
FUS Alters circRNA Metabolism in Human Motor Neurons Carrying the ALS-Linked P525L Mutation

 , , ,
, , ,
Abstract
1. Introduction
2. Results
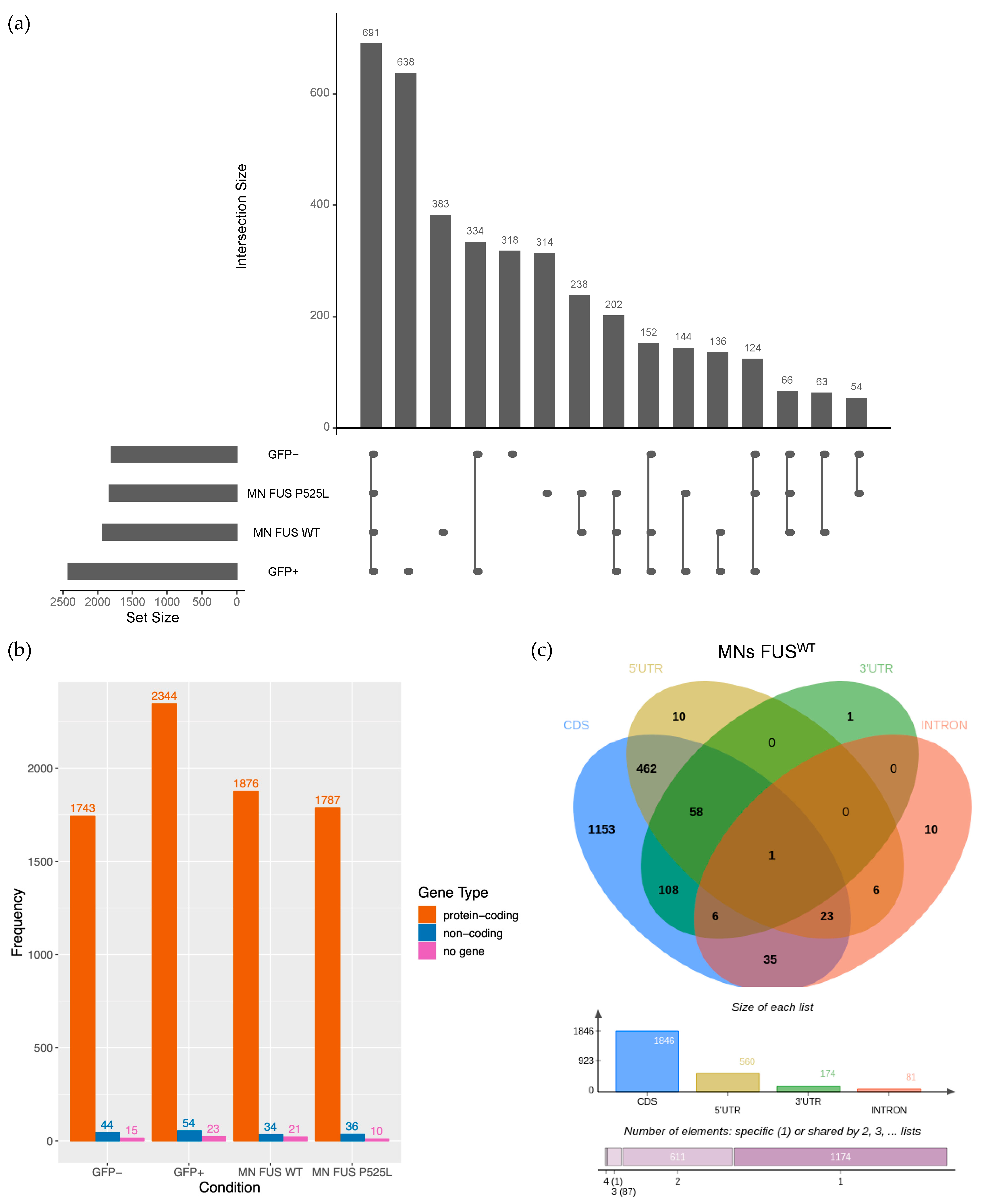
2.1. Identification of circRNAs Expressed in Human Motor Neurons
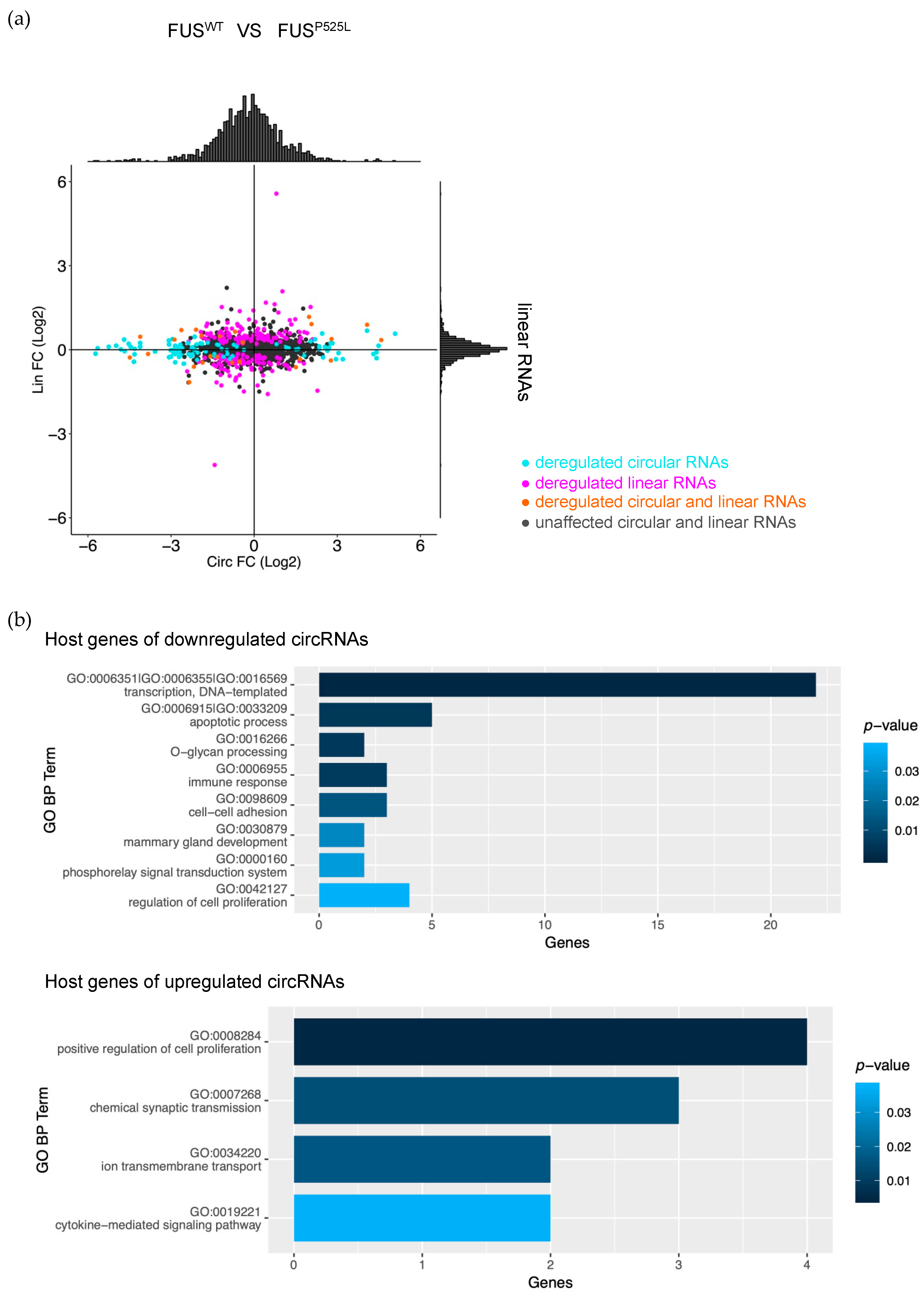
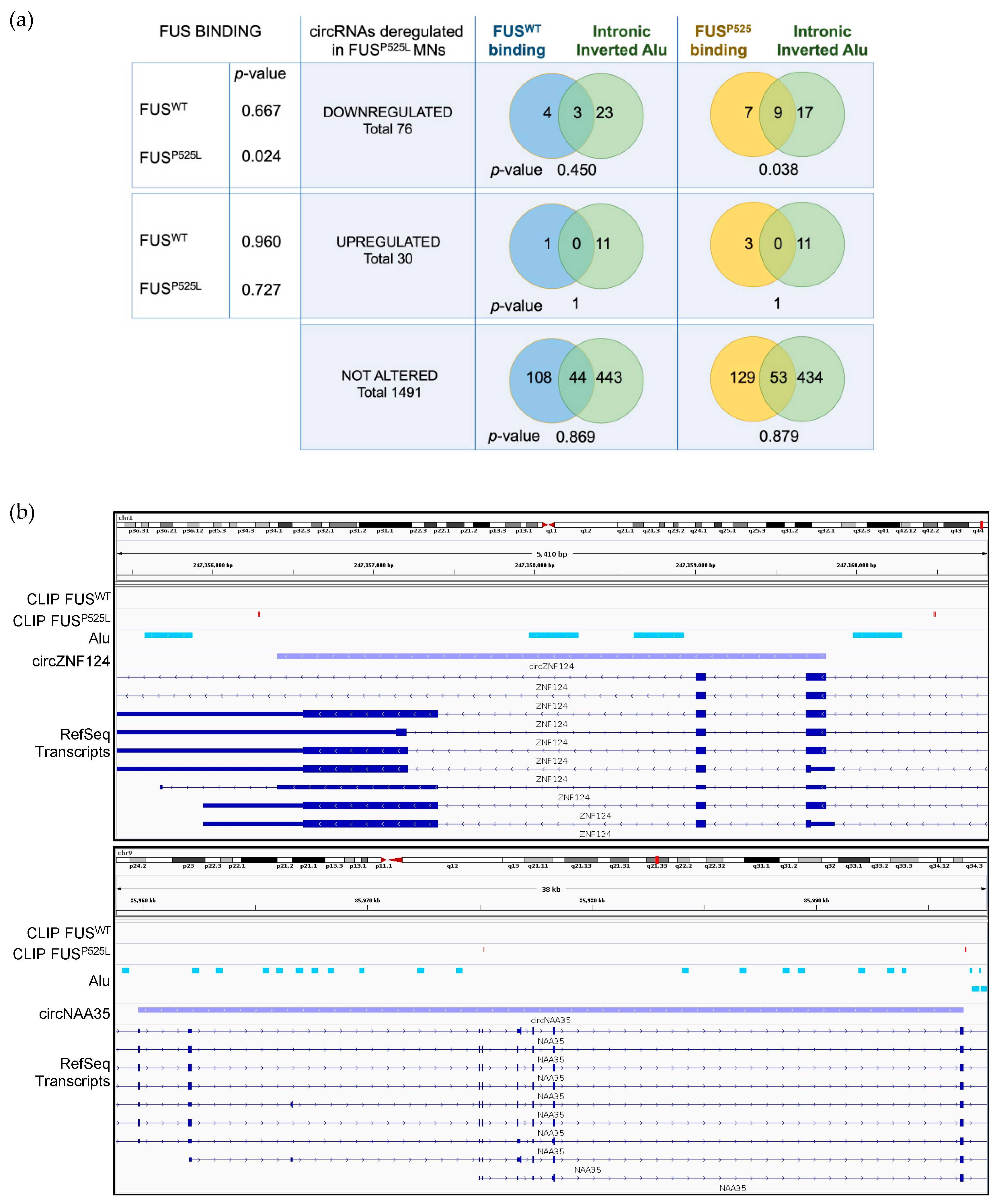
2.2. Mutant FUS Binds to Introns Containing Alu Repeats and Is Associated with Downregulated circRNAs
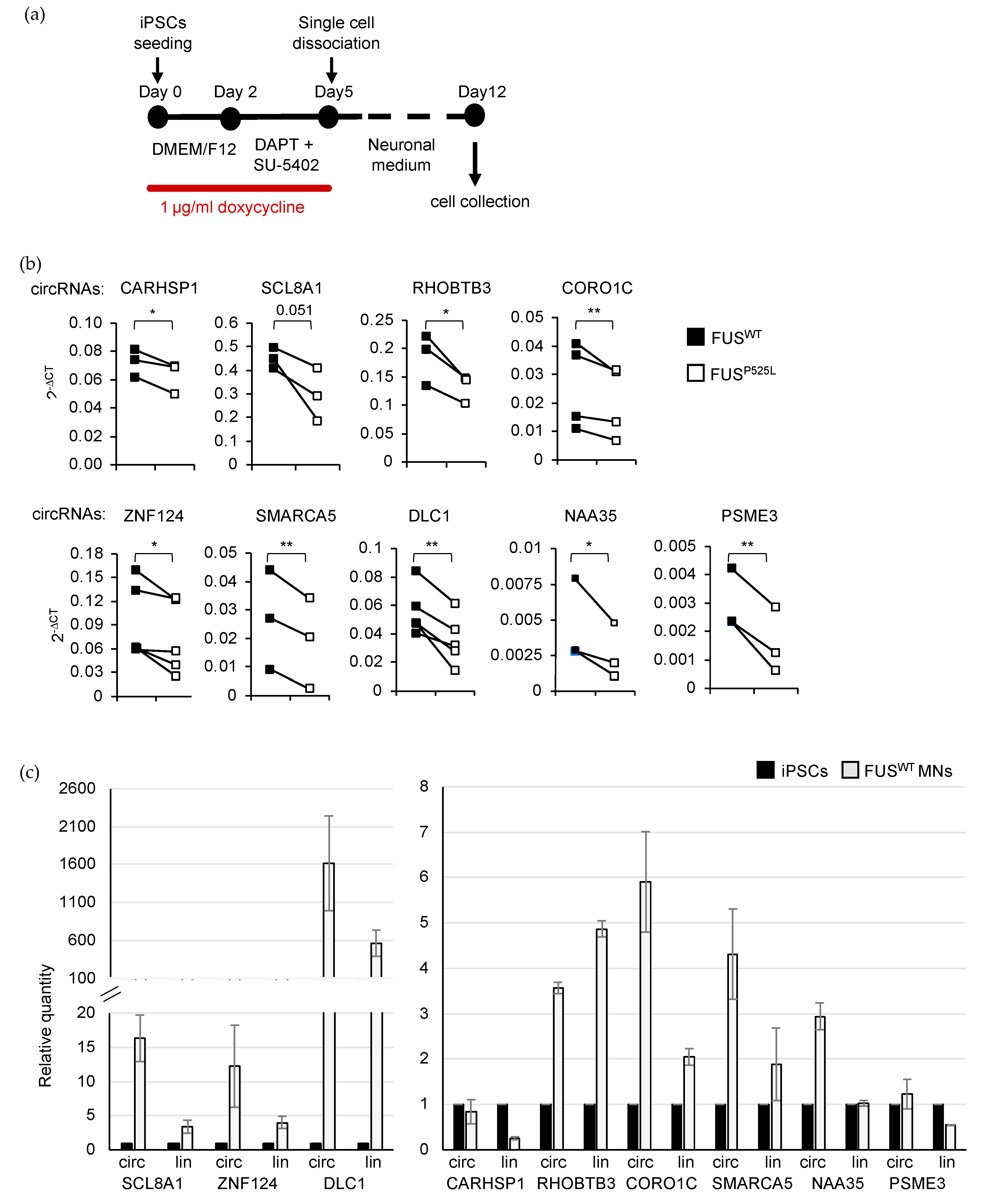
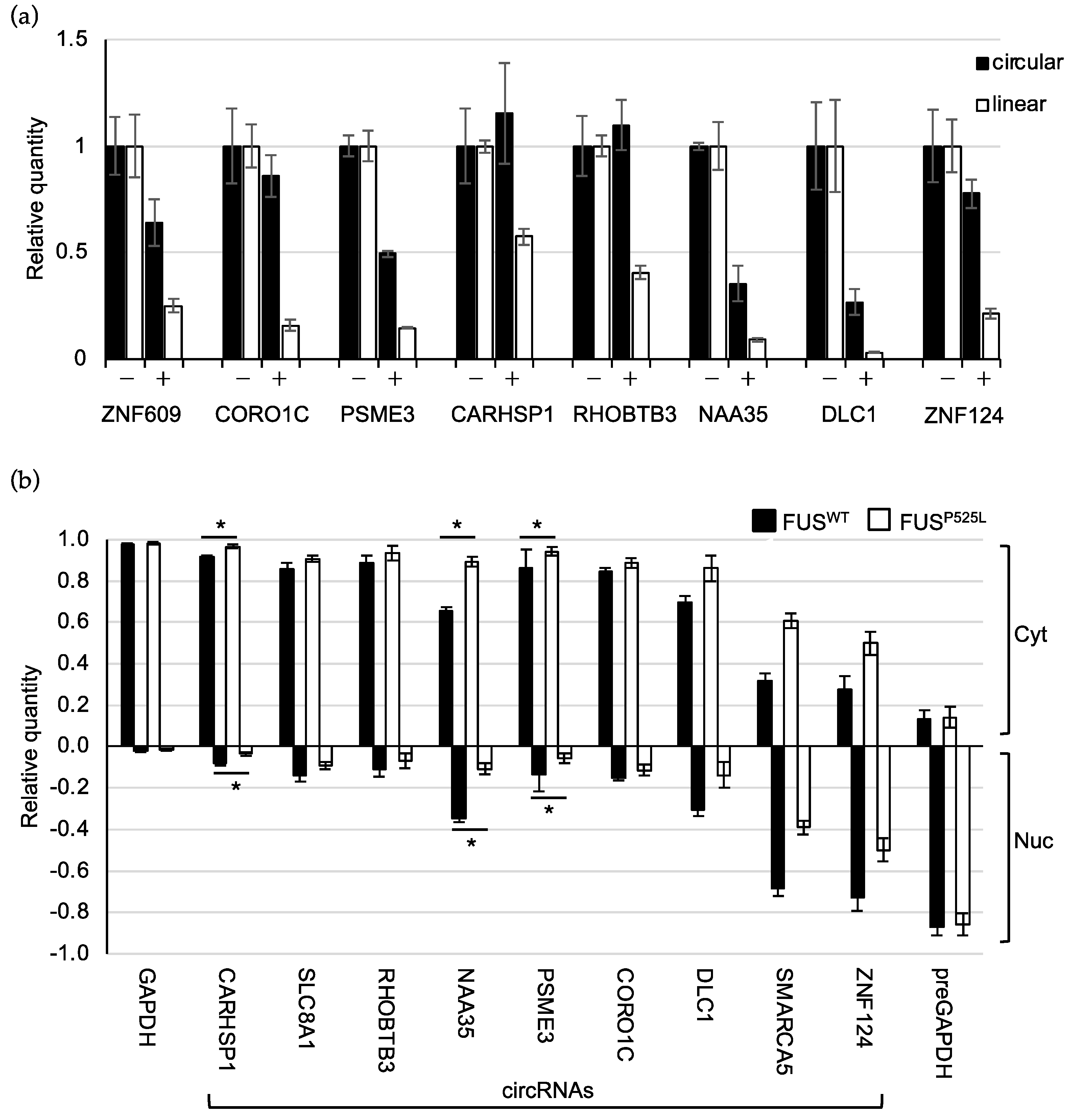
2.3. Validation of circRNA Expression in MNs
2.4. CircRNAs Have Preferential Cytoplasmic Localization
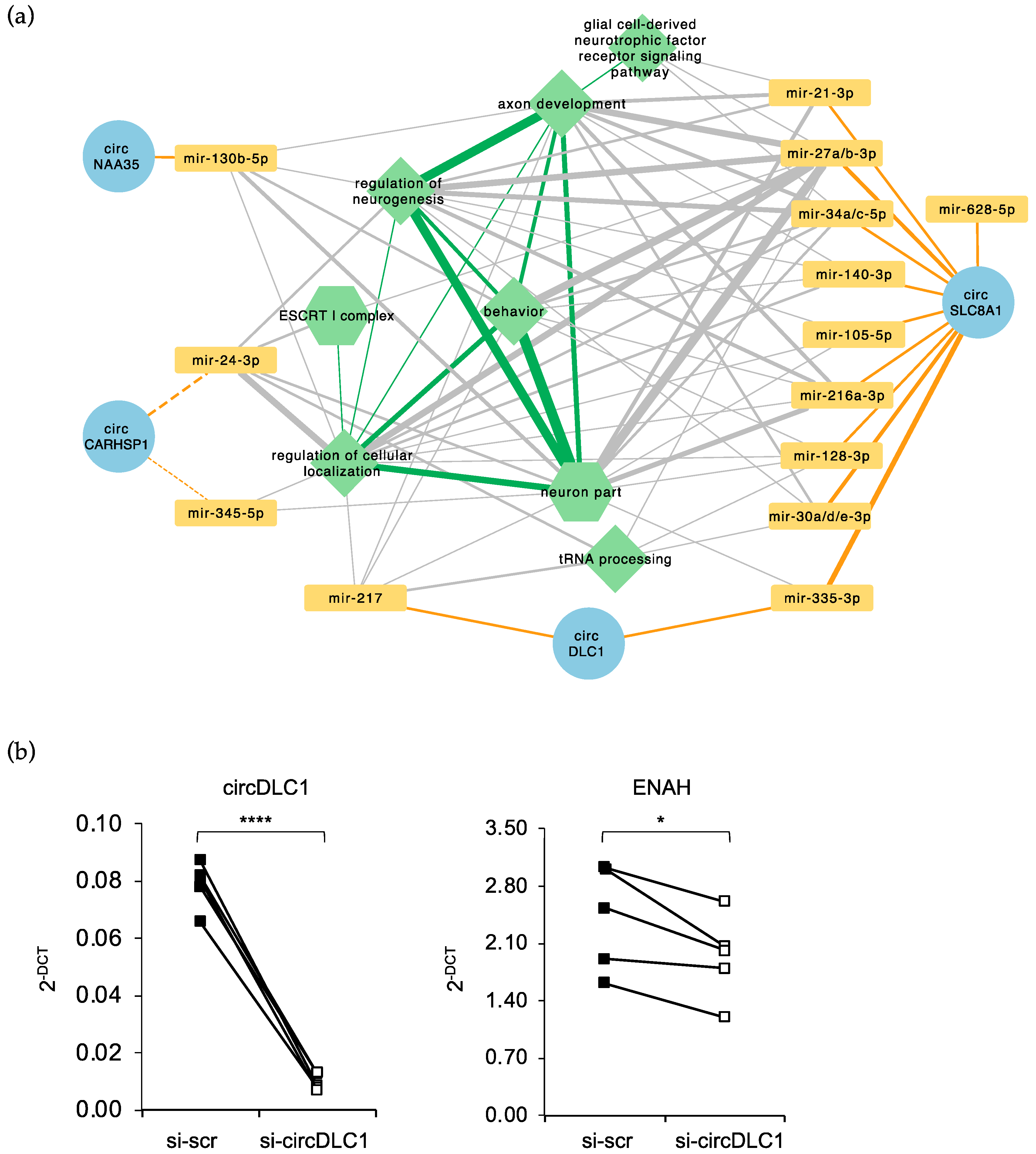
2.5. Prediction of a circRNA–miRNA–mRNA Network Altered in FUSP525L MNs
3. Discussion
4. Materials and Methods
4.1. Computational Identification of Back-Splicing Events
4.2. CircRNA Quantification and Differential Expression Analysis
4.3. Analysis of circRNA Flanking Intronic Regions
4.4. Inference of the circRNA–miRNA–mRNA Regulatory Network
4.5. iPSCs Maintenance, Spinal MN Differentiation and Treatment
4.6. Nucleus/Cytoplasm Fractionation
4.7. RNA Extraction and Analyses
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Alexiou, P.; Maragkakis, M.; Chang, A.; Mourelatos, Z. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA 2013, 19, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Sahadevan, S.; Hembach, K.M.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Megat, S.; Weber, J.; Schwarz, P.; Dupuis, L.; Robinson, M.D.; Tantardini, E.; et al. Synaptic FUS accumulation triggers early misregulation of synaptic RNAs in a mouse model of ALS. Nat. Commun. 2021, 12, 3027. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Marangi, G.; Traynor, B.J. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2014, 1607, 75–93. [Google Scholar] [CrossRef]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J., Jr.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H., Jr.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Deshpande, D.; Higelin, J.; Schoen, M.; Vomhof, T.; Boeckers, T.M.; Demestre, M.; Michaelis, J. Synaptic FUS localization during motoneuron development and its accumulation in human ALS synapses. Front. Cell Neurosci. 2019, 13, 256. [Google Scholar] [CrossRef]
- D’Ambra, E.; Santini, T.; Vitiello, E.; D’Uva, S.; Silenzi, V.; Morlando, M.; Bozzoni, I. Circ-Hdgfrp3 shuttles along neurites and is trapped in aggregates formed by ALS-associated mutant FUS. iScience 2021, 24, 103504. [Google Scholar] [CrossRef]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef] [PubMed]
