
Sigma-2 Receptors—From Basic Biology to Therapeutic Target: A Focus on Age-Related Degenerative Diseases
 , ,
, ,
Abstract
1. Introduction
2. Alzheimer’s Disease
2.1. Overview of the Disease
2.2. Currently Approved Alzheimer’s Disease Therapeutics
2.3. Therapeutic Approaches in Development to Treat the Underlying Disease Have Shown Modest Success
3. α-Synucleinopathies—Parkinson’s Disease and Dementia with Lewy Bodies
3.1. Overview of the Disease
3.2. Limitations of Current Treatments for α-Synucleinopathies
4. Retinal Diseases—Dry Age-Related Macular Degeneration
4.1. Overview of the Disease
4.2. Treatments for Dry AMD Are Limited
5. The Sigma-2 Receptor
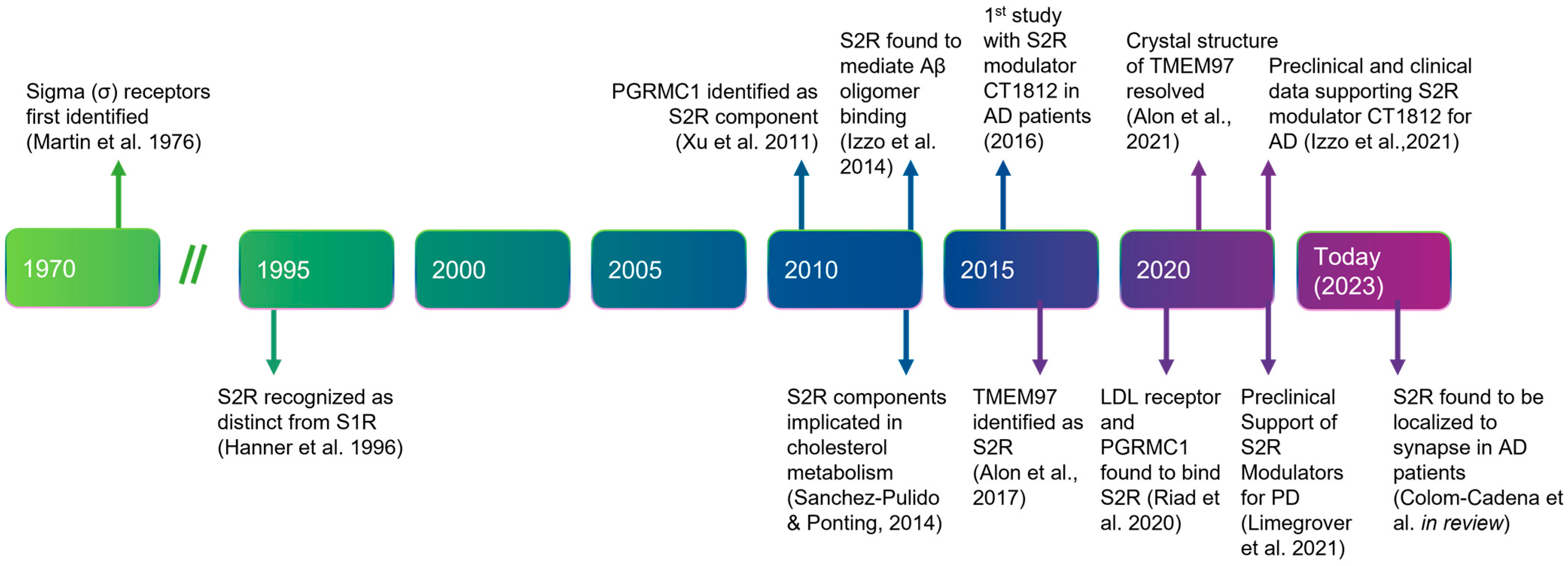
5.1. History, Structure, and Function of S2R
5.2. Expression and Regulation of S2R/TMEM97

5.3. Putatitve Endogenous Ligands
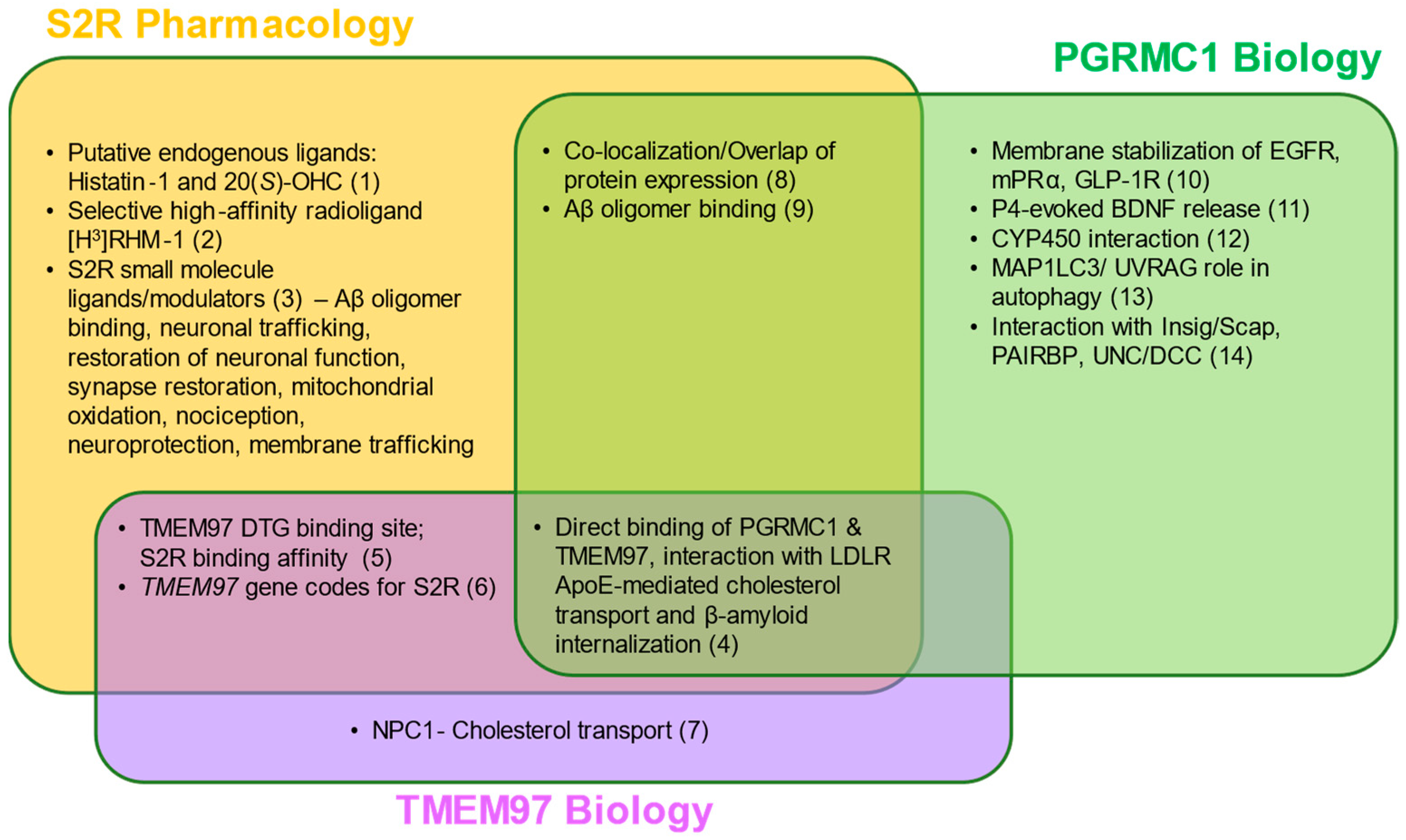
5.4. Protein Interactions Underlie the Functions of S2R
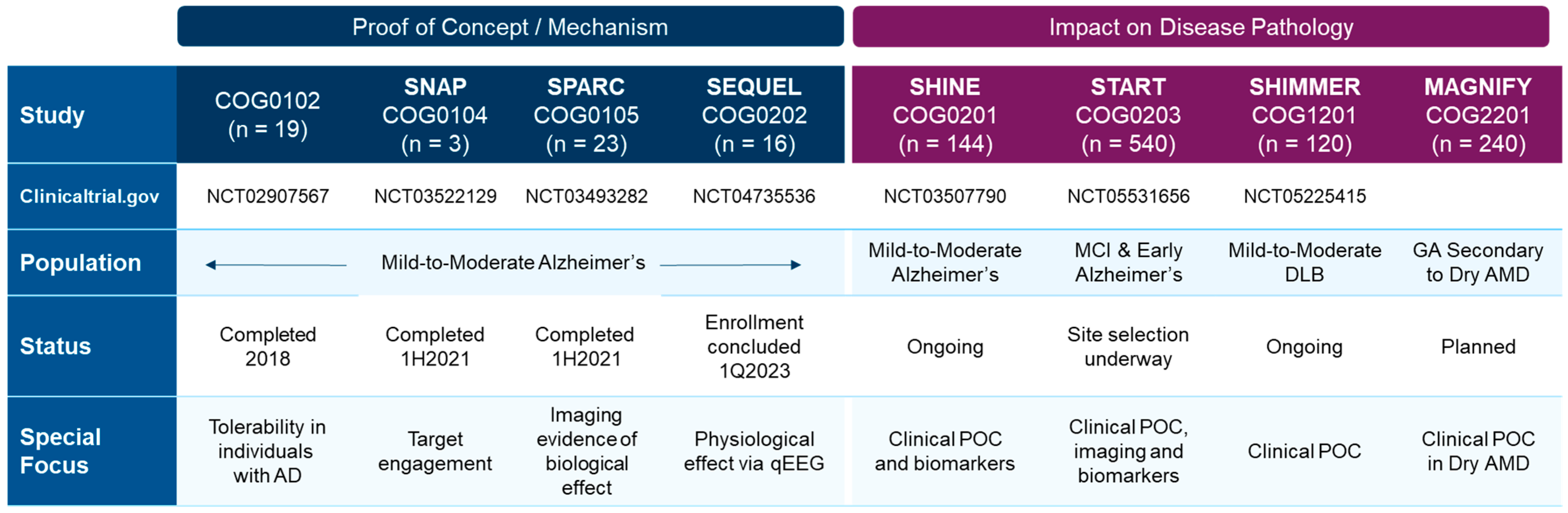
6. S2R Modulators as a Promising Therapeutic Approach for Age-Related Degenerative Diseases
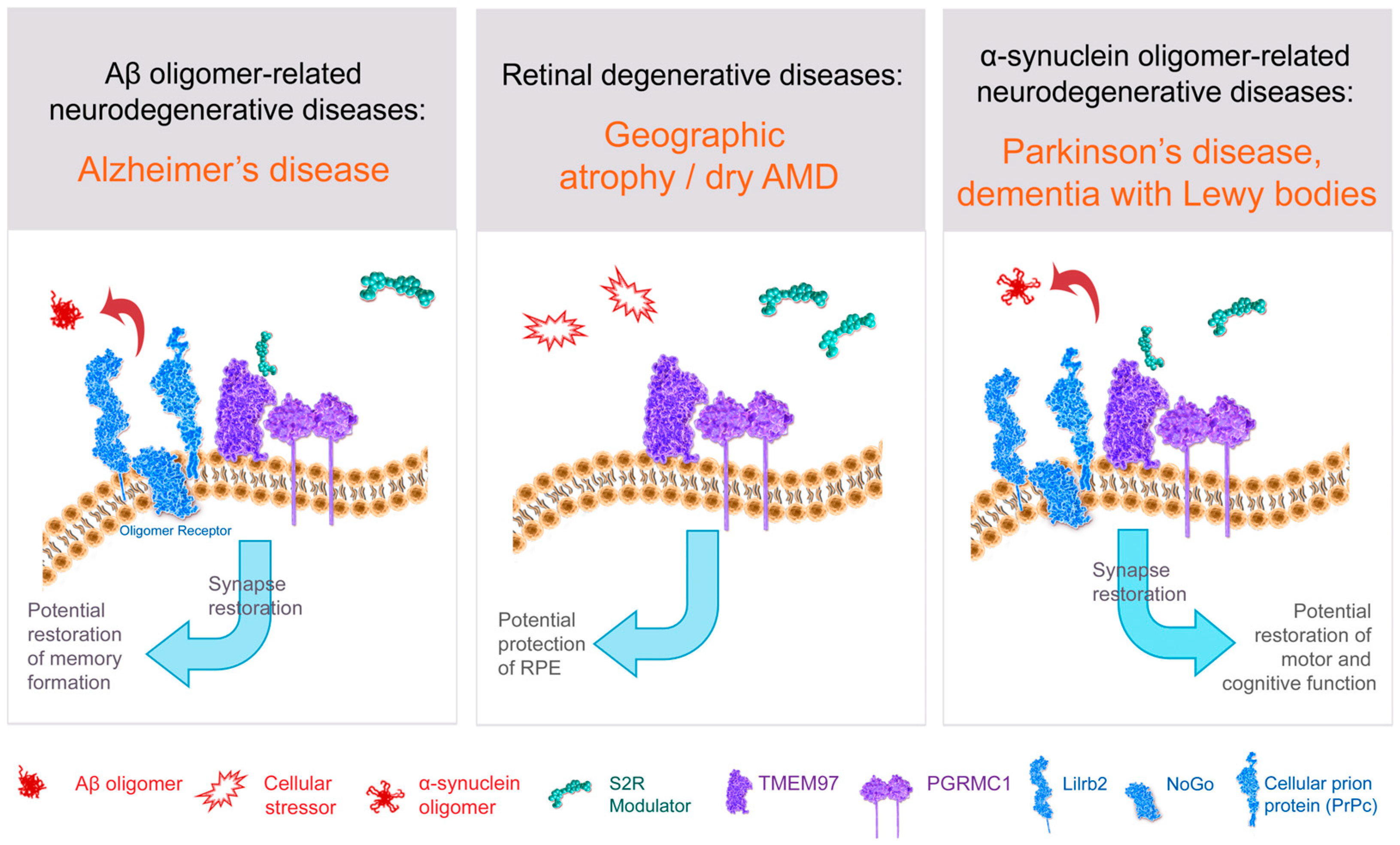
6.1. S2R in Alzheimer’s Disease: Synaptoprotection
6.2. Rationale for S2R Modulators for Alzheimer’s Disease: Synaptoprotective, Restoration of Function
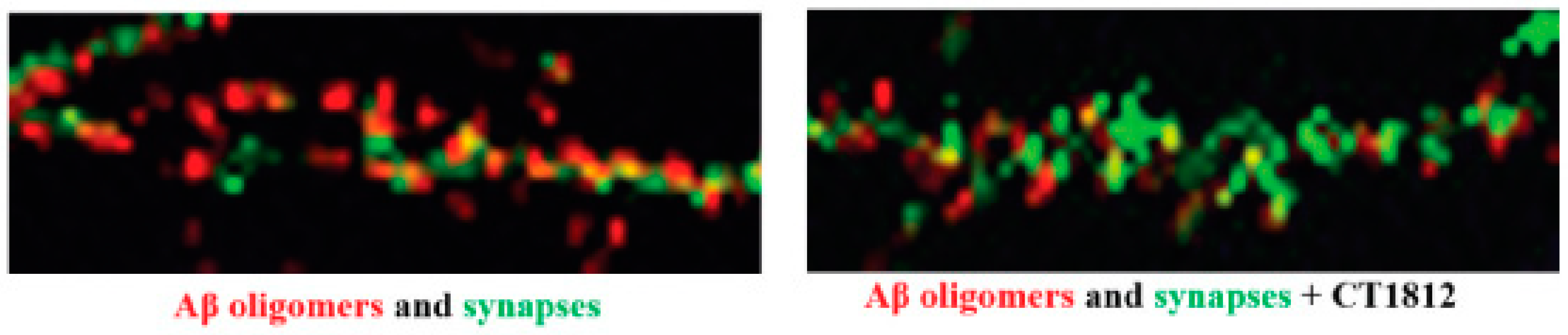
6.3. Evidence of Synaptoprotection through S2R Modulators Blocking Amyloid-β Oligomer Toxicity
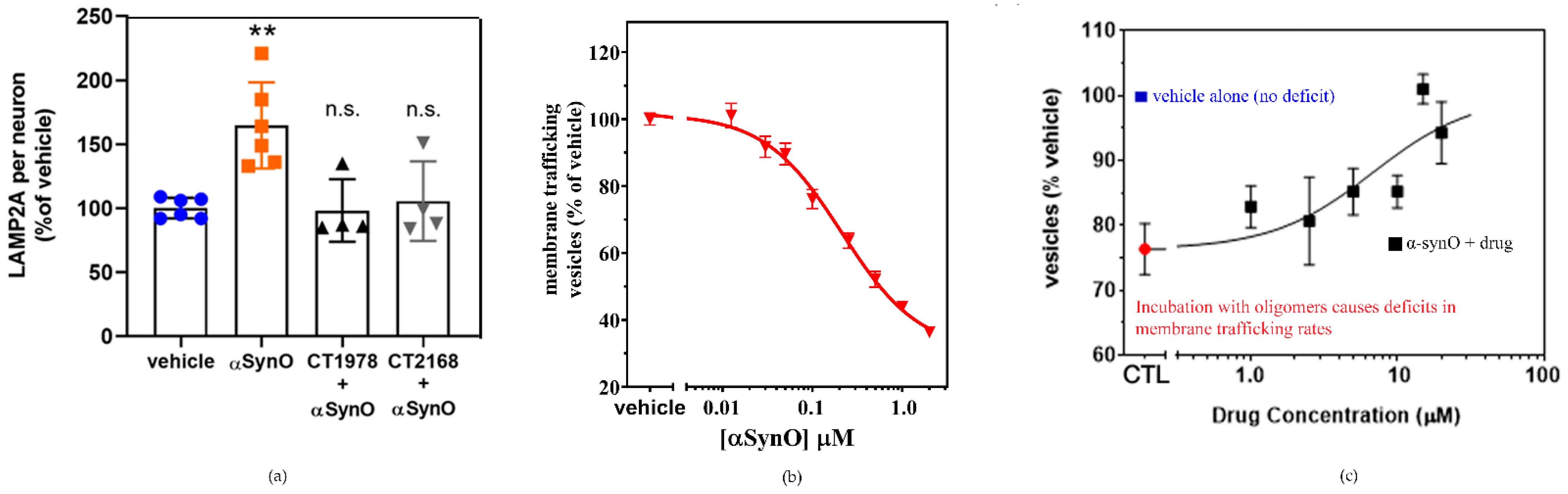
6.4. Preclinical Evidence for CT1812, Currently in Clinical Development for Alzheimer’s Disease
6.5. Rationale for Targeting S2R for α-Synucleinopathies
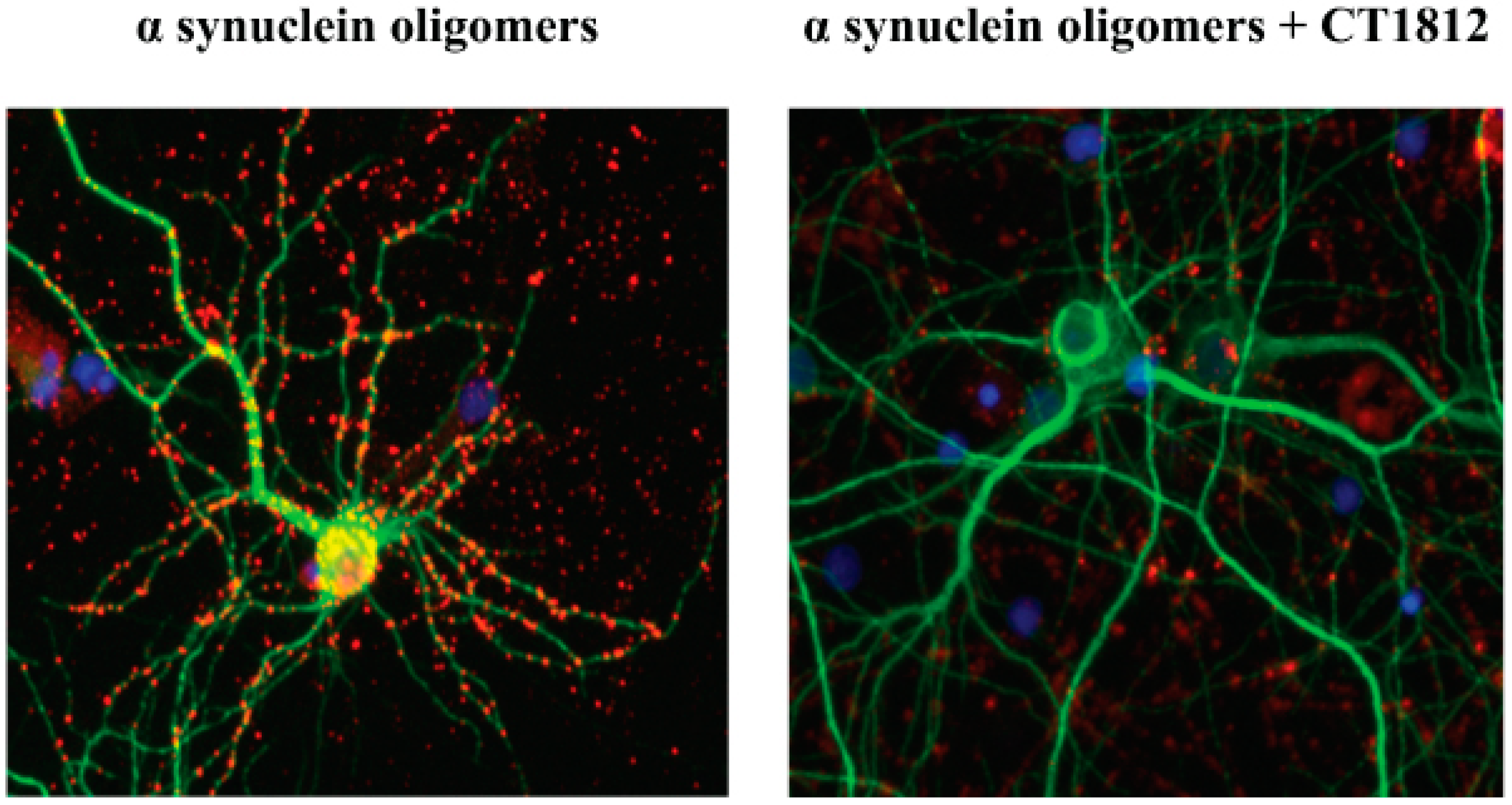
6.6. Small Molecules Targeting S2R for α-Synucleinopathies
6.7. Rationale for S2R in Dry AMD
6.8. Other Indications in Focus for S2R Therapeutic Intervention
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single–Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Izzo, N.J.; Staniszewski, A.; To, L.; Fa, M.; Teich, A.F.; Saeed, F.; Wostein, H.; Walko, T.; Vaswani, A.; Wardius, M.; et al. Alzheimer’s Therapeutics Targeting Amyloid Beta 1–42 Oligomers I: Abeta 42 Oligomer Binding to Specific Neuronal Receptors Is Displaced by Drug Candidates That Improve Cognitive Deficits. PLoS ONE 2014, 9, e111898. [Google Scholar] [CrossRef] [PubMed]
