
Sphingoid Bases Regulate the Sigma-1 Receptor—Sphingosine and N,N’-Dimethylsphingosine Are Endogenous Agonists

Abstract
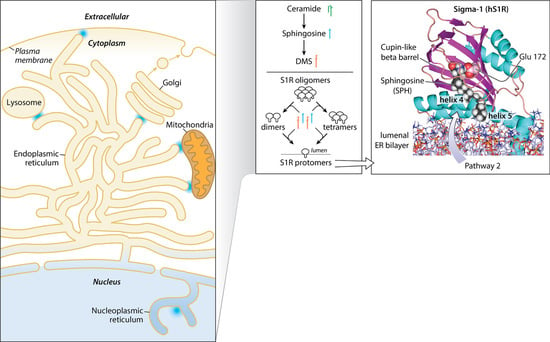
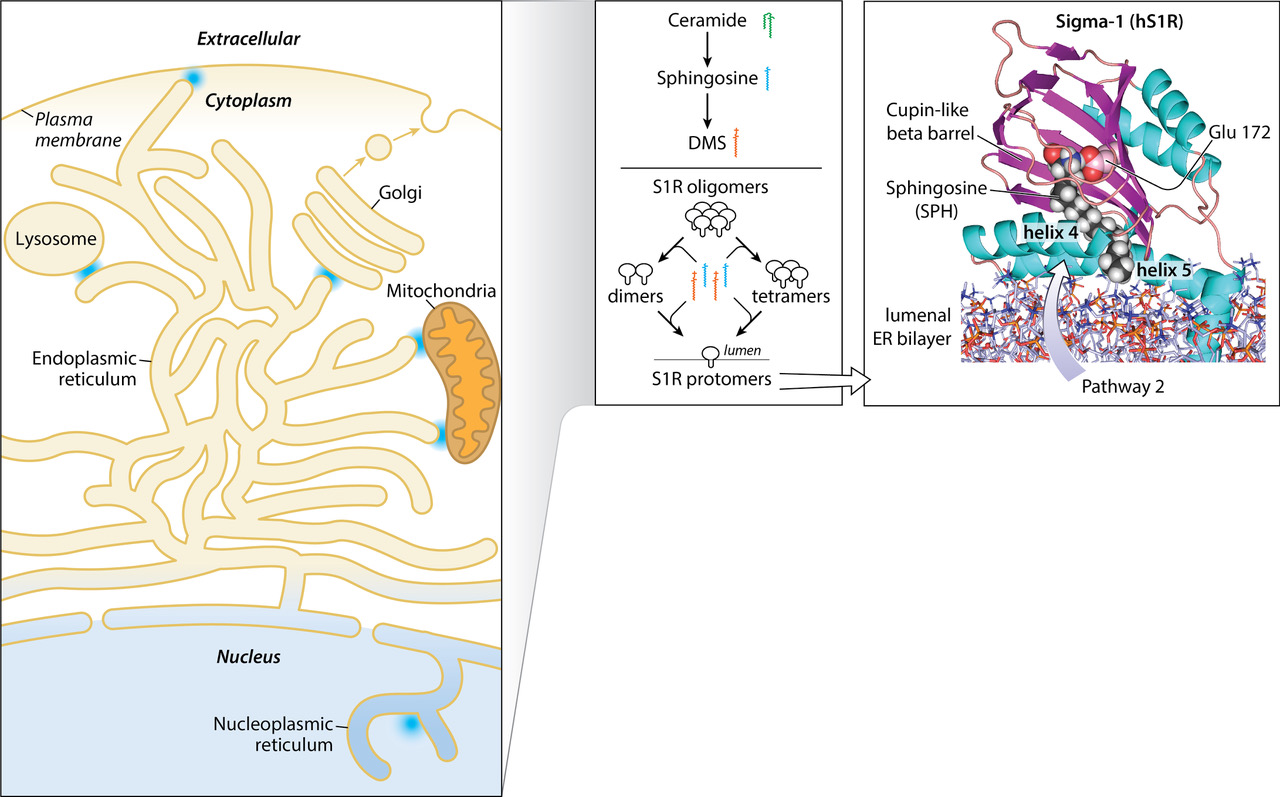{kind=link}
{kind=link}
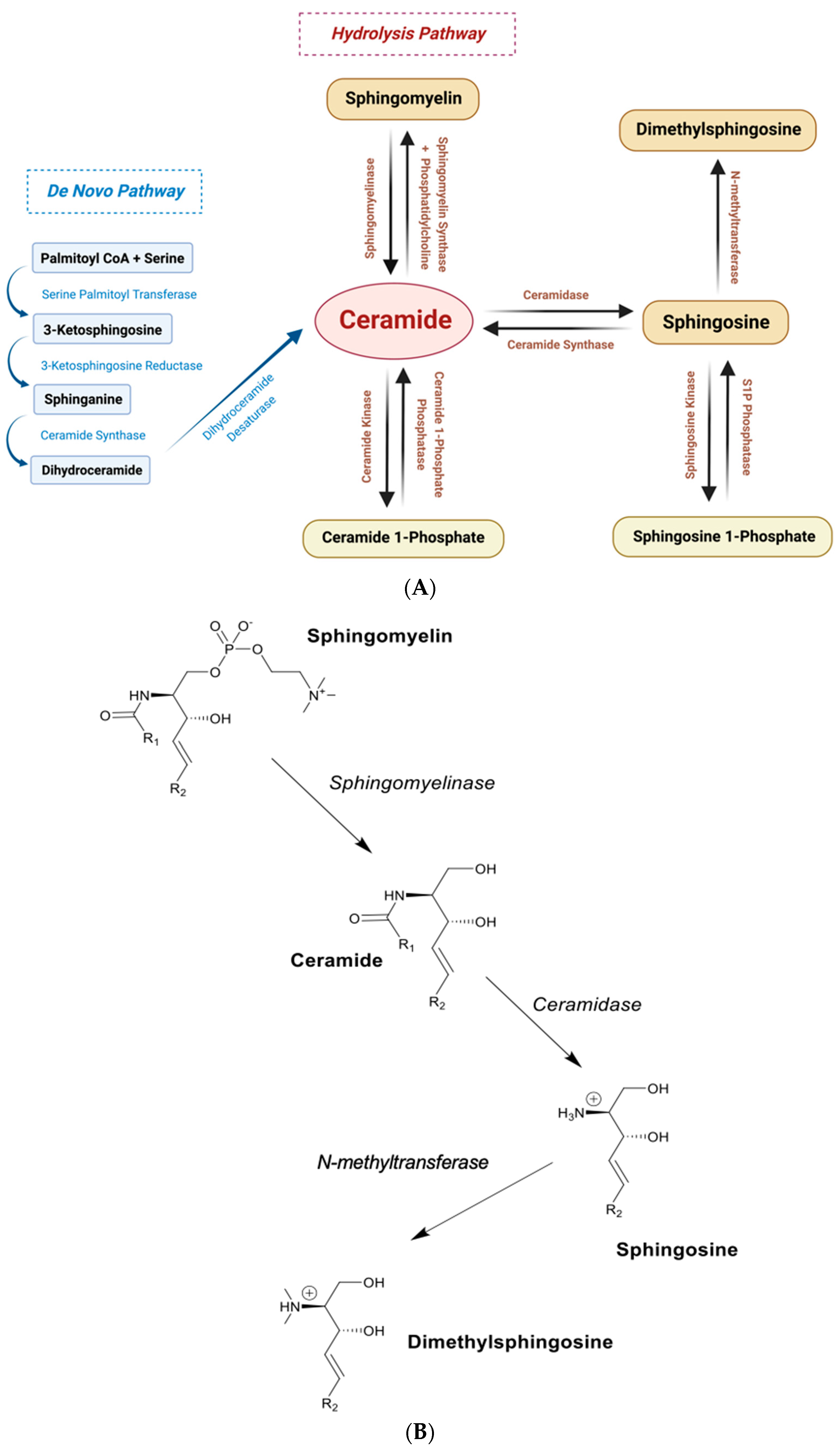{kind=link}
{kind=link}
{kind=link}
{kind=link}
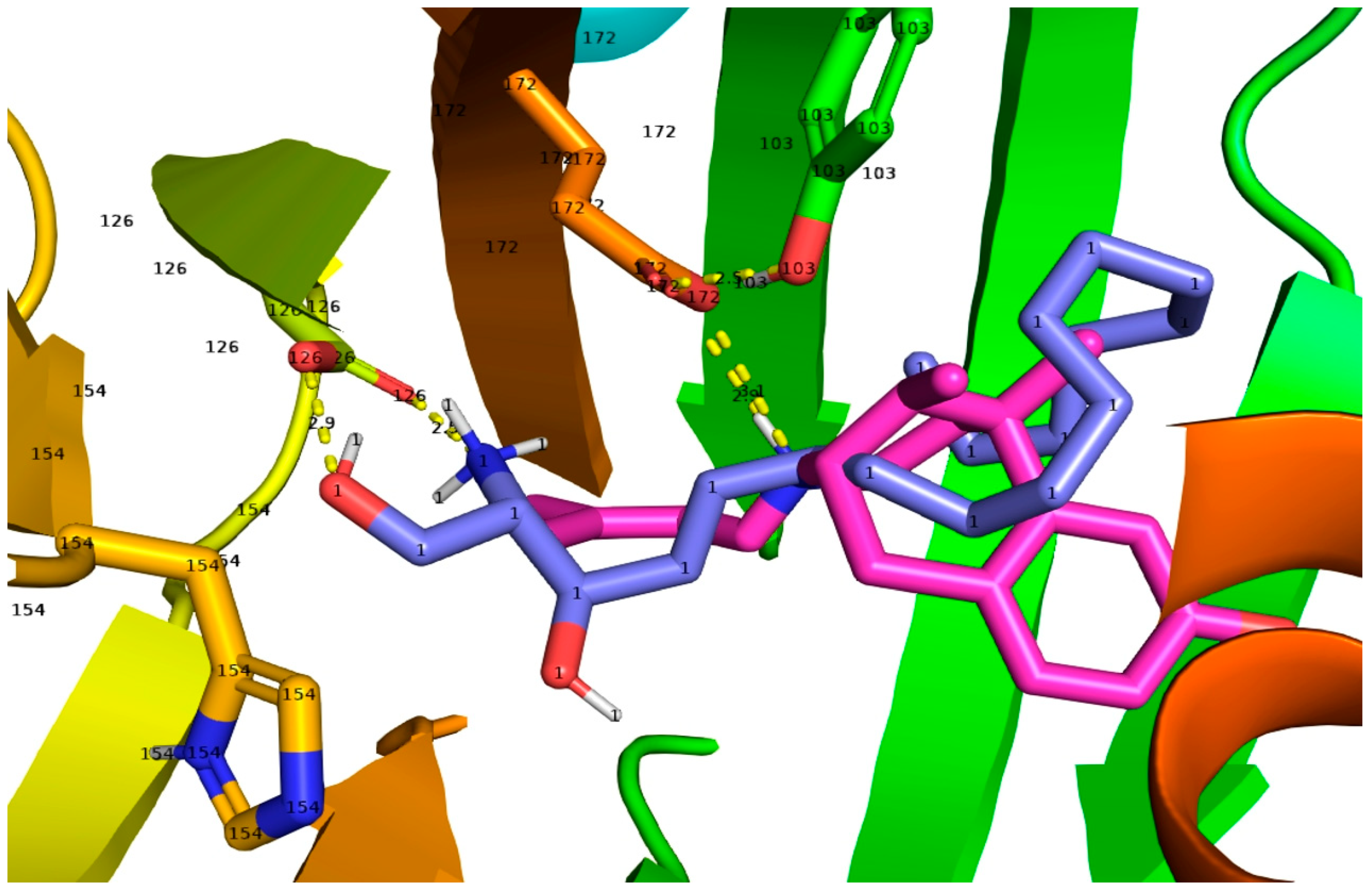{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. SPH and DMS Destabilize S1R Oligomers
2.2. SPH and DMS Dock to the S1R Cupin Beta Barrel
2.3. Molecular Dynamics of SPH Docking to the S1R
2.4. Molecular Dynamics of DMS Docking to the S1R
3. Discussion
3.1. SPH and DMS Are S1R Agonists
3.2. Docking of SPH and DMS to the S1R Cupin Beta Barrel
3.3. Sphingolipid Microdomains Are Potential Primary Sources of Endogenous SPH and DMS
3.4. SPH and DMS Access to the S1R Bindng Site—A Hypothesis
4. Materials and Methods
4.1. Wild-Type (WT) and S1R Knockout (KO) ARPE-19 Cell Lines
4.2. Cell Culture and Treatment with S1R Ligands
4.3. Non-Denaturing (Native) Poly Acrylamide Gel Electrophoresis (PAGE)
4.4. Immunoblotting
4.5. Statistical Analysis
4.6. SPH and DMS Dock to the S1R Protomer Binding Site
4.7. Molecular Dynamics (MD) Performed on Docked SPH and DMS in the Best Energy Position
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Aishwarya, R.; Abdullah, C.S.; Morshed, M.; Remex, N.S.; Bhuiyan, M.S. Sigmar1′s Molecular, Cellular, and Biological Functions in Regulating Cellular Pathophysiology. Front. Physiol. 2021, 12, 705575. [Google Scholar] [CrossRef] [PubMed]
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.M.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Mavlyutov, T.; Singh, D.R.; Biener, G.; Yang, J.; Oliver, J.A.; Ruoho, A.; Raicu, V. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 2015, 466, 263–271. [Google Scholar] [CrossRef]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016, 89, 142–153. [Google Scholar] [CrossRef]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2019, 294, 2121–2132. [Google Scholar] [CrossRef]
- Sun, H.; Wu, M.; Wang, M.; Zhang, X.; Zhu, J. The regulatory role of endoplasmic reticulum chaperone proteins in neurodevelopment. Front. Neurosci. 2022, 16, 1032607. [Google Scholar] [CrossRef]
- Janowska, M.K.; Baughman, H.E.R.; Woods, C.N.; Klevit, R.E. Mechanisms of Small Heat Shock Proteins. Cold Spring Harb. Perspect. Biol. 2019, 11, a034025. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Yang, H.; Epstein, M.L.; Ruoho, A.E.; Yang, J.; Guo, L.W. APEX2-enhanced electron microscopy distinguishes sigma-1 receptor localization in the nucleoplasmic reticulum. Oncotarget 2017, 8, 51317–51330. [Google Scholar] [CrossRef]
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef] [PubMed]
- Zhemkov, V.; Geva, M.; Hayden, M.R.; Bezprozvanny, I. Sigma-1 Receptor (S1R) Interaction with Cholesterol: Mechanisms of S1R Activation and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4082. [Google Scholar] [CrossRef] [PubMed]