- Garone, M.G.; Alfano, V.; Salvatori, B.; Braccia, C.; Peruzzi, G.; Colantoni, A.; Bozzoni, I.; Armirotti, A.; Rosa, A. Proteomics analysis of FUS mutant human motoneurons reveals altered regulation of cytoskeleton and other ALS-linked proteins via 3’UTR binding. Sci. Rep. 2020, 10, 11827. [Google Scholar] [CrossRef] [PubMed]
- Garone, M.G.; Birsa, N.; Rosito, M.; Salaris, F.; Mochi, M.; de Turris, V.; Nair, R.R.; Cunningham, T.J.; Fisher, E.M.C.; Morlando, M.; et al. ALS-related FUS mutations alter axon growth in motoneurons and affect HuD/ELAVL4 and FMRP activity. Commun. Biol. 2021, 4, 1025. [Google Scholar] [CrossRef]
- Morlando, M.; Dini Modigliani, S.; Torrelli, G.; Rosa, A.; Di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef]
- Yang, L.; Gal, J.; Chen, J.; Zhu, H. Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl. Acad. Sci. USA 2014, 111, 17809–17814. [Google Scholar] [CrossRef]
- Errichelli, L.; Dini Modigliani, S.; Laneve, P.; Colantoni, A.; Legnini, I.; Capauto, D.; Rosa, A.; De Santis, R.; Scarfò, R.; Peruzzi, G.; et al. FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons. Nat. Commun. 2017, 8, 14741. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Liu, G.; Oztu¨rk, A.; Hicks, G.G. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013, 9, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
- Humphrey, J.; Birsa, N.; Milioto, C.; McLaughlin, M.; Ule, A.M.; Robaldo, D.; Eberle, A.B.; Kräuchi, R.; Bentham, M.; Brown, A.L.; et al. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 2020, 48, 6889–6905. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless., N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, K.K.; Hansen, T.B.; Kjems, J. Insights into circular RNA biology. RNA Biol. 2017, 14, 1035–1045. [Google Scholar] [CrossRef]
- Wang, P.L.; Bao, Y.; Yee, M.C.; Barrett, S.P.; Hogan, G.J.; Olsen, M.N.; Dinneny, J.R.; Brown, P.O.; Salzman, J. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 2014, 9, e90859. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, X.O.; Jiang, T.; Cai, L.; Huang, X.; Liu, Q.; Li, D.; Lu, A.; Liu, Y.; Xue, W.; et al. Comprehensive identification of alternative back-splicing in human tissue transcriptomes. Nucleic Acids Res. 2020, 48, 1779–1789. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, D.; Verheijen, B.M.; Pasterkamp, R.J. Circular RNAs: Novel Regulators of Neuronal Development. Front. Mol. Neurosci. 2016, 9, 74. [Google Scholar] [CrossRef]
- Hanan, M.; Soreq, H.; Kadener, S. CircRNAs in the brain. RNA Biol. 2017, 14, 1028–1034. [Google Scholar] [CrossRef]
