pitfalls and possibilities. Aging Cell 2019, 18, e13009. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Towards a unified mechanistic theory of aging. Exp. Gerontol. 2019, 124, 110627. [Google Scholar] [CrossRef]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef]
- Valente, A.X.C.N.; Adilbayeva, A.; Tokay, T.; Rizvanov, A.A. The Universal Non-Neuronal Nature of Parkinson’s Disease: A Theory. Cent. Asian J. Glob. Health 2016, 5, 231. [Google Scholar] [CrossRef]
- Natarajan, V.; Chawla, R.; Mah, T.; Vivekanandan, R.; Tan, S.Y.; Sato, P.Y.; Mallilankaraman, K. Mitochondrial Dysfunction in Age-Related Metabolic Disorders. Proteomics 2020, 20, e1800404. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.H. The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging. Oxid. Med. Cell Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef]
- Santos-Otte, P.; Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems and Their Role in Cellular Senescence. Comput. Struct. Biotechnol. J. 2019, 17, 1265–1277. [Google Scholar] [CrossRef]
- Hendrickx, J.O.; van Gastel, J.; Leysen, H.; Martin, B.; Maudsley, S. High-dimensionality Data Analysis of Pharmacological Systems Associated with Complex Diseases. Pharmacol. Rev. 2020, 72, 191–217. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.; Ulven, T.; Murdoch, H.; Hudson, B.D. G-protein-coupled receptors for free fatty acids: Nutritional and therapeutic targets. Br. J. Nutr. 2014, 111, S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Martin, B.; Luttrell, L.M. The origins of diversity and specificity in g protein-coupled receptor signaling. J. Pharmacol. Exp. Ther. 2005, 314, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Cong, W.N.; Daimon, C.M.; Wang, R.; Tschöp, M.H.; Sévigny, J.; Martin, B.; Maudsley, S. Altered lipid and salt taste responsivity in ghrelin and GOAT null mice. PLoS ONE 2013, 8, e76553. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets, and indications. Nat. Rev. Drug. Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Vass, M.; Kooistra, A.J.; Yang, D.; Stevens, R.C.; Wang, M.W.; de Graaf, C. Chemical Diversity in the G Protein-Coupled Receptor Superfamily. Trends Pharmacol. Sci. 2018, 39, 494–512. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Cvicek, V.; Goddard, W.A., 3rd; Abrol, R. Structure-Based Sequence Alignment of the Transmembrane Domains of All Human GPCRs: Phylogenetic, Structural and Functional Implications. PLoS Comput. Biol. 2016, 12, e1004805. [Google Scholar] [CrossRef]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Maudsley, S.; Gesty-Palmer, D. Translating in vitro ligand bias into in vivo efficacy. Cell. Signal. 2018, 41, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Gradl, D.; Schambony, A.; Arenas, E.; Schulte, G. Beta-arrestin is a necessary component of Wnt/beta-catenin signaling in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Kim, J.; Whalen, E.J.; Ahn, S.; Chen, M.; Lefkowitz, R.J. Beta-arrestin-mediated signaling regulates protein synthesis. J. Biol. Chem. 2008, 283, 10611–10620. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, Y.; Wu, Y.; Luan, B.; Wang, Y.; Qu, B.; Pei, G. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol. Cell 2004, 14, 303–317. [Google Scholar] [CrossRef]
- Kang, J.; Shi, Y.; Xiang, B.; Qu, B.; Su, W.; Zhu, M.; Zhang, M.; Bao, G.; Wang, F.; Zhang, X.; et al. A nuclear function of beta-arrestin1 in GPCR signaling: Regulation of histone acetylation and gene transcription. Cell 2005, 123, 833–847. [Google Scholar] [CrossRef]
- Maudsley, S.; Devanarayan, V.; Martin, B.; Geerts, H.; Brain Health Modeling Initiative (BHMI). Intelligent and effective informatic deconvolution of “Big Data” and its future impact on the quantitative nature of neurodegenerative disease therapy. Alzheimers Dement. 2018, 14, 961–975. [Google Scholar] [CrossRef]
- Bossers, K.; Wirz, K.T.; Meerhoff, G.F.; Essing, A.H.; van Dongen, J.W.; Houba, P.; Kruse, C.G.; Verhaagen, J.; Swaab, D.F. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 2010, 133, 3699–3723. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, X.; Zeng, X.; Bossers, K.; Swaab, D.F.; Zhao, J.; Pei, G. β-arrestin1 regulates γ-secretase complex assembly and modulates amyloid-β pathology. Cell Res. 2013, 23, 351–365. [Google Scholar] [CrossRef]