- Duitama, M.; Vargas-López, V.; Casas, Z.; Albarracin, S.L.; Sutachan, J.J.; Torres, Y.P. TRP Channels Role in Pain Associated With Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 782. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, B.; Zamanillo, D.; Romero, L.; Carceller, A.; Vela, J.M.; Merlos, M.; Portillo-Salido, E. Comprehensive Preclinical Assessment of Sensory, Functional, Motivational-Affective, and Neurochemical Outcomes in Neuropathic Pain: The Case of the Sigma-1 Receptor. ACS Pharmacol. Transl. Sci. 2022, 5, 240–254. [Google Scholar] [CrossRef]
- Li, J.; Felix-Soriano, E.; Wright, K.R.; Shen, H.; Baer, L.A.; Stanford, K.I.; Guo, L.W. Differential Responses to Sigma-1 or Sigma-2 Receptor Ablation in Adiposity, Fat Oxidation, and Sexual Dimorphism. Int. J. Mol. Sci. 2022, 23, 10846. [Google Scholar] [CrossRef]
- Yang, H.; Shen, H.; Li, J.; Stanford, K.I.; Guo, L.W. Sigma-1 receptor ablation impedes adipocyte-like differentiation of mouse embryonic fibroblasts. Cell Signal. 2020, 75, 109732. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Wu, H.E.; Yasui, Y.; Geva, M.; Hayden, M.; Maurice, T.; Cozzolino, M.; Su, T.P. Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy 2022, 19, 126–151. [Google Scholar] [CrossRef]
- Yang, H.; Shen, H.; Li, J.; Guo, L.W. SIGMAR1/Sigma-1 receptor ablation impairs autophagosome clearance. Autophagy 2019, 15, 1539–1557. [Google Scholar] [CrossRef]
- Kumar, S.; Javed, R.; Mudd, M.; Pallikkuth, S.; Lidke, K.A.; Jain, A.; Tangavelou, K.; Gudmundsson, S.R.; Ye, C.; Rusten, T.E.; et al. Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell 2021, 184, 5950–5969.e22. [Google Scholar] [CrossRef]
- Smith, S.B.; Wang, J.; Cui, X.; Mysona, B.A.; Zhao, J.; Bollinger, K.E. Sigma 1 receptor: A novel therapeutic target in retinal disease. Prog. Retin. Eye Res. 2018, 67, 130–149. [Google Scholar] [CrossRef]
- Pabba, M.; Sibille, E. Sigma-1 and N-Methyl-d-Aspartate Receptors: A Partnership with Beneficial Outcomes. Mol. Neuropsychiatry 2015, 1, 47–51. [Google Scholar] [PubMed]
- Ortíz-Rentería, M.; Juárez-Contreras, R.; González-Ramírez, R.; Islas, L.D.; Sierra-Ramírez, F.; Llorente, I.; Simon, S.A.; Hiriart, M.; Rosenbaum, T.; Morales-Lázaro, S.L. TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain. Proc. Natl. Acad. Sci. USA 2018, 115, E1657–E1666. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Goguadze, N.; Kimura, Y.; Yasui, Y.; Pan, B.; Wang, T.Y.; Nakamura, Y.; Lin, Y.T.; Hogan, Q.H.; Wilson, K.L.; et al. Genomic Action of Sigma-1 Receptor Chaperone Relates to Neuropathic Pain. Mol. Neurobiol. 2021, 58, 2523–2541. [Google Scholar] [CrossRef]
- Kourrich, S.; Su, T.P.; Fujimoto, M.; Bonci, A. The sigma-1 receptor: Roles in neuronal plasticity and disease. Trends Neurosci. 2012, 35, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Su, T.P.; Su, T.C.; Nakamura, Y.; Tsai, S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef]
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptors. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef]