- Izzo, N.J.; Xu, J.; Zeng, C.; Kirk, M.J.; Mozzoni, K.; Silky, C.; Rehak, C.; Yurko, R.; Look, G.; Rishton, G.; et al. Alzheimer’s Therapeutics Targeting Amyloid Beta 1–42 Oligomers II: Sigma-2/PGRMC1 Receptors Mediate Abeta 42 Oligomer Binding and Synaptotoxicity. PLoS ONE 2014, 9, e111899. [Google Scholar] [CrossRef] [PubMed]
- Limegrover, C.S.; Yurko, R.; Izzo, N.J.; LaBarbera, K.M.; Rehak, C.; Look, G.; Rishton, G.; Safferstein, H.; Catalano, S.M. Sigma-2 Receptor Antagonists Rescue Neuronal Dysfunction Induced by Parkinson’s Patient Brain-derived A-synuclein. J. Neurosci. Res. 2021, 99, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Arbez, N.; Sahn, J.J.; Lu, Y.; Linkens, K.T.; Hodges, T.R.; Tang, A.; Wiseman, R.; Martin, S.F.; Ross, C.A. Neuroprotective Effects of σ 2 R/TMEM97 Receptor Modulators in the Neuronal Model of Huntington’s Disease. ACS Chem. Neurosci. 2022, 13, 2852–2862. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Peluso, J.J. Progesterone Receptor Membrane Component 1 and Its Role in Ovarian Follicle Growth. Front. Neurosci. 2013, 7, 99. [Google Scholar] [CrossRef]
- Peluso, J.J.; Pru, J.K. Non-Canonical Progesterone Signaling in Granulosa Cell Function. Reproduction 2014, 147, 169–178. [Google Scholar] [CrossRef]
- Thomas, P. Characteristics of Membrane Progestin Receptor Alpha (MPRα) and Progesterone Membrane Receptor Component 1 (PGMRC1) and Their Roles in Mediating Rapid Progestin Actions. Front. Neuroendocrinol. 2008, 29, 292–312. [Google Scholar] [CrossRef]
- Zeng, C.; Riad, A.; Mach, R.H. The Biological Function of Sigma-2 Receptor/Tmem97 and Its Utility in Pet Imaging Studies in Cancer. Cancers 2020, 12, 1877. [Google Scholar] [CrossRef]
- Cahill, M.A.; Jazayeri, J.A.; Catalano, S.M.; Toyokuni, S.; Kovacevic, Z.; Richardson, D.R. The Emerging Role of Progesterone Receptor Membrane Component 1 (PGRMC1) in Cancer Biology. Biochim. Biophys. Acta 2016, 1866, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Zhang, Y.; Huang, Y. Imaging Sigma Receptors in the Brain: New Opportunities for Diagnosis of Alzheimer’s Disease and Therapeutic Development. Neurosci. Lett. 2018, 691, 3–10. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Amyloid β-Protein and beyond: The Path Forward in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Piechal, A.; Jakimiuk, A.; Mirowska-Guzel, D. Sigma Receptors and Neurological Disorders. Pharmacol. Rep. 2021, 73, 1582–1594. [Google Scholar] [CrossRef]
- Terada, K.; Migita, K.; Matsushima, Y.; Kamei, C. Sigma-2 Receptor as a Potential Therapeutic Target for Treating Central Nervous System Disorders. Neural Regen. Res. 2019, 14, 1893. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.S.; Klein, K.; Zanger, U.M. Membrane Associated Progesterone Receptors: Promiscuous Proteins with Pleiotropic Functions—Focus on Interactions with Cytochromes P450. Front. Pharmacol. 2017, 8, 159. [Google Scholar] [CrossRef]
- Cahill, M.A.; Medlock, A.E. Thoughts on Interactions between PGRMC1 and Diverse Attested and Potential Hydrophobic Ligands. J. Steroid Biochem. Mol. Biol. 2017, 171, 11–33. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- World Health Organization. Global Action Plan on the Public Health Response to Dementia 2017–2025; World Health Organization: Geneva, Switzerland, 2017; 52p. [Google Scholar]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical Trials and Late-Stage Drug Development for Alzheimer’s Disease: An Appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- Herring, W.J.; Ceesay, P.; Snyder, E.; Bliwise, D.; Budd, K.; Hutzelmann, J.; Stevens, J.; Lines, C.; Michelson, D. Polysomnographic Assessment of Suvorexant in Patients with Probable Alzheimer’s Disease Dementia and Insomnia: A Randomized Trial. Alzheimer’s Dement. 2020, 16, 541–551. [Google Scholar] [CrossRef] [PubMed]
- van Waarde, A.; Ramakrishnan, N.K.; Rybczynska, A.A.; Elsinga, P.H.; Ishiwata, K.; Nijholt, I.M.; Luiten, P.G.M.; Dierckx, R.A. The Cholinergic System, Sigma-1 Receptors and Cognition. Behav. Brain Res. 2011, 221, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Veroniki, A.A.; Ashoor, H.M.; Rios, P.; Seitidis, G.; Stewart, L.; Clarke, M.; Tudur-Smith, C.; Mavridis, D.; Hemmelgarn, B.R.; Holroyd-Leduc, J.; et al. Comparative Safety and Efficacy of Cognitive Enhancers for Alzheimer’s Dementia: A Systematic Review with Individual Patient Data Network Meta-Analysis. BMJ Open 2022, 12, e053012. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.E.Z.N.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2022. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12295. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimer’s Dis. 2022, 87, 83–100. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Lozupone, M.; Watling, M.; Panza, F. Discontinued Disease-Modifying Therapies for Alzheimer’s Disease: Status and Future Perspectives. Expert Opin. Investig. Drugs 2020, 29, 919–934. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Low-n Oligomers as Therapeutic Targets of Alzheimer’s Disease. J. Neurochem. 2011, 117, 19–28. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Larson, M.E.; Lesné, S.E. Soluble Aβ Oligomer Production and Toxicity. J. Neurochem. 2012, 120 (Suppl. S1), 125–139. [Google Scholar] [CrossRef]
- Karpinar, D.P.; Balija, M.B.G.; Kügler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.-Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-Fibrillar α-Synuclein Variants with Impaired β-Structure Increase Neurotoxicity in Parkinson’s Disease Models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Putcha, P.; Tetzlaff, J.E.; Spoelgen, R.; Koker, M.; Carvalho, F.; Hyman, B.T.; McLean, P.J. Formation of Toxic Oligomeric Alpha-Synuclein Species in Living Cells. PLoS ONE 2008, 3, e1867. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers-Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef]
- Du, X.; Xie, X.; Liu, R. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef]
- Hogan, D.B.; Fiest, K.M.; Roberts, J.I.; Maxwell, C.J.; Dykeman, J.; Pringsheim, T.; Steeves, T.; Smith, E.E.; Pearson, D.; Jetté, N. The Prevalence and Incidence of Dementia with Lewy Bodies: A Systematic Review. Can. J. Neurol. Sci. 2016, 43, S83–S95. [Google Scholar] [CrossRef]