- Hanan, M.; Simchovitz, A.; Yayon, N.; Vaknine, S.; Cohen-Fultheim, R.; Karmon, M.; Madrer, N.; Rohrlich, T.M.; Maman, M.; Bennett, E.R.; et al. A Parkinson’s disease CircRNAs Resource reveals a link between circSLC8A1 and oxidative stress. EMBO Mol. Med. 2020, 12, e11942. [Google Scholar] [CrossRef]
- Li, J.; Sun, C.; Cui, H.; Sun, J.; Zhou, P. Role of circRNAs in neurodevelopment and neurodegenerative diseases. J. Mol. Neurosci. 2021, 71, 1743–1751. [Google Scholar] [CrossRef]
- Vakili, O.; Asili, P.; Babaei, Z.; Mirahmad, M.; Keshavarzmotamed, A.; Asemi, Z.; Mafi, A. Circular RNAs in Alzheimer’s Disease: A New Perspective of Diagnostic and Therapeutic Targets. CNS Neurol. Disord. Drug Targets 2022. [Google Scholar] [CrossRef]
- D’Ambra, E.; Capauto, D.; Morlando, M. Exploring the Regulatory Role of Circular RNAs in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 5477. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Das, A.; Sinha, T.; Shyamal, S.; Panda, A.C. Emerging Role of Circular RNA-Protein Interactions. Noncoding RNA 2021, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37.e9. [Google Scholar] [CrossRef] [PubMed]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, J.; Tian, Y.; Gao, Y.; Dong, X.; Chen, W.; Yuan, X.; Yin, W.; Xu, J.; Chen, K.; et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol. Cancer 2020, 19, 128. [Google Scholar] [CrossRef]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, J.; De Santis, R.; de Turris, V.; Morlando, M.; Laneve, P.; Calvo, A.; Caliendo, V.; Chiò, A.; Rosa, A.; Bozzoni, I. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Model. Mech. 2015, 8, 755–766. [Google Scholar] [CrossRef]
- De Santis, R.; Alfano, V.; de Turris, V.; Colantoni, A.; Santini, L.; Garone, M.G.; Antonacci, G.; Peruzzi, G.; Sudria-Lopez, E.; Wyler, E.; et al. Mutant FUS and ELAVL4 (HuD) Aberrant Crosstalk in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 27, 3818–3831.e5. [Google Scholar] [CrossRef]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- De Santis, R.; Santini, L.; Colantoni, A.; Peruzzi, G.; de Turris, V.; Alfano, V.; Bozzoni, I.; Rosa, A. FUS Mutant Human Motoneurons Display Altered Transcriptome and microRNA Pathways with Implications for ALS Pathogenesis. Stem Cell Rep. 2017, 9, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Shinoe, T.; Wanaka, A.; Nikaido, T.; Kanazawa, K.Y.; Shimizu, J.; Imaizumi, K.; Kanazawa, I. Upregulation of the pro-apoptotic BH3-only peptide harakiri in spinal neurons of amyotrophic lateral sclerosis patients. Neurosci. Lett. 2001, 313, 153–157. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef]