- Bergkemper, M.; Kronenberg, E.; Thum, S.; Börgel, F.; Daniliuc, C.; Schepmann, D.; Nieto, F.R.; Brust, P.; Reinoso, R.F.; Alvarez, I.; et al. Synthesis, Receptor Affinity, and Antiallodynic Activity of Spirocyclic σ Receptor Ligands with Exocyclic Amino Moiety. J. Med. Chem. 2018, 61, 9666–9690. [Google Scholar] [CrossRef]
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.C. Small Molecules Selectively Targeting Sigma-1 Receptor for the Treatment of Neurological Diseases. J. Med. Chem. 2020, 63, 15187–15217. [Google Scholar] [CrossRef]
- Romeo, G.; Bonanno, F.; Wilson, L.L.; Arena, E.; Modica, M.N.; Pittalà, V.; Salerno, L.; Prezzavento, O.; McLaughlin, J.P.; Intagliata, S. Development of New Benzylpiperazine Derivatives as σ(1) Receptor Ligands with in Vivo Antinociceptive and Anti-Allodynic Effects. ACS Chem. Neurosci. 2021, 12, 2003–2012. [Google Scholar] [CrossRef]
- Greenfield, D.A.; Schmidt, H.R.; Skiba, M.A.; Mandler, M.D.; Anderson, J.R.; Sliz, P.; Kruse, A.C. Virtual Screening for Ligand Discovery at the σ(1) Receptor. ACS Med. Chem. Lett. 2020, 11, 1555–1561. [Google Scholar] [CrossRef]
- Abatematteo, F.S.; Niso, M.; Contino, M.; Leopoldo, M.; Abate, C. Multi-Target Directed Ligands (MTDLs) Binding the σ1 Receptor as Promising Therapeutics: State of the Art and Perspectives. Int. J. Mol. Sci. 2021, 22, 6359. [Google Scholar] [CrossRef] [PubMed]
- Keuler, T.; Lemke, C.; Elsinghorst, P.W.; Iriepa, I.; Chioua, M.; Martínez-Grau, M.A.; Beadle, C.D.; Vetman, T.; López-Muñoz, F.; Wille, T.; et al. The Chemotype of Chromanones as a Privileged Scaffold for Multineurotarget Anti-Alzheimer Agents. ACS Pharmacol. Transl. Sci. 2022, 5, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Guitart, X.; Codony, X.; Monroy, X. Sigma receptors: Biology and therapeutic potential. Psychopharmacology 2004, 174, 301–319. [Google Scholar] [CrossRef]
- Maurice, T. Bi-phasic dose response in the preclinical and clinical developments of sigma-1 receptor ligands for the treatment of neurodegenerative disorders. Expert. Opin. Drug Discov. 2021, 16, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Fontanilla, D.; Johannessen, M.; Hajipour, A.R.; Cozzi, N.V.; Jackson, M.B.; Ruoho, A.E. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 2009, 323, 934–937. [Google Scholar] [CrossRef]
- Tsai, S.Y.; Chuang, J.Y.; Tsai, M.S.; Wang, X.F.; Xi, Z.X.; Hung, J.J.; Chang, W.C.; Bonci, A.; Su, T.P. Sigma-1 receptor mediates cocaine-induced transcriptional regulation by recruiting chromatin-remodeling factors at the nuclear envelope. Proc. Natl. Acad. Sci. USA 2015, 112, E6562–E6570. [Google Scholar] [CrossRef]
- Waterhouse, R.N.; Chang, R.C.; Atuehene, N.; Collier, T.L. In vitro and in vivo binding of neuroactive steroids to the sigma-1 receptor as measured with the positron emission tomography radioligand [18F]FPS. Synapse 2007, 61, 540–546. [Google Scholar] [CrossRef]