- Zhu, C.W.; Scarmeas, N.; Stavitsky, K.; Albert, M.; Brandt, J.; Blacker, D.; Sano, M.; Stern, Y. Comparison of Costs of Care between Patients with Alzheimer’s Disease and Dementia with Lewy Bodies. Alzheimer’s Dement. 2008, 4, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Raymick, J.; Imam, S. Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. Int. J. Mol. Sci. 2016, 17, 904. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2022 Update. J. Parkinsons. Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Thom, T.; Schmitz, M.; Fischer, A.L.; Correia, A.; Correia, S.; Llorens, F.; Pique, A.V.; Möbius, W.; Domingues, R.; Zafar, S.; et al. Cellular Prion Protein Mediates α-Synuclein Uptake, Localization, and Toxicity In Vitro and In Vivo. Mov. Disord. 2022, 37, 39–51. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-Related Macular Degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef]
- Schultz, N.M.; Bhardwaj, S.; Barclay, C.; Gaspar, L.; Schwartz, J. Global Burden of Dry Age-Related Macular Degeneration: A Targeted Literature Review. Clin. Ther. 2021, 43, 1792–1818. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-Related Macular Degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Wei, Y.; Liao, H.; Ye, J. Therapeutic Effects of Various Therapeutic Strategies on Non-Exudative Age-Related Macular Degeneration. Medicine 2018, 97, e10422. [Google Scholar] [CrossRef]
- Lambert, N.G.; ElShelmani, H.; Singh, M.K.; Mansergh, F.C.; Wride, M.A.; Padilla, M.; Keegan, D.; Hogg, R.E.; Ambati, B.K. Risk Factors and Biomarkers of Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2016, 54, 64–102. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, J.; Galante, K. Iveric Bio Announces Positive Topline Data from Zimura® GATHER2 Phase 3 Clinical Trial in Geographic Atrophy. Available online: https://www.businesswire.com/news/home/20220905005451/en/ (accessed on 14 February 2023).
- Pavluk, L.; Kaya, M. FDA Approves SYFOVRETM (Pegcetacoplan Injection) as the First and Only Treatment for Geographic Atrophy (GA), a Leading Cause of Blindness. Available online: https://investors.apellis.com/news-releases/news-release-details/fda-approves-syfovretm-pegcetacoplan-injection-first-and-only (accessed on 17 March 2023).
- Kansteiner, F. Apellis Wins FDA Approval for First Geographic Atrophy Drug. Available online: https://www.fiercepharma.com/pharma/apellis-wins-fda-approval-first-geographic-atrophy-drug (accessed on 17 March 2023).