- Zhou, Z.; Liao, J.M.; Zhang, P.; Fan, J.B.; Chen, J.; Liang, Y. Parkinson disease drug screening based on the interaction between D(2) dopamine receptor and beta-arrestin 2 detected by capillary zone electrophoresis. Protein Cell 2011, 2, 899–905. [Google Scholar] [CrossRef]
- Urs, N.M.; Daigle, T.L.; Caron, M.G. A dopamine D1 receptor-dependent β-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology 2011, 36, 551–558. [Google Scholar] [CrossRef]
- Urs, N.M.; Bido, S.; Peterson, S.M.; Daigle, T.L.; Bass, C.E.; Gainetdinov, R.R.; Bezard, E.; Caron, M.G. Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, E2517–E2526. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Zhao, J.; Wu, H.; Duan, B.; Shu, G.; Wang, X.; Li, D.; Jia, W.; Kang, J.; Pei, G. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature 2009, 457, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Al-Sabah, S.; Al-Fulaij, M.; Shaaban, G.; Ahmed, H.A.; Mann, R.J.; Donnelly, D.; Bünemann, M.; Krasel, C. The GIP receptor displays higher basal activity than the GLP-1 receptor but does not recruit GRK2 or arrestin3 effectively. PLoS ONE 2014, 9, e106890. [Google Scholar] [CrossRef]
- Pang, Y.; Zhu, H.; Xu, J.; Yang, L.; Liu, L.; Li, J. β-arrestin-2 is involved in irisin induced glucose metabolism in type 2 diabetes via p38 MAPK signaling. Exp. Cell Res. 2017, 360, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Gesty-Palmer, D.; Flannery, P.; Yuan, L.; Corsino, L.; Spurney, R.; Lefkowitz, R.J.; Luttrell, L.M. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci. Transl. Med. 2009, 1, 1ra1. [Google Scholar] [CrossRef]
- Urs, N.M.; Peterson, S.M.; Caron, M.G. New Concepts in Dopamine D2 Receptor Biased Signaling and Implications for Schizophrenia Therapy. Biol. Psychiatry 2017, 81, 78–85. [Google Scholar] [CrossRef]
- Park, S.M.; Chen, M.; Schmerberg, C.M.; Dulman, R.S.; Rodriguiz, R.M.; Caron, M.G.; Jin, J.; Wetsel, W.C. Effects of β-Arrestin-Biased Dopamine D2 Receptor Ligands on Schizophrenia-Like Behavior in Hypoglutamatergic Mice. Neuropsychopharmacology 2016, 41, 704–715. [Google Scholar] [CrossRef]
- Anckaerts, C.; van Gastel, J.; Leysen, V.; Hinz, R.; Azmi, A.; Simoens, P.; Shah, D.; Kara, F.; Langbeen, A.; Bols, P.; et al. Image-guided phenotyping of ovariectomized mice: Altered functional connectivity, cognition, myelination, and dopaminergic functionality. Neurobiol. Aging 2019, 74, 77–89. [Google Scholar] [CrossRef]
- Chadwick, W.; Zhou, Y.; Park, S.S.; Wang, L.; Mitchell, N.; Stone, M.D.; Becker, K.G.; Martin, B.; Maudsley, S. Minimal peroxide exposure of neuronal cells induces multifaceted adaptive responses. PLoS ONE 2010, 5, e14352. [Google Scholar] [CrossRef]
- Rasheed, N.; Wang, X.; Niu, Q.T.; Yeh, J.; Li, B. Atm-deficient mice: An osteoporosis model with defective osteoblast differentiation and increased osteoclastogenesis. Hum. Mol. Genet. 2006, 15, 1938–1948. [Google Scholar] [CrossRef]
- Wang, X.; Liao, S.; Nelson, E.R.; Schmalzigaug, R.; Spurney, R.F.; Guilak, F.; Premont, R.T.; Gesty-Palmer, D. The cytoskeletal regulatory scaffold protein GIT2 modulates mesenchymal stem cell differentiation and osteoblastogenesis. Biochem. Biophys. Res. Commun. 2012, 425, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Martin, B.; Veenker, L.; Beuning, S.; Coppens, V.; Morrens, M.; Maudsley, S. Enhanced Molecular Appreciation of Psychiatric Disorders Through High-Dimensionality Data Acquisition and Analytics. Methods Mol. Biol. 2019, 2011, 671–723. [Google Scholar] [PubMed]
- Lee, S.A.; Huang, K.C. Epigenetic profiling of human brain differential DNA methylation networks in schizophrenia. BMC Med. Genom. 2016, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Appleton, K.M.; Lee, M.H.; Alele, C.; Alele, C.; Luttrell, D.K.; Peterson, Y.K.; Morinelli, T.A.; Luttrell, L.M. Biasing the parathyroid hormone receptor: Relating in vitro ligand efficacy to in vivo biological activity. Methods Enzymol. 2013, 522, 229–262. [Google Scholar]