- Brailoiu, E.; Chakraborty, S.; Brailoiu, G.C.; Zhao, P.; Barr, J.L.; Ilies, M.A.; Unterwald, E.M.; Abood, M.E.; Taylor, C.W. Choline Is an Intracellular Messenger Linking Extracellular Stimuli to IP3-Evoked Ca2+ Signals through Sigma-1 Receptors. Cell Rep. 2019, 26, 330–337.e4. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Meng, F.; Xiao, Y.; Ji, Y.; Sun, Z.; Zhou, X. An open-like conformation of the sigma-1 receptor reveals its ligand entry pathway. Nat. Commun. 2022, 13, 1267. [Google Scholar] [CrossRef]
- Mavlyutov, T.; Chen, X.; Guo, L.; Yang, J. APEX2-tagging of Sigma 1-receptor indicates subcellular protein topology with cytosolic N-terminus and ER luminal C-terminus. Protein Cell 2018, 9, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Patel, C.; Shenkman, M.; Kessel, A.; Ben-Tal, N.; Lederkremer, G.Z. The Sigma-1 receptor is an ER-localized type II membrane protein. J. Biol. Chem. 2021, 297, 101299. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Roldan, J.L.; Ossa, F.; Schnell, J.R. Characterization of the human sigma-1 receptor chaperone domain structure and binding immunoglobulin protein (BiP) interactions. J. Biol. Chem. 2013, 288, 21448–21457. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Hajipour, A.R.; Fontanilla, D.; Ramachandran, S.; Chu, U.B.; Mavlyutov, T.; Ruoho, A.E. Identification of regions of the sigma-1 receptor ligand binding site using a novel photoprobe. Mol. Pharmacol. 2007, 72, 921–933. [Google Scholar] [CrossRef]
- Fontanilla, D.; Hajipour, A.R.; Pal, A.; Chu, U.B.; Arbabian, M.; Ruoho, A.E. Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry 2008, 47, 7205–7217. [Google Scholar] [CrossRef]
- McCann, D.J.; Su, T.P. Solubilization and characterization of haloperidol-sensitive (+)-[3H]SKF-10,047 binding sites (sigma sites) from rat liver membranes. J. Pharmacol. Exp. Ther. 1991, 257, 547–554. [Google Scholar]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef]
- Rossino, G.; Orellana, I.; Caballero, J.; Schepmann, D.; Wünsch, B.; Rui, M.; Rossi, D.; González-Avendaño, M.; Collina, S.; Vergara-Jaque, A. New Insights into the Opening of the Occluded Ligand-Binding Pocket of Sigma1 Receptor: Binding of a Novel Bivalent RC-33 Derivative. J. Chem. Inf. Model. 2020, 60, 756–765. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural basis for sigma1 receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef]
- Ruoho, A.E.; Chu, U.B.; Ramachandran, S.; Fontanilla, D.; Mavlyutov, T.; Hajipour, A.R. The ligand binding region of the sigma-1 receptor: Studies utilizing photoaffinity probes, sphingosine and N-alkylamines. Curr. Pharm. Des. 2012, 18, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Brimson, J.M.; Safrany, S.T.; Qassam, H.; Tencomnao, T. Dipentylammonium Binds to the Sigma-1 Receptor and Protects Against Glutamate Toxicity, Attenuates Dopamine Toxicity and Potentiates Neurite Outgrowth in Various Cultured Cell Lines. Neurotox. Res. 2018, 34, 263–272. [Google Scholar] [CrossRef]
- Brimson, J.M.; Akula, K.K.; Abbas, H.; Ferry, D.R.; Kulkarni, S.K.; Russell, S.T.; Tisdale, M.J.; Tencomnao, T.; Safrany, S.T. Simple ammonium salts acting on sigma-1 receptors yield potential treatments for cancer and depression. Sci. Rep. 2020, 10, 9251. [Google Scholar] [CrossRef] [PubMed]