- Hellewell, S.B.; Bowen, W.D. A Sigma-like Binding Site in Rat Pheochromocytoma (PC12) Cells: Decreased Affinity for (+)-Benzomorphans and Lower Molecular Weight Suggest a Different Sigma Receptor Form from That of Guinea Pig Brain. Brain Res. 1990, 527, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Hellewell, S.B.; Bruce, A.; Feinstein, G.; Orringer, J.; Williams, W.; Bowen, W.D. Rat Liver and Kidney Contain High Densities of Sigma 1 and Sigma 2 Receptors: Characterization by Ligand Binding and Photoaffinity Labeling. Eur. J. Pharmacol. 1994, 268, 9–18. [Google Scholar] [CrossRef]
- Zeng, C.; Garg, N.; Mach, R.H. The PGRMC1 Protein Level Correlates with the Binding Activity of a Sigma-2 Fluorescent Probe (SW120) in Rat Brain Cells. Mol. Imaging Biol. 2016, 18, 172–179. [Google Scholar] [CrossRef]
- Nicholson, H.; Mesangeau, C.; McCurdy, C.R.; Bowen, W.D. Sigma-2 Receptors Play a Role in Cellular Metabolism: Stimulation of Glycolytic Hallmarks by CM764 in Human SK-N-SH Neuroblastomas. J. Pharmacol. Exp. Ther. 2016, 356, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Niso, M.; Infantino, V.; Menga, A.; Berardi, F. Elements in Support of the ‘ Non-Identity ’ of the PGRMC1 Protein with the σ 2 Receptor. Eur. J. Pharmacol. 2015, 758, 16–23. [Google Scholar] [CrossRef]
- Chu, U.B.; Mavlyutov, T.A.; Chu, M.; Yang, H.; Schulman, A.; Mesangeau, C.; McCurdy, C.R.; Guo, L.; Ruoho, A.E. The Sigma-2 Receptor and Progesterone Receptor Membrane Component 1 Are Different Binding Sites Derived From Independent Genes. EBioMedicine 2015, 2, 1806–1813. [Google Scholar] [CrossRef]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The Effects of Morphine- and Nalorphine-like Drugs in the Nondependent and Morphine-Dependent Chronic Spinal Dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, Molecular Cloning, and Expression of the Mammalian Sigma1-Binding Site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef]
- Xu, J.; Zeng, C.; Chu, W.; Pan, F.; Rothfuss, J.M.; Zhang, F.; Tu, Z.; Zhou, D.; Zeng, D.; Vangveravong, S.; et al. Identification of the PGRMC1 Protein Complex as the Putative Sigma-2 Receptor Binding Site. Nat. Commun. 2011, 2, 380. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pulido, L.; Ponting, C.P. TM6SF2 and MAC30, New Enzyme Homologs in Sterol Metabolism and Common Metabolic Disease. Front. Genet. 2014, 5, 439. [Google Scholar] [CrossRef] [PubMed]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the Gene That Codes for the σ 2 Receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 7160–7165. [Google Scholar] [CrossRef]
- Riad, A.; Lengyel-Zhand, Z.; Zeng, C.; Weng, C.-C.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mach, R.H. The Sigma-2 Receptor/TMEM97, PGRMC1, and LDL Receptor Complex Are Responsible for the Cellular Uptake of Aβ42 and Its Protein Aggregates. Mol. Neurobiol. 2020, 57, 3803–3813. [Google Scholar] [CrossRef]
- Alon, A.; Lyu, J.; Braz, J.M.; Tummino, T.A.; Craik, V.; O’Meara, M.J.; Webb, C.M.; Radchenko, D.S.; Moroz, Y.S.; Huang, X.; et al. Structures of the Σ2 Receptor Enable Docking for Bioactive Ligand Discovery. Nature 2021, 600, 759–764. [Google Scholar] [CrossRef]
- Izzo, N.J.; Yuede, C.M.; LaBarbera, K.M.; Limegrover, C.S.; Rehak, C.; Yurko, R.; Waybright, L.; Look, G.; Rishton, G.; Safferstein, H.; et al. Preclinical and Clinical Biomarker Studies of CT1812: A Novel Approach to Alzheimer’s Disease Modification. Alzheimer’s Dement. 2021, 17, 1365–1382. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Tulloch, J.; Jackson, R.J.; Catterson, J.H.; Rose, J.; Davies, C.; Hooley, M.; Anton-Fernandez, A.; Dunnett, S.; Tempelaar, R.; et al. TMEM97 Increases in Synapses and Is a Potential Synaptic Aβ Binding Partner in Human Alzheimer’s Disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Riad, A.; Zeng, C.; Weng, C.-C.C.; Winters, H.; Xu, K.; Makvandi, M.; Metz, T.; Carlin, S.; Mach, R.H. Sigma-2 Receptor/TMEM97 and PGRMC-1 Increase the Rate of Internalization of LDL by LDL Receptor through the Formation of a Ternary Complex. Sci. Rep. 2018, 8, 16845. [Google Scholar] [CrossRef] [PubMed]
- Cantonero, C.; Camello, P.J.; Abate, C.; Berardi, F.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. NO1, a New Sigma 2 Receptor/TMEM97 Fluorescent Ligand, Downregulates SOCE and Promotes Apoptosis in the Triple Negative Breast Cancer Cell Lines. Cancers 2020, 12, 257. [Google Scholar] [CrossRef] [PubMed]
- Langa, F.; Codony, X.; Tovar, V.; Lavado, A.; Gimenez, E.; Cozar, P.; Cantero, M.; Dordal, A.; Hernandez, E.; Perez, R.; et al. Generation and Phenotypic Analysis of Sigma Receptor Type I (Sigma1) Knockout Mice. Eur. J. Neurosci. 2003, 18, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.; Hurtado, M.L.; Jackson, R.J.; Eaton, S.L.; Herrmann, A.G.; Colom-Cadena, M.; Tzioras, M.; King, D.; Rose, J.; Tulloch, J.; et al. Comparative Profiling of the Synaptic Proteome from Alzheimer’s Disease Patients with Focus on the APOE Genotype. Acta Neuropathol. Commun. 2019, 7, 214. [Google Scholar] [CrossRef]
- Wang, J.-H.; Urrutia-Cabrera, D.; Mora, S.M.; Nguyen, T.; Hung, S.; Hewitt, A.W.; Edwards, T.L.; Wong, R.C.B. Functional Study of the AMD-Associated Gene TMEM97 in Retinal Pigmented Epithelium Using CRISPR Interference. bioRxiv 2020, 2020-07. [Google Scholar] [CrossRef]
- Shanmugam, A.K.; Mysona, B.A.; Wang, J.; Zhao, J.; Tawfik, A.; Sanders, A.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Bollinger, K.E.; et al. Progesterone Receptor Membrane Component 1 (PGRMC1) Expression in Murine Retina. Curr. Eye Res. 2016, 41, 1105–1112. [Google Scholar] [CrossRef]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal Transcriptome and EQTL Analyses Identify Genes Associated with Age-Related Macular Degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef]