- Gesty-Palmer, D.; Luttrell, L.M. Refining efficacy: Exploiting functional selectivity for drug discovery. Adv. Pharmacol. 2011, 62, 79–107. [Google Scholar]
- O’Dowd, B.F.; Nguyen, T.; Lynch, K.R.; Kolakowski, L.F., Jr.; Thompson, M.; Cheng, R.; Marchese, A.; Ng, G.; Heng, H.H.; George, S.R. A novel gene codes for a putative G protein-coupled receptor with an abundant expression in brain. FEBS Lett. 1996, 394, 325–329. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Montpetit, A.; Sinnett, D. Physical mapping of the G-protein coupled receptor 19 (GPR19) in the chromosome 12p12.3 region frequently rearranged in cancer cells. Hum. Genet. 1999, 105, 162–164. [Google Scholar] [CrossRef]
- Davenport, A.P.; Alexander, S.P.; Sharman, J.L.; Pawson, A.J.; Benson, H.E.; Monaghan, A.E.; Liew, W.C.; Mpamhanga, C.P.; Bonner, T.I.; Neubig, R.R.; et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: Recommendations for new pairings with cognate ligands. Pharmacol. Rev. 2013, 65, 967–986. [Google Scholar] [CrossRef]
- Rao, A.; Herr, D.R. G protein-coupled receptor GPR19 regulates E-cadherin expression and invasion of breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1318–1327. [Google Scholar] [CrossRef]
- Stein, L.M.; Yosten, G.L.; Samson, W.K. Adropin acts in brain to inhibit water drinking: Potential interaction with the orphan G protein-coupled receptor, GPR19. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R476–R480. [Google Scholar] [CrossRef] [PubMed]
- Thapa, D.; Stoner, M.W.; Zhang, M.; Xie, B.; Manning, J.R.; Guimaraes, D.; Shiva, S.; Jurczak, M.J.; Scott, I. Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR-MAPK-PDK4 signaling pathway. Redox Biol. 2018, 18, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.R.; Hauser, A.S.; Vedel, L.; Strachan, R.T.; Huang, X.P.; Gavin, A.C.; Shah, S.D.; Nayak, A.P.; Haugaard-Kedström, L.M.; Penn, R.B.; et al. Discovery of Human Signaling Systems: Pairing Peptides to G Protein-Coupled Receptors. Cell 2019, 179, 895–908. [Google Scholar] [CrossRef]
- Kakarala, K.K.; Jamil, K. Sequence-structure based phylogeny of GPCR Class A Rhodopsin receptors. Mol. Phylogenet. Evol. 2014, 74, 66–96. [Google Scholar] [CrossRef] [PubMed]
- Reinscheid, R.K.; Xu, Y.L. Neuropeptide S and its receptor: A newly deorphanized G protein-coupled receptor system. Neuroscientist 2005, 11, 532–538. [Google Scholar] [CrossRef]
- Bresnick, J.N.; Skynner, H.A.; Chapman, K.L.; Jack, A.D.; Zamiara, E.; Negulescu, P.; Beaumont, K.; Patel, S.; McAllister, G. Identification of signal transduction pathways used by orphan g protein-coupled receptors. Assay Drug Dev. Technol. 2003, 1, 239–249. [Google Scholar] [CrossRef]
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS ONE 2015, 10, e0138463. [Google Scholar] [CrossRef]
- Southern, C.; Cook, J.M.; Neetoo-Isseljee, Z.; Taylor, D.L.; Kettleborough, C.A.; Merritt, A.; Bassoni, D.L.; Raab, W.J.; Quinn, E.; Wehrman, T.S.; et al. Screening β-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J. Biomol. Screen. 2013, 18, 599–609. [Google Scholar] [CrossRef]
- Huang, L.; Guo, B.; Liu, S.; Miao, C.; Li, Y. Inhibition of the LncRNA Gpr19 attenuates ischemia-reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR-324-5p/Mtfr1 axis. IUBMB Life 2020, 72, 373–383. [Google Scholar] [CrossRef]
- Jasaszwili, M.; Billert, M.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. Adropin as A Fat-Burning Hormone with Multiple Functions-Review of a Decade of Research. Molecules 2020, 25, 549. [Google Scholar] [CrossRef]
- Yang, C.; DeMars, K.M.; Candelario-Jalil, E. Age-Dependent Decrease in Adropin is Associated with Reduced Levels of Endothelial Nitric Oxide Synthase and Increased Oxidative Stress in the Rat Brain. Aging Dis. 2018, 9, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ghoshal, S.; Girardet, C.; DeMars, K.M.; Yang, C.; Niehoff, M.L.; Nguyen, A.D.; Jayanth, P.; Hoelscher, B.A.; Xu, F.; et al. Adropin correlates with aging-related neuropathology in humans and improves cognitive function in aging mice. NPJ Aging Mech. Dis. 2021, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.; Trevaskis, J.L.; Lam, D.D.; Sutton, G.M.; Koza, R.A.; Chouljenko, V.N.; Kousoulas, K.G.; Rogers, P.M.; Kesterson, R.A.; Thearle, M.; et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 2008, 8, 468–481. [Google Scholar] [CrossRef]