- Chu, U.B.; Hajipour, A.R.; Ramachandran, S.; Ruoho, A.E. Characterization of interactions of 4-nitrophenylpropyl-N-alkylamine with ς receptors. Biochemistry 2011, 50, 7568–7578. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Chu, U.B.; Mavlyutov, T.A.; Pal, A.; Pyne, S.; Ruoho, A.E. The sigma1 receptor interacts with N-alkyl amines and endogenous sphingolipids. Eur. J. Pharmacol. 2009, 609, 19–26. [Google Scholar] [CrossRef]
- Kahoun, J.R.; Ruoho, A.E. (125I)iodoazidococaine, a photoaffinity label for the haloperidol-sensitive sigma receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 1393–1397. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Spassieva, S.; Bieberich, E. Lysosphingolipids and sphingolipidoses: Psychosine in Krabbe’s disease. J. Neurosci. Res. 2016, 94, 974–981. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Pelled, D.; Riebeling, C.; Bodennec, J.; de-Morgan, A.; Waller, H.; Schiffmann, R.; Futerman, A.H. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 2003, 278, 23594–23599. [Google Scholar] [CrossRef]
- Adams, D.R.; Pyne, S.; Pyne, N.J. Sphingosine Kinases: Emerging Structure-Function Insights. Trends Biochem. Sci. 2016, 41, 395–409. [Google Scholar] [CrossRef]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A house divided: Ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 2006, 1758, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Welch, S.P.; Sim-Selley, L.J.; Selley, D.E. Sphingosine-1-phosphate receptors as emerging targets for treatment of pain. Biochem. Pharmacol. 2012, 84, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Doyle, T.; Kress, M.; Nicol, G. Therapeutic targeting of the ceramide-to-sphingosine 1-phosphate pathway in pain. Trends Pharmacol. Sci. 2013, 34, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef]
- Igarashi, Y.; Hakomori, S. Enzymatic synthesis of N,N-dimethyl-sphingosine: Demonstration of the sphingosine: N-methyltransferase in mouse brain. Biochem. Biophys. Res. Commun. 1989, 164, 1411–1416. [Google Scholar] [CrossRef]
- Malta, I.; Moraes, T.; Rodrigues, G.; Franco, P.; Galdino, G. The role of oligodendrocytes in chronic pain: Cellular and molecular mechanisms. J. Physiol. Pharmacol. 2019, 70, 299–309. [Google Scholar]
- Chen, Y.J.; Hill, S.; Huang, H.; Taraboletti, A.; Cho, K.; Gallo, R.; Manchester, M.; Shriver, L.P.; Patti, G.J. Inflammation triggers production of dimethylsphingosine from oligodendrocytes. Neuroscience 2014, 279, 113–121. [Google Scholar] [CrossRef]
- de Costa, B.R.; Bowen, W.D.; Hellewell, S.B.; Walker, J.M.; Thurkauf, A.; Jacobson, A.E.; Rice, K.C. Synthesis and evaluation of optically pure [3H]-(+)-pentazocine, a highly potent and selective radioligand for sigma receptors. FEBS Lett. 1989, 251, 53–58. [Google Scholar] [CrossRef]
- Johnson, C.H.; Patti, G.J.; Courade, J.P.; Shriver, L.P.; Hoang, L.T.; Manchester, M.; Siuzdak, G. Alterations in Spinal Cord Metabolism during Treatment of Neuropathic Pain. J. Neuroimmune Pharmacol. 2015, 10, 396–401. [Google Scholar] [CrossRef]