- Vaz, A.R.; Cunha, C.; Gomes, C.; Schmucki, N.; Barbosa, M.; Brites, D. Glycoursodeoxycholic Acid Reduces Matrix Metalloproteinase-9 and Caspase-9 Activation in a Cellular Model of Superoxide Dismutase-1 Neurodegeneration. Mol. Neurobiol. 2015, 51, 864–877. [Google Scholar] [CrossRef]
- Martínez, H.R.; Escamilla-Ocañas, C.E.; Tenorio-Pedraza, J.M.; Gómez-Almaguer, D.; Jaime-Perez, J.C.; Olguín-Ramírez, L.A.; Salazar-Marioni, S.; González-Garza, M.T. Altered CSF cytokine network in amyotrophic lateral sclerosis patients: A pathway-based statistical analysis. Cytokine 2017, 90, 1–5. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, E.; Hickman, J.J.; Guo, X. Ion channel dysfunction and altered motoneuron excitability in ALS. Neurol. Disord. Epilepsy J. 2019, 3, 124. [Google Scholar] [PubMed]
- Howells, J.; Matamala, J.M.; Park, S.B.; Garg, N.; Vucic, S.; Bostock, H.; Burke, D.; Kiernan, M.C. In vivo evidence for reduced ion channel expression in motor axons of patients with amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 5379–5396. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef]
- Osenberg, S.; Paz Yaacov, N.; Safran, M.; Moshkovitz, S.; Shtrichman, R.; Sherf, O.; Jacob-Hirsch, J.; Keshet, G.; Amariglio, N.; Itskovitz-Eldor, J.; et al. Alu sequences in undifferentiated human embryonic stem cells display high levels of A-to-I RNA editing. PLoS ONE 2010, 5, e11173. [Google Scholar] [CrossRef]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods 2012, 9, 579–581. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Garone, M.G.; de Turris, V.; Soloperto, A.; Brighi, C.; De Santis, R.; Pagani, F.; Di Angelantonio, S.; Rosa, A. Conversion of Human Induced Pluripotent Stem Cells (iPSCs) into Functional Spinal and Cranial Motor Neurons Using PiggyBac Vectors. J. Vis. Exp. 2019, 147, e59321. [Google Scholar] [CrossRef]
- Hawkins, S.; Namboori, S.C.; Tariq, A.; Blaker, C.; Flaxman, C.; Dey, N.S.; Henley, P.; Randall, A.; Rosa, A.; Stanton, L.W.; et al. Upregulation of β-catenin due to loss of miR-139 contributes to motor neuron death in amyotrophic lateral sclerosis. Stem Cell Rep. 2022, 17, 1650–1665. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef]
- Tanaka, H.; Shimazawa, M.; Kimura, M.; Takata, K.; Tsuruma, M.; Yamada, H.; Takahashi, I.; Hozumi, J.; Niwa, Y.; Iguchi, T.; et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci. Rep. 2012, 2, 573. [Google Scholar] [CrossRef]