- Thapa, D.; Xie, B.; Manning, J.R.; Zhang, M.; Stoner, M.W.; Huckestein, B.R.; Edmunds, L.R.; Zhang, X.; Dedousis, N.L.; O’Doherty, R.M.; et al. Adropin reduces blood glucose levels in mice by limiting hepatic glucose production. Physiol. Rep. 2019, 7, e14043. [Google Scholar] [CrossRef]
- Gao, S.; McMillan, R.P.; Zhu, Q.; Lopaschuk, G.D.; Hulver, M.W.; Butler, A.A. Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol. Metab. 2015, 4, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.R.; Kearney, M.L.; St-Onge, M.P.; Stanhope, K.L.; Havel, P.J.; Kanaley, J.A.; Thyfault, J.P.; Weiss, E.P.; Butler, A.A. Inverse association between carbohydrate consumption and plasma adropin concentrations in humans. Obesity 2016, 24, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Jiang, F.; Cheng, X.; Xu, H.; Hu, X. Serum adropin levels are decreased in Chinese type 2 diabetic patients and negatively correlated with body mass index. Endocr. J. 2018, 65, 685–691. [Google Scholar] [CrossRef]
- Wu, L.; Fang, J.; Chen, L.; Zhao, Z.; Luo, Y.; Lin, C.; Fan, L. Low serum adropin is associated with coronary atherosclerosis in type 2 diabetic and non-diabetic patients. Clin. Chem. Lab. Med. 2014, 52, 751–758. [Google Scholar] [CrossRef]
- Kastner, S.; Voss, T.; Keuerleber, S.; Glöckel, C.; Freissmuth, M.; Sommergruber, W. Expression of G protein-coupled receptor 19 in human lung cancer cells is triggered by entry into S-phase and supports G(2)-M cell-cycle progression. Mol. Cancer Res. 2012, 10, 1343–1358. [Google Scholar] [CrossRef]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 13. [Google Scholar] [CrossRef]
- Rossi, P.; Dolci, S.; Sette, C.; Capolunghi, F.; Pellegrini, M.; Loiarro, M.; Di Agostino, S.; Paronetto, M.P.; Grimaldi, P.; Merico, D.; et al. Analysis of the gene expression profile of mouse male meiotic germ cells. Gene Expr. Patterns 2004, 4, 267–281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed]
- Hossain, M.S.; Mineno, K.; Katafuchi, T. Neuronal Orphan G-Protein Coupled Receptor Proteins Mediate Plasmalogens-Induced Activation of ERK and Akt Signaling. PLoS ONE 2016, 11, e0150846. [Google Scholar] [CrossRef] [PubMed]
- Jasaszwili, M.; Wojciechowicz, T.; Billert, M.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. Effects of adropin on proliferation and differentiation of 3T3-L1 cells and rat primary preadipocytes. Mol. Cell. Endocrinol. 2019, 496, 110532. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Al-Omran, M.; Teoh, H.; Verma, S. Adropin is a novel regulator of endothelial function. Circulation 2010, 122, S185–S192. [Google Scholar] [CrossRef]
- Shahjouei, S.; Ansari, S.; Pourmotabbed, T.; Zand, R. Potential Roles of Adropin in Central Nervous System: Review of Current Literature. Front. Mol. Biosci. 2016, 3, 25. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- Kolben, Y.; Weksler-Zangen, S.; Ilan, Y. Adropin as a potential mediator of the metabolic system-autonomic nervous system-chronobiology axis: Implementing a personalized signature-based platform for chronotherapy. Obes. Rev. 2021, 22, e13108. [Google Scholar] [CrossRef]
- Wang, B.; Xue, Y.; Shang, F.; Ni, S.; Liu, X.; Fan, B.; Wang, H. Association of serum adropin with the presence of atrial fibrillation and atrial remodeling. J. Clin. Lab. Anal. 2019, 33, e22672. [Google Scholar] [CrossRef]
- Williams, L.M.; Lago, B.A.; McArthur, A.G.; Raphenya, A.R.; Pray, N.; Saleem, N.; Salas, S.; Paulson, K.; Mangar, R.S.; Liu, Y.; et al. The transcription factor, Nuclear factor, erythroid 2 (Nfe2), is a regulator of the oxidative stress response during Danio rerio development. Aquat. Toxicol. 2016, 180, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.; Seo, E.J.; Efferth, T. Novel molecular mechanisms for the adaptogenic effects of herbal extracts on isolated brain cells using systems biology. Phytomedicine 2018, 50, 257–284. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Havel, P.J. Adropin and insulin resistance: Integration of endocrine, circadian, and stress signals regulating glucose metabolism. Obesity 2021, 29, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar]