- Wei, H.; Chen, Z.; Koivisto, A.; Pertovaara, A. Spinal mechanisms contributing to the development of pain hypersensitivity induced by sphingolipids in the rat. Pharmacol. Rep. 2021, 73, 672–679. [Google Scholar] [CrossRef]
- Grimm, C.; Kraft, R.; Schultz, G.; Harteneck, C. Activation of the melastatin-related cation channel TRPM3 by D-erythro-sphingosine. Mol. Pharmacol. 2005, 67, 798–805, Erratum in Mol. Pharmacol. 2005, 67, 1382. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y. Possible Roles of Sphingosine and N,N-Dimethylsphingosine as Modulators in Membrane Signal Transduction Systems. Trends Glycosci. Glycotechnol. 1990, 2, 319–332. [Google Scholar] [CrossRef]
- Yatomi, Y.; Ruan, F.; Megidish, T.; Toyokuni, T.; Hakomori, S.; Igarashi, Y. N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry 1996, 35, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Kitamura, K.; Toyokuni, T.; Dean, B.; Fenderson, B.; Ogawass, T.; Hakomori, S. A specific enhancing effect of N,N-dimethylsphingosine on epidermal growth factor receptor autophosphorylation. Demonstration of its endogenous occurrence (and the virtual absence of unsubstituted sphingosine) in human epidermoid carcinoma A431 cells. J. Biol. Chem. 1990, 265, 5385–5389. [Google Scholar] [CrossRef]
- Jo, J.Y.; Kim, H.L.; Lee, Y.K.; Tomura, H.; Bae, Y.S.; Okajima, F.; Im, D.S. N,N-Dimethyl-D-erythro-sphingosine inhibits store-operated Ca2+ entry in U937 monocytes. J. Pharmacol. Sci. 2008, 107, 303–307. [Google Scholar] [CrossRef]
- Hong, W.C. Distinct Regulation of sigma (1) Receptor Multimerization by Its Agonists and Antagonists in Transfected Cells and Rat Liver Membranes. J. Pharmacol. Exp. Ther. 2020, 373, 290–301. [Google Scholar] [CrossRef]
- Yano, H.; Bonifazi, A.; Xu, M.; Guthrie, D.A.; Schneck, S.N.; Abramyan, A.M.; Fant, A.D.; Hong, W.C.; Newman, A.H.; Shi, L. Pharmacological profiling of sigma 1 receptor ligands by novel receptor homomer assays. Neuropharmacology 2018, 133, 264–275. [Google Scholar] [CrossRef]
- Hong, W.C.; Yano, H.; Hiranita, T.; Chin, F.T.; McCurdy, C.R.; Su, T.P.; Amara, S.G.; Katz, J.L. The sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats. J. Biol. Chem. 2017, 292, 11250–11261. [Google Scholar] [CrossRef]
- Matsumoto, R.R.; Bowen, W.D.; Tom, M.A.; Vo, V.N.; Truong, D.D.; De Costa, B.R. Characterization of two novel sigma receptor ligands: Antidystonic effects in rats suggest sigma receptor antagonism. Eur. J. Pharmacol. 1995, 280, 301–310. [Google Scholar] [CrossRef]
- Ola, M.S.; Moore, P.; El-Sherbeny, A.; Roon, P.; Agarwal, N.; Sarthy, V.P.; Casellas, P.; Ganapathy, V.; Smith, S.B. Expression pattern of sigma receptor 1 mRNA and protein in mammalian retina. Brain Res. Mol. Brain Res. 2001, 95, 86–95. [Google Scholar]
- Matsumoto, R.R.; Nguyen, L.; Kaushal, N.; Robson, M.J. Sigma (sigma) receptors as potential therapeutic targets to mitigate psychostimulant effects. Adv. Pharmacol. 2014, 69, 323–386. [Google Scholar] [PubMed]