- Wang, H.; Peng, Z.; Li, Y.; Sahn, J.J.; Hodges, T.R.; Chou, T.H.; Liu, Q.; Zhou, X.; Jiao, S.; Porciatti, V.; et al. Σ2R/TMEM97 in Retinal Ganglion Cell Degeneration. Sci. Rep. 2022, 12, 20753. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, J.Y.; Hsieh, C.-J.; Riad, A.; Izzo, N.J.; Catalano, S.M.; Graham, T.J.A.; Mach, R.H. Screening of σ 2 Receptor Ligands and In Vivo Evaluation of 11C-Labeled 6,7-Dimethoxy-2-[4-(4-Methoxyphenyl)Butan-2-Yl]-1,2,3,4-Tetrahydroisoquinoline for Potential Use as a σ2 Receptor Brain PET Tracer. J. Med. Chem. 2022, 65, 6261–6272. [Google Scholar] [CrossRef]
- Ahmed, I.S.A.; Chamberlain, C.; Craven, R.J. S2R(Pgrmc1): The Cytochrome-Related Sigma-2 Receptor That Regulates Lipid and Drug Metabolism and Hormone Signaling. Expert Opin. Drug Metab. Toxicol. 2012, 8, 361–370. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwon, S.W.; Kaya, P.; Lee, Y.H.; Lee, J.G.; Kim, G.; Lee, G.-S.; Baek, I.-J.; Hong, E.-J. Loss of Progesterone Receptor Membrane Component 1 Promotes Hepatic Steatosis via the Induced de Novo Lipogenesis. Sci. Rep. 2018, 8, 15711. [Google Scholar] [CrossRef]
- Cai, H.L.; Tan, Q.Y.; Jiang, P.; Dang, R.L.; Xue, Y.; Tang, M.M.; Xu, P.; Deng, Y.; Li, H.D.; Yao, J.K. A Potential Mechanism Underlying Atypical Antipsychotics-Induced Lipid Disturbances. Transl. Psychiatry 2015, 5, e661. [Google Scholar] [CrossRef]
- Hughes, A.L.; Powell, D.W.; Bard, M.; Eckstein, J.; Barbuch, R.; Link, A.J.; Espenshade, P.J. Dap1/PGRMC1 Binds and Regulates Cytochrome P450 Enzymes. Cell Metab. 2007, 5, 143–149. [Google Scholar] [CrossRef]
- Lafyatis, R.; Mantero, J.C.; Gordon, J.; Kishore, N.; Carns, M.; Dittrich, H.; Spiera, R.; Simms, R.W.; Varga, J. Inhibition of β-Catenin Signaling in the Skin Rescues Cutaneous Adipogenesis in Systemic Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Trial of C-82. J. Investig. Dermatol. 2017, 137, 2473–2483. [Google Scholar] [CrossRef]
- Son, K.; Lee, H.; Shah, D.; Kalmodia, S.; Miller, R.C.; Ali, M.; Balasubramaniam, A.; Cologna, S.M.; Kong, H.; Shukla, D.; et al. Histatin-1 Is an Endogenous Ligand of the Sigma-2 Receptor. FEBS J. 2021, 288, 6815–6827. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, T.; Ma, X.; Pratuangtham, S.; Zhang, G.C.; Ondrus, A.A.; Mafi, A.; Lomenick, B.; Jones, J.J.; Ondrus, A.E. A Proteome-Wide Map of 20(S)-Hydroxycholesterol Interactors in Cell Membranes. Nat. Chem. Biol. 2021, 17, 1271–1280. [Google Scholar] [CrossRef]
- Mach, R.H.; Wheeler, K.T. Development of Molecular Probes for Imaging Sigma-2 Receptors in Vitro and in Vivo. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 230–245. [Google Scholar] [CrossRef]
- Turgutalp, B.; Bhattarai, P.; Ercetin, T.; Luise, C.; Reis, R.; Gurdal, E.E.; Isaak, A.; Biriken, D.; Dinter, E.; Sipahi, H.; et al. Discovery of Potent Cholinesterase Inhibition-Based Multi-Target- Directed Lead Compounds for Synaptoprotection in Alzheimer’s Disease. J. Med. Chem. 2022, 65, 12292–12318. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Li, J.; Liu, X.; Shen, H.; Yang, H.; McCurdy, C.R.; Pattnaik, B.; Guo, L.W. Retinal Photoreceptor Protection in an AMD-Related Mouse Model by Selective Sigma-1 or Sigma-2 Receptor Modulation. Genes 2022, 13, 2386. [Google Scholar] [CrossRef]
- Vilner, B.J.; Bowen, W.D. Modulation of Cellular Calcium by Sigma-2 Receptors: Release from Intracellular Stores in Human SK-N-SH Neuroblastoma Cells. J. Pharmacol. Exp. Ther. 2000, 292, 900–911. [Google Scholar]
- Cassano, G.; Gasparre, G.; Contino, M.; Niso, M.; Berardi, F.; Perrone, R.; Colabufo, N.A. The Sigma-2 Receptor Agonist PB28 Inhibits Calcium Release from the Endoplasmic Reticulum of SK-N-SH Neuroblastoma Cells. Cell Calcium 2006, 40, 23–28. [Google Scholar] [CrossRef]
- Guo, L.; Zhen, X. Sigma-2 Receptor Ligands: Neurobiological Effects. Curr. Med. Chem. 2015, 22, 989–1003. [Google Scholar] [CrossRef]
- Mach, R.H.; Zeng, C.; Hawkins, W.G. The σ2 Receptor: A Novel Protein for the Imaging and Treatment of Cancer. J. Med. Chem. 2013, 56, 7137–7160. [Google Scholar] [CrossRef]
- Zeng, C.; Weng, C.-C.C.; Schneider, M.E.; Puentes, L.; Riad, A.; Xu, K.; Makvandi, M.; Jin, L.; Hawkins, W.G.; Mach, R.H. TMEM97 and PGRMC1 Do Not Mediate Sigma-2 Ligand-Induced Cell Death. Cell Death Discov. 2019, 5, 58. [Google Scholar] [CrossRef]
- Pati, M.L.; Hornick, J.R.; Niso, M.; Berardi, F.; Spitzer, D.; Abate, C.; Hawkins, W. Sigma-2 Receptor Agonist Derivatives of 1-Cyclohexyl-4-[3-(5-Methoxy-1,2,3,4-Tetrahydronaphthalen-1-Yl)Propyl]Piperazine (PB28) Induce Cell Death via Mitochondrial Superoxide Production and Caspase Activation in Pancreatic Cancer. BMC Cancer 2017, 17, 51. [Google Scholar] [CrossRef]
- Quirion, R.; Bowen, W.D.; Itzhak, Y.; Junien, J.L.; Musacchio, J.M.; Rothman, R.B.; Su, T.P.; Tam, S.W.; Taylor, D.P. A Proposal for the Classification of Sigma Binding Sites. Trends Pharmacol. Sci. 1992, 13, 85–86. [Google Scholar] [CrossRef]
- Vilner, B.J.; John, C.S.; Bowen, W.D. Sigma-1 and Sigma-2 Receptors Are Expressed in a Wide Variety of Human and Rodent Tumor Cell Lines. Cancer Res. 1995, 55, 408–413. [Google Scholar]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of Cholesterol-Regulating Genes by Targeted RNAi Screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Wahlster, L.; Bartz, F.; Werenbeck-Ueding, J.; Praggastis, M.; Zhang, J.; Joggerst-Thomalla, B.; Theiss, S.; Grimm, D.; Ory, D.S.; et al. Reduction of TMEM97 Increases NPC1 Protein Levels and Restores Cholesterol Trafficking in Niemann-Pick Type C1 Disease Cells. Hum. Mol. Genet. 2015, 25, 3588–3599. [Google Scholar] [CrossRef]
- Intlekofer, K.A.; Petersen, S.L. Distribution of MRNAs Encoding Classical Progestin Receptor, Progesterone Membrane Components 1 and 2, Serpine MRNA Binding Protein 1, and Progestin and ADIPOQ Receptor Family Members 7 and 8 in Rat Forebrain. Neuroscience 2011, 172, 55–65. [Google Scholar] [CrossRef]