- Fafián-Labora, J.; Carpintero-Fernández, P.; Jordan, S.J.D.; Shikh-Bahaei, T.; Abdullah, S.M.; Mahenthiran, M.; Rodríguez-Navarro, J.A.; Niklison-Chirou, M.V.; O’Loghlen, A. FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis. 2019, 10, 318. [Google Scholar] [CrossRef]
- Bradshaw, P.C. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef]
- Lehallier, B.; Shokhirev, M.N.; Wyss-Coray, T.; Johnson, A.A. Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell 2020, 19, e13256. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wong, C.C.; Zhang, X.; Kang, W.; Nakatsu, G.; Zhao, Q.; Chen, H.; Go, M.Y.Y.; Chiu, P.W.Y.; Wang, X.; et al. CAB39L elicited an anti-Warburg effect via a LKB1-AMPK-PGC1α axis to inhibit gastric tumorigenesis. Oncogene 2018, 37, 6383–6398. [Google Scholar] [CrossRef]
- Doig, C.L.; Zielinska, A.E.; Fletcher, R.S.; Oakey, L.A.; Elhassan, Y.S.; Garten, A.; Cartwright, D.; Heising, S.; Alsheri, A.; Watson, D.G.; et al. Induction of the nicotinamide riboside kinase NAD+ salvage pathway in a model of sarcoplasmic reticulum dysfunction. Skelet. Muscle 2020, 10, 5. [Google Scholar] [CrossRef]
- Kwan, V.; Rosa, E.; Xing, S.; Murtaza, N.; Singh, K.; Holzapfel, N.T.; Berg, T.; Lu, Y.; Singh, K.K. Proteomic Analysis Reveals Autism-Associated Gene DIXDC1 Regulates Proteins Associated with Mitochondrial Organization and Function. J. Proteome Res. 2021, 20, 1052–1062. [Google Scholar] [CrossRef]
- Martínez-Carreres, L.; Puyal, J.; Leal-Esteban, L.C.; Orpinell, M.; Castillo-Armengol, J.; Giralt, A.; Dergai, O.; Moret, C.; Barquissau, V.; Nasrallah, A.; et al. CDK4 Regulates Lysosomal Function and mTORC1 Activation to Promote Cancer Cell Survival. Cancer Res. 2019, 79, 5245–5259. [Google Scholar] [CrossRef] [PubMed]
- Weeks, R.J.; Ludgate, J.L.; Le Mée, G.; Khanal, R.; Mehta, S.; Williams, G.; Slatter, T.L.; Braithwaite, A.W.; Morison, I.M. Silencing of Testin expression is a frequent event in spontaneous lymphomas from Trp53-mutant mice. Sci. Rep. 2020, 10, 16255. [Google Scholar] [CrossRef] [PubMed]
- Herrema, H.; Guan, D.; Choi, J.W.; Feng, X.; Salazar Hernandez, M.A.; Faruk, F.; Auen, T.; Boudett, E.; Tao, R.; Chun, H.; et al. FKBP11 rewires UPR signaling to promote glucose homeostasis in type 2 diabetes and obesity. Cell Metab. 2022, 34, 1004–1022. [Google Scholar] [CrossRef]
- Makboul, R.; Abdelkawi, I.F.; Badary, D.M.; Hussein, M.R.A.; Rhim, J.S.; Toraih, E.A.; Zerfaoui, M.; Abd Elmageed, Z.Y. Transmembrane and Tetratricopeptide Repeat Containing 4 Is a Novel Diagnostic Marker for Prostate Cancer with High Specificity and Sensitivity. Cells 2021, 10, 1029. [Google Scholar] [CrossRef]
- Xie, J.; Wen, M.; Zhang, J.; Wang, Z.; Wang, M.; Qiu, Y.; Zhao, W.; Zhu, F.; Yao, M.; Rong, Z.; et al. The Roles of RNA Helicases in DNA Damage Repair and Tumorigenesis Reveal Precision Therapeutic Strategies. Cancer Res. 2022, 82, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, J.; Guo, Y.; Yang, Y.; Teng, T.; Yu, Q.; Wang, T.; Zhou, M.; Zhu, Q.; Wang, W.; et al. Genome-Wide Detection of CNVs and Association With Body Weight in Sheep Based on 600K SNP Arrays. Front. Genet. 2020, 11, 558. [Google Scholar] [CrossRef]
- Blengini, C.S.; Nguyen, A.L.; Aboelenain, M.; Schindler, K. Age-dependent integrity of the meiotic spindle assembly checkpoint in females requires Aurora kinase B. Aging Cell 2021, 20, e13489. [Google Scholar] [CrossRef]
- Lachapelle, S.; Gagné, J.P.; Garand, C.; Desbiens, M.; Coulombe, Y.; Bohr, V.A.; Hendzel, M.J.; Masson, J.Y.; Poirier, G.G.; Lebel, M. Proteome-wide identification of WRN-interacting proteins in untreated and nuclease-treated samples. J. Proteome Res. 2011, 10, 1216–1227. [Google Scholar] [CrossRef]
- Su, H.; Fan, Y.; Wang, Z.; Jiang, L. A comprehensive investigation on pan-cancer impacts of constitutive centromere associated network gene family by integrating multi-omics data: A CONSORT-compliant article. Medicine 2022, 101, e28821. [Google Scholar] [CrossRef]