- Brindley, R.L.; Bauer, M.B.; Hartley, N.D.; Horning, K.J.; Currie, K.P.M. Sigma-1 receptor ligands inhibit catecholamine secretion from adrenal chromaffin cells due to block of nicotinic acetylcholine receptors. J. Neurochem. 2017, 143, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Yamamoto, H.; Atta, M.S.; Mahmoud, S.; Oh-hashi, K.; Kiuchi, K. Chloroquine inhibits glutamate-induced death of a neuronal cell line by reducing reactive oxygen species through sigma-1 receptor. J. Neurochem. 2011, 119, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Brimson, J.M.; Brown, C.A.; Safrany, S.T. Antagonists show GTP-sensitive high-affinity binding to the sigma-1 receptor. Br. J. Pharmacol. 2011, 164, 772–780. [Google Scholar] [CrossRef]
- Su, T.P.; Wu, X.Z.; Cone, E.J.; Shukla, K.; Gund, T.M.; Dodge, A.L.; Parish, D.W. Sigma compounds derived from phencyclidine: Identification of PRE-084, a new, selective sigma ligand. J. Pharmacol. Exp. Ther. 1991, 259, 543–550. [Google Scholar] [PubMed]
- Yano, H.; Liu, L.; Naing, S.; Shi, L. The Effects of Terminal Tagging on Homomeric Interactions of the Sigma 1 Receptor. Front. Neurosci. 2019, 13, 1356. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Pokrass, M.J.; Klauer, N.R.; Nohara, H.; Su, T.P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. USA 2015, 112, 6742–6747. [Google Scholar] [CrossRef]
- Su, T.P.; London, E.D.; Jaffe, J.H. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science 1988, 240, 219–221. [Google Scholar] [CrossRef]
- Monnet, F.P.; Maurice, T. The sigma1 protein as a target for the non-genomic effects of neuro(active)steroids: Molecular, physiological, and behavioral aspects. J. Pharmacol. Sci. 2006, 100, 93–118. [Google Scholar] [CrossRef]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar] [CrossRef]
- Xu, R.; Sun, W.; Jin, J.; Obeid, L.M.; Mao, C. Role of alkaline ceramidases in the generation of sphingosine and its phosphate in erythrocytes. FASEB J. 2010, 24, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Zelnik, I.D.; Ventura, A.E.; Kim, J.L.; Silva, L.C.; Futerman, A.H. The role of ceramide in regulating endoplasmic reticulum function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158489. [Google Scholar] [CrossRef] [PubMed]
- Mehendale, N.; Mallik, R.; Kamat, S.S. Mapping Sphingolipid Metabolism Pathways during Phagosomal Maturation. ACS Chem. Biol. 2021, 16, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Contreras, F.X.; Ernst, A.M.; Haberkant, P.; Bjorkholm, P.; Lindahl, E.; Gonen, B.; Tischer, C.; Elofsson, A.; von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 2012, 481, 525–529. [Google Scholar] [CrossRef]
- Ernst, A.M.; Brugger, B. Sphingolipids as modulators of membrane proteins. Biochim. Biophys. Acta 2014, 1841, 665–670. [Google Scholar] [CrossRef]
- Bjorkholm, P.; Ernst, A.M.; Hacke, M.; Wieland, F.; Brugger, B.; von Heijne, G. Identification of novel sphingolipid-binding motifs in mammalian membrane proteins. Biochim. Biophys. Acta 2014, 1838, 2066–2070. [Google Scholar] [CrossRef]
- Pannwitt, S.; Stangl, M.; Schneider, D. Lipid Binding Controls Dimerization of the Coat Protein p24 Transmembrane Helix. Biophys. J. 2019, 117, 1554–1562. [Google Scholar] [CrossRef]