- Helmer, R.A.; Foreman, O.; Dertien, J.S.; Panchoo, M.; Bhakta, S.M.; Chilton, B.S. Role of helicase-like transcription factor (hltf) in the G2/m transition and apoptosis in brain. PLoS ONE 2013, 8, e66799. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Chan, N.P.; Wan, T.S.; Lam, L.Y.; Cheung, C.H.; Wong, T.H.; Ip, R.K.; Wong, R.S.; Ng, M.H. Helicase-like transcription factor is a RUNX1 target whose downregulation promotes genomic instability and correlates with complex cytogenetic features in acute myeloid leukemia. Haematologica 2016, 101, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, S.; Gotoh, N.; Matsumoto, H.; Murayama, C.; Suzuki, T.; Yamamoto, T. Expression of sorting nexin 18 (SNX18) is dynamically regulated in developing spinal motor neurons. J. Histochem. Cytochem. 2011, 59, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Bernard-Marissal, N.; Vourc’h, P.; Guettard, Y.O.; Sunyach, C.; Augereau, O.; Khederchah, J.; Mouzat, K.; Antar, C.; Gordon, P.H.; et al. A rare motor neuron deleterious missense mutation in the DPYSL3 (CRMP4) gene is associated with ALS. Hum. Mutat. 2013, 34, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Huang, X.; Talbot, K.; Chan, H.Y.E. A fine balance between Prpf19 and Exoc7 in achieving degradation of aggregated protein and suppression of cell death in spinocerebellar ataxia type 3. Cell Death Dis. 2021, 12, 136. [Google Scholar] [CrossRef]
- Wang, J.; Schultz, P.G.; Johnson, K.A. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. USA 2018, 115, E4604–E4612. [Google Scholar] [CrossRef]
- Park, M.; Jung, H.-G.; Kweon, H.-J.; Kim, Y.-E.; Park, J.-Y.; Hwang, E.M. The E3 ubiquitin ligase, NEDD4L (NEDD4-2) regulates bestrophin-1 (BEST1) by ubiquitin-dependent proteolysis. Biochem. Biophys. Res. Commun. 2019, 514, 344–350. [Google Scholar] [CrossRef]
- Mori, K.; Nihei, Y.; Arzberger, T.; Zhou, Q.; Mackenzie, I.R.; Hermann, A.; Hanisch, F.; German Consortium for Frontotemporal Lobar Degeneration; Bavarian Brain Banking Alliance; Kamp, F.; et al. Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide-repeat protein deposition. EMBO Rep. 2016, 17, 1314–1325. [Google Scholar] [CrossRef]
- An, H.; Litscher, G.; Watanabe, N.; Wei, W.; Hashimoto, T.; Iwatsubo, T.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked cytoplasmic FUS assemblies are compositionally different from physiological stress granules and sequester hnRNPA3, a novel modifier of FUS toxicity. Neurobiol. Dis. 2022, 162, 105585. [Google Scholar] [CrossRef] [PubMed]
- Calini, D.; Corrado, L.; Del Bo, R.; Gagliardi, S.; Pensato, V.; Verde, F.; Corti, S.; Mazzini, L.; Milani, P.; Castellotti, B.; et al. Analysis of hnRNPA1, A2/B1, and A3 genes in patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, e11–e12. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ma, N.; Chen, X.; Wang, Y.; Zhang, W.; Wan, J. CircRtn4 Acts as the Sponge of miR-24-3p to Promote Neurite Growth by Regulating CHD5. Front. Mol. Neurosci. 