- Li, J.; Zhao, R.; Zhao, H.; Chen, G.; Jiang, Y.; Lyu, X.; Wu, T. Reduction of Aging-Induced Oxidative Stress and Activation of Autophagy by Bilberry Anthocyanin Supplementation via the AMPK-mTOR Signaling Pathway in Aged Female Rats. J. Agric. Food Chem. 2019, 67, 7832–7843. [Google Scholar] [CrossRef]
- Zhao, L.N.; Björklund, M.; Caldez, M.J.; Zheng, J.; Kaldis, P. Therapeutic targeting of the mitochondrial one-carbon pathway: Perspectives, pitfalls, and potential. Oncogene 2021, 40, 2339–2354. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.; Kienesberger, P.C. Autotaxin-LPA-LPP3 Axis in Energy Metabolism and Metabolic Disease. Int. J. Mol. Sci. 2021, 22, 9575. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Wang, K.; Marshall, J.D.; Naggert, J.K.; Rethmeyer, J.A.; Gunewardena, S.S.; Manzardo, A.M. Coding and noncoding expression patterns associated with rare obesity-related disorders: Prader-Willi and Alström syndromes. Adv. Genom. Genet. 2015, 2015, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Kloske, C.M.; Dugan, A.J.; Weekman, E.M.; Winder, Z.; Patel, E.; Nelson, P.T.; Fardo, D.W.; Wilcock, D.M. Inflammatory Pathways Are Impaired in Alzheimer Disease and Differentially Associated With Apolipoprotein E Status. J. Neuropathol. Exp. Neurol. 2021, 80, 922–932. [Google Scholar] [CrossRef]
- Albanese, F.; Mercatelli, D.; Finetti, L.; Lamonaca, G.; Pizzi, S.; Shimshek, D.R.; Bernacchia, G.; Morari, M. Constitutive silencing of LRRK2 kinase activity leads to early glucocerebrosidase deregulation and late impairment of autophagy in vivo. Neurobiol. Dis 2021, 159, 105487. [Google Scholar] [CrossRef]
- Zhou, J.; Nie, W.; Yuan, J.; Zhang, Z.; Mi, L.; Wang, C.; Huang, R. GSG2 knockdown suppresses cholangiocarcinoma progression by regulating cell proliferation, apoptosis, and migration. Oncol. Rep. 2021, 45, 91. [Google Scholar] [CrossRef]
- Florentinus-Mefailoski, A.; Bowden, P.; Scheltens, P.; Killestein, J.; Teunissen, C.; Marshall, J.G. The plasma peptides of Alzheimer’s disease. Clin. Proteom. 2021, 18, 17. [Google Scholar] [CrossRef]
- De Medina, P.; Silvente-Poirot, S.; Poirot, M. Oxysterols are potential physiological regulators of ageing. Ageing Res. Rev. 2022, 77, 101615. [Google Scholar] [CrossRef]
- Che, Y.; Yang, J.; Tang, F.; Wei, Z.; Chao, Y.; Li, N.; Li, H.; Wu, S.; Dong, X. New Function of Cholesterol Oxidation Products Involved in Osteoporosis Pathogenesis. Int. J. Mol. Sci. 2022, 23, 2020. [Google Scholar] [CrossRef]
- Maioli, S.; Leander, K.; Nilsson, P.; Nalvarte, I. Estrogen receptors and the aging brain. Essays Biochem. 2021, 65, 913–925. [Google Scholar] [CrossRef]
- Willis, C.M.; Nicaise, A.M.; Krzak, G.; Ionescu, R.B.; Pappa, V.; D’Angelo, A.; Agarwal, R.; Repollés-de-Dalmau, M.; Peruzzotti-Jametti, L.; Pluchino, S. Soluble factors influencing the neural stem cell niche in brain physiology, inflammation, and aging. Exp. Neurol. 2022, 355, 114124. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Goodell, M.A.; Rando, T.A. Ageing and rejuvenation of tissue stem cells and their niches. Nat. Rev. Mol. Cell Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Li, X.; Tian, Y. Mitochondrial-to-nuclear communication in aging: An epigenetic perspective. Trends Biochem. Sci. 2022, 47, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.; Garcia, G.; Higuchi-Sanabria, R. Hijacking Cellular Stress Responses to Promote Lifespan. Front. Aging 2022, 3, 860404. [Google Scholar] [CrossRef]
- Singla, R.; Mishra, A.; Cao, R. The trilateral interactions between mammalian target of rapamycin (mTOR) signaling, the circadian clock, and psychiatric disorders: An emerging model. Transl. Psychiatry 2022, 12, 355. [Google Scholar] [CrossRef]
- Tomtheelnganbee, E.; Sah, P.; Sharma, R. Mitochondrial function, and nutrient sensing pathways in ageing: Enhancing longevity through dietary interventions. Biogerontology 2022. [Google Scholar] [CrossRef]
- Mattson, M.P.; Maudsley, S.; Martin, B. A neural signaling triumvirate that influences ageing and age-related disease: Insulin/IGF-1, BDNF and serotonin. Ageing Res. Rev. 2004, 3, 445–464. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Zhou, Y.; Martin, B.; Maudsley, S. Pharmacomimetics of exercise: Novel approaches for hippocampally-targeted neuroprotective agents. Curr. Med. Chem. 2009, 16, 4668–4678. [Google Scholar] [CrossRef]