- Zhemkov, V.; Ditlev, J.A.; Lee, W.R.; Wilson, M.; Liou, J.; Rosen, M.K.; Bezprozvanny, I. The role of sigma 1 receptor in organization of endoplasmic reticulum signaling microdomains. eLife 2021, 10, e65192. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Tanaka, Y.; Seto, M.; Kakegawa, K.; Takami, K.; Kikuchi, F.; Yamamoto, T.; Nakamura, M.; Daini, M.; Murakami, M.; Ohashi, T.; et al. Discovery of Brain-Penetrant Glucosylceramide Synthase Inhibitors with a Novel Pharmacophore. J. Med. Chem. 2022, 65, 4270–4290. [Google Scholar] [CrossRef]
- Saied, E.M.; Arenz, C. Small molecule inhibitors of ceramidases. Cell Physiol. Biochem. 2014, 34, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Bielawska, A.; Greenberg, M.S.; Perry, D.; Jayadev, S.; Shayman, J.A.; McKay, C.; Hannun, Y.A. (1S,2R)-D-erythro-2-(N-myristoylamino)-1-phenyl-1-propanol as an inhibitor of ceramidase. J. Biol. Chem. 1996, 271, 12646–12654. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 2003, 306, 718–725. [Google Scholar] [CrossRef]
- Hayashi, T.; Hayashi, E.; Fujimoto, M.; Sprong, H.; Su, T.P. The lifetime of UDP-galactose:ceramide galactosyltransferase is controlled by a distinct endoplasmic reticulum-associated degradation (ERAD) regulated by sigma-1 receptor chaperones. J. Biol. Chem. 2012, 287, 43156–43169. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14949–14954. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ramachandran, S.; Lu, H.; Prabhu, U.; Ruoho, A.E. Purification and characterization of the guinea pig sigma-1 receptor functionally expressed in Escherichia coli. Protein Expr. Purif. 2007, 51, 283–292. [Google Scholar] [CrossRef]
- Kumagai, K.; Shono, K.; Nakayama, H.; Ohno, Y.; Saji, I. (2R-trans)-2-butyl-5-heptylpyrrolidine as a potent sigma receptor ligand produced by Streptomyces longispororuber. J. Antibiot. 2000, 53, 467–473. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Satyshur, K.A.; Guo, L.-W.; Ruoho, A.E. Sphingoid Bases Regulate the Sigma-1 Receptor—Sphingosine and N,N’-Dimethylsphingosine Are Endogenous Agonists. Int. J. Mol. Sci. 2023, 24, 3103. https://doi.org/10.3390/ijms24043103
Li J, Satyshur KA, Guo L-W, Ruoho AE. Sphingoid Bases Regulate the Sigma-1 Receptor—Sphingosine and N,N’-Dimethylsphingosine Are Endogenous Agonists. International Journal of Molecular Sciences. 2023; 24(4):3103. https://doi.org/10.3390/ijms24043103
Chicago/Turabian StyleLi, Jing, Kenneth A. Satyshur, Lian-Wang Guo, and Arnold E. Ruoho. 2023. "Sphingoid Bases Regulate the Sigma-1 Receptor—Sphingosine and N,N’-Dimethylsphingosine Are Endogenous Agonists" International Journal of Molecular Sciences 24, no. 4: 3103. https://doi.org/10.3390/ijms24043103
APA StyleLi, J., Satyshur, K. A., Guo, L.-W., & Ruoho, A. E. (2023). Sphingoid Bases Regulate the Sigma-1 Receptor—Sphingosine and N,N’-Dimethylsphingosine Are Endogenous Agonists. International Journal of Molecular Sciences, 24(4), 3103. https://doi.org/10.3390/ijms24043103