2021, 14, 660429. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Park, S.Y.; Han, J.S. MicroRNA-24-3p regulates neuronal differentiation by controlling hippocalcin expression. Cell Mol. Life Sci. 2019, 76, 4569–4580. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.; Wang, X.; Dong, J. MicroRNA-345-5p regulates depression by targeting suppressor of cytokine signaling 1. Brain Behav. 2020, 10, e01653. [Google Scholar] [CrossRef] [PubMed]
- Kmetzsch, V.; Anquetil, V.; Saracino, D.; Rinaldi, D.; Camuzat, A.; Gareau, T.; Jornea, L.; Forlani, S.; Couratier, P.; Wallon, D.; et al. Plasma microRNA signature in presymptomatic and symptomatic subjects with C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 485–493. [Google Scholar] [CrossRef]
- Memczak, S.; Papavasileiou, P.; Peters, O.; Rajewsky, N. Identification and Characterization of Circular RNAs As a New Class of Putative Biomarkers in Human Blood. PLoS ONE 2015, 10, e0141214. [Google Scholar] [CrossRef]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef]
- Dolinar, A.; Koritnik, B.; Glavač, D.; Ravnik-Glavač, M. Circular RNAs as Potential Blood Biomarkers in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 8052–8062. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Ishigaki, S.; Masuda, A.; Iguchi, Y.; Udagawa, T.; Watanabe, H.; Katsuno, M.; Ohno, K.; Sobue, G. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci. Rep. 2013, 3, 2388. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Okada, S.; Sakurai, M. Adenosine-to-inosine RNA editing in neurological development and disease. RNA Biol. 2021, 18, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Dastidar, S.G.; Tokunaga, S.; Ho, W.Y.; Lim, K.; Ilieva, H.; Parone, P.A.; Tyan, S.H.; Tse, T.M.; Chang, J.C.; et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. eLife 2019, 8, e40811. [Google Scholar] [CrossRef]
- Grassano, M.; Brodini, G.; De Marco, G.; Casale, F.; Fuda, G.; Salamone, P.; Brunetti, M.; Sbaiz, L.; Gallone, S.; Cugnasco, P.; et al. Phenotype Analysis of Fused in Sarcoma Mutations in Amyotrophic Lateral Sclerosis. Neurol. Genet. 2022, 8, e200011. [Google Scholar] [CrossRef]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.; Chian, R.J.; Boggio, K.J.; Quaresma, A.J.; Nickerson, J.A.; Bosco, D.A. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef]
- Salta, E.; De Strooper, B. Noncoding RNAs in neurodegeneration. Nat. Rev. Neurosci. 2017, 18, 627–640. [Google Scholar] [CrossRef]
- Hur, J.; Paez-Colasante, X.; Figueroa-Romero, C.; Lo, T.W.; Barmada, S.J.; Paulsen, M.; Ljungman, M.; Alakwaa, F.M.; Savelieff, M.G.; Goutman, S.A.; et al. miRNA analysis reveals novel dysregulated pathways in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2022, ddac250. [Google Scholar] [CrossRef]