- Crochemore, C.; Cimmaruta, C.; Fernández-Molina, C.; Ricchetti, M. Reactive Species in Progeroid Syndromes and Aging-Related Processes. Antioxid. Redox Signal. 2022, 37, 208–228. [Google Scholar] [CrossRef]
- Sengupta, D.; Sengupta, K. Lamin A, and telomere maintenance in aging: Two to Tango. Mutat. Res. 2022, 825, 111788. [Google Scholar] [CrossRef]
- Chen, H.; Martin, B.; Daimon, C.M.; Maudsley, S. Effective use of latent semantic indexing and computational linguistics in biological and biomedical applications. Front. Physiol. 2013, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Martin, B.; Daimon, C.M.; Siddiqui, S.; Luttrell, L.M.; Maudsley, S. Textrous!: Extracting semantic textual meaning from gene sets. PLoS ONE 2013, 8, e62665. [Google Scholar] [CrossRef] [PubMed]
- Van Meenen, J.; Leysen, H.; Chen, H.; Baccarne, R.; Walter, D.; Martin, B.; Maudsley, S. Making Biomedical Sciences publications more accessible for machines. Med. Health Care. Philos. 2022, 25, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Jourquin, J.; Duncan, D.; Shi, Z.; Zhang, B. GLAD4U: Deriving and prioritizing gene lists from PubMed literature. BMC Genom. 2012, 13, S20. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.P.; Venkatraman, V.; Xing, Y.; Lau, E.; Cao, Q.; Ng, D.C.; Su, A.I.; Ge, J.; van Eyk, J.E.; Ping, P. Data-Driven Approach To Determine Popular Proteins for Targeted Proteomics Translation of Six Organ Systems. J. Proteome Res. 2016, 15, 4126–4134. [Google Scholar] [CrossRef]
- Lachmann, A.; Schilder, B.M.; Wojciechowicz, M.L.; Torre, D.; Kuleshov, M.V.; Keenan, A.B.; Ma’ayan, A. Geneshot: Search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019, 47, W571–W577. [Google Scholar] [CrossRef]
- Annus, T.; Müller, D.; Jezsó, B.; Ullaga, G.; Németh, B.; Harami, G.M.; Orbán, L.; Kovács, M.; Varga, M. Bloom syndrome helicase contributes to germ line development and longevity in zebrafish. Cell Death Dis. 2022, 13, 363. [Google Scholar] [CrossRef]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Pizzo, R.; Alvente, S.; Berteotti, C.; Lupori, L.; Sagona, G.; Galvani, G.; Gurgone., A.; et al. Age-Related Cognitive and Motor Decline in a Mouse Model of CDKL5 Deficiency Disorder is Associated with Increased Neuronal Senescence and Death. Aging Dis. 2021, 12, 764–785. [Google Scholar] [CrossRef]
- Lu, H.; Guan, J.; Wang, S.Y.; Li, G.M.; Bohr, V.A.; Davis, A.J. DNA-PKcs-dependent phosphorylation of RECQL4 promotes NHEJ by stabilizing the NHEJ machinery at DNA double-strand breaks. Nucleic Acids Res. 2022, 50, 5635–5651. [Google Scholar] [CrossRef]
- Xu, M.D.; Liu, S.L.; Zheng, B.B.; Wu, J.; Wu, M.Y.; Zhang, Y.; Gong, F.R.; Tao, M.; Zhang, J.; Li, W. The radiotherapy-sensitization effect of cantharidin: Mechanisms involving cell cycle regulation, enhanced DNA damage, and inhibited DNA damage repair. Pancreatology 2018, 18, 822–832. [Google Scholar] [CrossRef]
- Francica, P.; Nisa, L.; Aebersold, D.M.; Langer, R.; Bladt, F.; Blaukat, A.; Stroka, D.; Martínez, M.R.; Zimmer, Y.; Medová, M. Depletion of FOXM1 via MET Targeting Underlies Establishment of a DNA Damage-Induced Senescence Program in Gastric Cancer. Clin. Cancer Res. 2016, 22, 5322–5336. [Google Scholar] [CrossRef]
- Maranon, D.G.; Sharma, N.; Huang, Y.; Selemenakis, P.; Wang, M.; Altina, N.; Zhao, W.; Wiese, C. NUCKS1 promotes RAD54 activity in homologous recombination DNA repair. J. Cell Biol. 2020, 219, e201911049. [Google Scholar] [CrossRef] [PubMed]
- Billard, P.; Poncet, D.A. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B. Uncoupling protein-2 and the potential link between metabolism and longevity. Curr. Aging Sci. 2010, 3, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Szántó, M.; Gupte, R.; Kraus, W.L.; Pacher, P.; Bai, P. PARPs in lipid metabolism and related diseases. Prog. Lipid Res. 2021, 84, 101117. [Google Scholar] [CrossRef]
- Truong, V.L.; Jun, M.; Jeong, W.S. Role of resveratrol in regulation of cellular defense systems against oxidative stress. Biofactors 2018, 44, 36–49. [Google Scholar] [CrossRef]