- Kwiatkowski, A.V.; Rubinson, D.; Dent, E.W.; Edward van Veen, J.; Leslie, J.D.; Zhang, J.; Mebane, L.M.; Philippar, U.; Pinheiro, E.M.; Burds, A.a.; et al. Ena/VASP Is Required for neuritogenesis in the developing cortex. Neuron 2007, 56, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Lei, Y.T.; Hong, C.J.; Hsueh, Y.P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841. [Google Scholar] [CrossRef]
- McConnell, R.E.; Van Veen, J.E.; Vidaki, M.; Kwiatkowski, A.V.; Meyer, A.S.; Gertler, F.B. A requirement for filopodia extension toward Slit during Robo-mediated axon repulsion. J. Cell Biol. 2016, 213, 261–274. [Google Scholar] [CrossRef]
- Vidaki, M.; Drees, F.; Saxena, T.; Lanslots, E.; Taliaferro, M.J.; Tatarakis, A.; Burge, C.B.; Wang, E.T.; Gertler, F.B. A Requirement for Mena, an Actin Regulator, in Local mRNA Translation in Developing Neurons. Neuron 2017, 95, 608–622.e5. [Google Scholar] [CrossRef]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.S.; Taverne, T.; Girard, A.; et al. A novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Jin, N.; Shi, J.; Yin, X.; Jin, X.; Wang, S.; Cao, M.; Iqbal, K.; Gong, C.X.; Liu, F. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) enhances tau expression. J. Alzheimers Dis. 2013, 37, 529–538. [Google Scholar] [CrossRef]
- Salpietro, V.; Malintan, N.T.; Llano-Rivas, I.; Spaeth, C.G.; Efthymiou, S.; Striano, P.; Vandrovcova, J.; Cutrupi, M.C.; Chimenz, R.; David, E.; et al. Mutations in the Neuronal Vesicular SNARE VAMP2 Affect Synaptic Membrane Fusion and Impair Human Neurodevelopment. Am. J. Hum. Genet. 2019, 104, 721–730. [Google Scholar] [CrossRef]
- Laszlo, Z.I.; Hindley, N.; Sanchez Avila, A.; Kline, R.A.; Eaton, S.L.; Lamont, D.J.; Smith, C.; Spires-Jones, T.L.; Wishart, T.M.; Henstridge, C.M. Synaptic proteomics reveal distinct molecular signatures of cognitive change and C9ORF72 repeat expansion in the human ALS cortex. Acta Neuropathol. Commun. 2022, 10, 156. [Google Scholar] [CrossRef]
- Lee, J.A.; Gao, F.B. Neuronal Functions of ESCRTs. Exp. Neurobiol. 2012, 21, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648, 571–579. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel. B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 4 June 2014).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S. Ensembl 2014. Nucleic Acids Res. 2014, 42, D749–D755. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinformatics 2014, 15, 293. [Google Scholar] [CrossRef]
- Oliveros, J.C. 2007–2015, Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 22 September 2020).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 4, 4–10. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.H.; Levanon, E.Y.; Eisenberg, E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat. Methods 2019, 16, 1131–1138. [Google Scholar] [CrossRef]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Hu, H. TarPmiR: A new approach for microRNA target site prediction. Bioinformatics 2016, 32, 2768–2775. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- De Santis, R.; Garone, M.G.; Pagani, F.; de Turris, V.; Di Angelantonio, S.; Rosa, A. Direct conversion of human pluripotent stem cells into cranial motor neurons using a piggyBac vector. Stem Cell Res. 2018, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GRCh38 Coordinates | Host Gene Name | Log2(FC) | p-Value |
|---|---|---|---|
| 16:8858350-8859335_- | CARHSP1 | −2.043424531 | 0.018454 |
| 5:95755396-95763620_+ | RHOBTB3 | −1.099679428 | 0.008357 |
| 12:108652272 108654410_- | CORO1C | −0.986079226 | 0.002601 |
| 1:247156406-247159813_- | ZNF124 | −0.548012424 | 0.008117 |
| 4:143543509-143543972_+ | SMARCA5 | −0.368885114 | 0.023003 |
| 9:85959793-85996577_+ | NAA35 | −4.693947572 | 0.003701 |
| 17:42838731-42839380_+ | PSME3 | −4.31708731 | 0.008135 |
| 2:40428473-40430304_- | SLC8A1 | −0.520421943 | 0.019316 |
| 8:13499049-13500196_- | DLC1 | −0.409393486 | 0.053325 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colantoni, A.; Capauto, D.; Alfano, V.; D’Ambra, E.; D’Uva, S.; Tartaglia, G.G.; Morlando, M. FUS Alters circRNA Metabolism in Human Motor Neurons Carrying the ALS-Linked P525L Mutation. Int. J. Mol. Sci. 2023, 24, 3181. https://doi.org/10.3390/ijms24043181
Colantoni A, Capauto D, Alfano V, D’Ambra E, D’Uva S, Tartaglia GG, Morlando M. FUS Alters circRNA Metabolism in Human Motor Neurons Carrying the ALS-Linked P525L Mutation. International Journal of Molecular Sciences. 2023; 24(4):3181. https://doi.org/10.3390/ijms24043181
Chicago/Turabian StyleColantoni, Alessio, Davide Capauto, Vincenzo Alfano, Eleonora D’Ambra, Sara D’Uva, Gian Gaetano Tartaglia, and Mariangela Morlando. 2023. "FUS Alters circRNA Metabolism in Human Motor Neurons Carrying the ALS-Linked P525L Mutation" International Journal of Molecular Sciences 24, no. 4: 3181. https://doi.org/10.3390/ijms24043181
APA StyleColantoni, A., Capauto, D., Alfano, V., D’Ambra, E., D’Uva, S., Tartaglia, G. G., & Morlando, M. (2023). FUS Alters circRNA Metabolism in Human Motor Neurons Carrying the ALS-Linked P525L Mutation. International Journal of Molecular Sciences, 24(4), 3181. https://doi.org/10.3390/ijms24043181