- Huang, J.; Liu, C.; Ming, X.F.; Yang, Z. Inhibition of p38mapk Reduces Adipose Tissue Inflammation in Aging Mediated by Arginase-II. Pharmacology 2020, 105, 491–504. [Google Scholar] [CrossRef]
- Marmentini, C.; Soares, G.M.; Bronczek, G.A.; Piovan, S.; Mareze-Costa, C.E.; Carneiro, E.M.; Boschero, A.C.; Kurauti, M.A. Aging Reduces Insulin Clearance in Mice. Front. Endocrinol. 2021, 12, 679492. [Google Scholar] [CrossRef]
- Gravina, S.; Lescai, F.; Hurteau, G.; Brock, G.J.; Saramaki, A.; Salvioli, S.; Franceschi, C.; Roninson, I.B. Identification of single nucleotide polymorphisms in the p21 (CDKN1A) gene and correlations with longevity in the Italian population. Aging 2009, 1, 470–480. [Google Scholar] [CrossRef]
- Connell-Crowley, L.; Vo, D.; Luke, L.; Giniger, E. Drosophila lacking the Cdk5 activator, p35, display defective axon guidance, age-dependent behavioral deficits, and reduced lifespan. Mech. Dev. 2007, 124, 341–349. [Google Scholar] [CrossRef]
- Schuck, N.W.; Petok, J.R.; Meeter, M.; Schjeide, B.M.; Schröder, J.; Bertram, L.; Gluck, M.A.; Li, S.C. Aging and a genetic KIBRA polymorphism interactively affect feedback- and observation-based probabilistic classification learning. Neurobiol. Aging 2018, 61, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Funabiki, H. Integrity of the human centromere DNA repeats is protected by CENP-A, CENP-C, and CENP-T. Proc. Natl. Acad. Sci. USA 2017, 114, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Marcelot, A.; Worman, H.J.; Zinn-Justin, S. Protein structural and mechanistic basis of progeroid laminopathies. FEBS J. 2021, 288, 2757–2772. [Google Scholar] [CrossRef]
- Cai, G.; Lin, F.; Wu, D.; Lin, C.; Chen, H.; Wei, Y.; Weng, H.; Chen, Z.; Wu, M.; Huang, E.; et al. Rosmarinic Acid Inhibits Mitochondrial Damage by Alleviating Unfolded Protein Response. Front. Pharmacol. 2022, 13, 859978. [Google Scholar] [CrossRef] [PubMed]
- Qian, H. Loss of SQSTM1/p62 Induces Obesity and Exacerbates Alcohol-induced Liver Injury in Aged Mice. FASEB J. 2022, 36, S1. [Google Scholar] [CrossRef]
- Shinoda, Y.; Fujita, K.; Saito, S.; Matsui, H.; Kanto, Y.; Nagaura, Y.; Fukunaga, K.; Tamura, S.; Kobayashi, T. Acyl-CoA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER-Golgi membrane contact sites. FEBS Lett. 2012, 586, 3024–3029. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Jacobsen, R.; Nygaard, M.; Soerensen, M.; Mengel-From, J.; Christiansen, L.; Christensen, K. Cohort Differences in the Associations of Selected Candidate Genes With Risk of All-Cause Mortality at Advanced Ages. Am. J. Epidemiol. 2020, 189, 708–716. [Google Scholar] [CrossRef]
- Chadwick, W.; Boyle, J.P.; Zhou, Y.; Wang, L.; Park, S.S.; Martin, B.; Wang, R.; Becker, K.G.; Wood, W.H., 3rd; Zhang, Y.; et al. Multiple oxygen tension environments reveal diverse patterns of transcriptional regulation in primary astrocytes. PLoS ONE 2011, 6, e21638. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maudsley, S.; Walter, D.; Schrauwen, C.; Van Loon, N.; Harputluoğlu, İ.; Lenaerts, J.; McDonald, P. Intersection of the Orphan G Protein-Coupled Receptor, GPR19, with the Aging Process. Int. J. Mol. Sci. 2022, 23, 13598. https://doi.org/10.3390/ijms232113598
Maudsley S, Walter D, Schrauwen C, Van Loon N, Harputluoğlu İ, Lenaerts J, McDonald P. Intersection of the Orphan G Protein-Coupled Receptor, GPR19, with the Aging Process. International Journal of Molecular Sciences. 2022; 23(21):13598. https://doi.org/10.3390/ijms232113598
Chicago/Turabian StyleMaudsley, Stuart, Deborah Walter, Claudia Schrauwen, Nore Van Loon, İrem Harputluoğlu, Julia Lenaerts, and Patricia McDonald. 2022. "Intersection of the Orphan G Protein-Coupled Receptor, GPR19, with the Aging Process" International Journal of Molecular Sciences 23, no. 21: 13598. https://doi.org/10.3390/ijms232113598
APA StyleMaudsley, S., Walter, D., Schrauwen, C., Van Loon, N., Harputluoğlu, İ., Lenaerts, J., & McDonald, P. (2022). Intersection of the Orphan G Protein-Coupled Receptor, GPR19, with the Aging Process. International Journal of Molecular Sciences, 23(21), 13598. https://doi.org/10.3390/ijms232113598