- Catalano, S.M.; Mozzoni, K.; Rehak, C.; Waybright, L.; Sadlek, K.; Safferstein, H.; Watto, E.; Izzo, N.J.; Grundman, M.; Dekosky, S.; et al. CT1812 Demonstrates Evidence of Synapse Preservation in Alzheimer’s Disease Patients and Abeta Oligomer Displacement in Preclinical Models; Presentation 200.15/B19; Society for Neuroscience: Chicago, IL, USA, 2019. [Google Scholar]
- Ahmed, I.S.; Rohe, H.J.; Twist, K.E.; Mattingly, M.N.; Craven, R.J. Progesterone Receptor Membrane Component 1 (Pgrmc1): A Heme-1 Domain Protein That Promotes Tumorigenesis and Is Inhibited by a Small Molecule. J. Pharmacol. Exp. Ther. 2010, 333, 564–573. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Dong, J. Enhancement of Cell Surface Expression and Receptor Functions of Membrane Progestin Receptor α (MPRα) by Progesterone Receptor Membrane Component 1 (PGRMC1): Evidence for a Role of PGRMC1 as an Adaptor Protein for Steroid Receptors. Endocrinology 2014, 155, 1107–1119. [Google Scholar] [CrossRef]
- Zhang, M.; Robitaille, M.; Showalter, A.D.; Huang, X.; Liu, Y.; Bhattacharjee, A.; Willard, F.S.; Han, J.; Froese, S.; Wei, L.; et al. Progesterone Receptor Membrane Component 1 Is a Functional Part of the GLP-1 Receptor Complex in Pancreatic Beta Cells. Mol. Cell. Proteom. 2014, 1, 3049–3062. [Google Scholar] [CrossRef]
- Su, C.; Cunningham, R.L.; Rybalchenko, N.; Singh, M. Progesterone Increases the Release of Brain-Derived Neurotrophic Factor from Glia via Progesterone Receptor Membrane Component 1 (Pgrmc1)-Dependent ERK5 Signaling. Endocrinology 2012, 153, 4389–4400. [Google Scholar] [CrossRef]
- Mir, S.U.R.; Schwarze, S.R.; Jin, L.; Zhang, J.; Friend, W.; Miriyala, S.; Clair, D.S.; Craven, R.J.; St Clair, D.; Craven, R.J. Progesterone Receptor Membrane Component 1/ Sigma-2 Receptor Associates with MAP1LC3B and Promotes Autophagy. Autophagy 2013, 9, 1566–1578. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network Organization of the Human Autophagy System. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef]
- Rohe, H.J.; Ahmed, I.S.; Twist, K.E.; Craven, R.J. PGRMC1 (Progesterone Receptor Membrane Component 1): A Targetable Protein with Multiple Functions in Steroid Signaling, P450 Activation and Drug Binding. Pharmacol. Ther. 2009, 121, 14–19. [Google Scholar] [CrossRef]
- Peluso, J.J.; Yuan, A.; Liu, X.; Lodde, V. Plasminogen Activator Inhibitor 1 RNA-Binding Protein Interacts with Progesterone Receptor Membrane Component 1 to Regulate Progesterone’s Ability to Maintain the Viability of Spontaneously Immortalized Granulosa Cells and Rat Granulosa Cells. Biol. Reprod. 2013, 88, 20. [Google Scholar] [CrossRef][Green Version]
- Runko, E.; Kaprielian, Z. Expression of Vema in the Developing Mouse Spinal Cord and Optic Chiasm. J. Comp. Neurol. 2002, 451, 289–299. [Google Scholar] [CrossRef]
- Runko, E.; Kaprielian, Z. Caenorhabditis Elegans VEM-1, a Novel Membrane Protein, Regulates the Guidance of Ventral Nerve Cord-Associated Axons. J. Neurosci. 2004, 24, 9015–9026. [Google Scholar] [CrossRef]
- Contreras, P.C.; Dimaggio, D.A.; O’Donohue, T.L. An Endogenous Ligand for the Sigma Opioid Binding Site. Synapse 1987, 1, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Connor, M.A.; Chavkin, C. Ionic Zinc May Function as an Endogenous Ligand for the Haloperidol-Sensitive Sigma 2 Receptor in Rat Brain. Mol. Pharmacol. 1992, 42, 471–479. [Google Scholar] [PubMed]
- Oudhoff, M.J.; Bolscher, J.G.M.; Nazmi, K.; Kalay, H.; Hof, W.; Amerongen, A.V.N.; Veerman, E.C.I. Histatins Are the Major Wound-closure Stimulating Factors in Human Saliva as Identified in a Cell Culture Assay. FASEB J. 2008, 22, 3805–3812. [Google Scholar] [CrossRef]
- Torres, P.; Hernández, N.; Mateluna, C.; Silva, P.; Reyes, M.; Solano, L.; Venegas, S.; Criollo, A.; Nazmi, K.; Bikker, F.J.; et al. Histatin-1 Is a Novel Osteogenic Factor That Promotes Bone Cell Adhesion, Migration, and Differentiation. J. Tissue Eng. Regen. Med. 2021, 15, 336–346. [Google Scholar] [CrossRef]
- Kalmodia, S.; Son, K.N.; Cao, D.; Lee, B.S.; Surenkhuu, B.; Shah, D.; Ali, M.; Balasubramaniam, A.; Jain, S.; Aakalu, V.K. Presence of Histatin-1 in Human Tears and Association with Aqueous Deficient Dry Eye Diagnosis: A Preliminary Study. Sci. Rep. 2019, 9, 10304. [Google Scholar] [CrossRef]
- Ridgway, N.D. 25-Hydroxycholesterol Stimulates Sphingomyelin Synthesis in Chinese Hamster Ovary Cells. J. Lipid Res. 1995, 36, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Hsieh, C.-J.; Lee, J.Y.; Riad, A.; Izzo, N.J.; Look, G.; Catalano, S.; Mach, R.H. Exploration of Diazaspiro Cores as Piperazine Bioisosteres in the Development of σ2 Receptor Ligands. Int. J. Mol. Sci. 2022, 23, 8259. [Google Scholar] [CrossRef]
- Xu, X.; Ruan, X.; Ju, R.; Wang, Z.; Yang, Y.; Cheng, J.; Gu, M.; Mueck, A.O. Progesterone Receptor Membrane Component-1 May Promote Survival of Human Brain Microvascular Endothelial Cells in Alzheimer’s Disease. Am. J. Alzheimers. Dis. Other Demen. 2022, 37. [Google Scholar] [CrossRef] [PubMed]
- Kline, R.A.; Kaifer, K.A.; Osman, E.Y.; Carella, F.; Tiberi, A.; Ross, J.; Pennetta, G.; Lorson, C.L.; Murray, L.M. Comparison of Independent Screens on Differentially Vulnerable Motor Neurons Reveals Alpha-Synuclein as a Common Modifier in Motor Neuron Diseases. PLoS Genet. 2017, 13, e1006680. [Google Scholar] [CrossRef]
- Wyse Jackson, A.C.; Roche, S.L.; Byrne, A.M.; Ruiz-Lopez, A.M.; Cotter, T.G. Progesterone Receptor Signalling in Retinal Photoreceptor Neuroprotection. J. Neurochem. 2016, 136, 63–77. [Google Scholar] [CrossRef]
- Cahill, M.A.; Jazayeri, J.A.; Kovacevic, Z.; Richardson, D.R. PGRMC1 Regulation by Phosphorylation: Potential New Insights in Controlling Biological Activity! Oncotarget 2016, 67, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Rothfuss, J.; Zhang, J.; Chu, W.; Vangveravong, S.; Tu, Z.; Pan, F.; Chang, K.C.; Hotchkiss, R.; Mach, R.H. Sigma-2 Ligands Induce Tumour Cell Death by Multiple Signalling Pathways. Br. J. Cancer 2012, 106, 693–701. [Google Scholar] [CrossRef]
- Shen, H.; Li, J.; Heisler-Taylor, T.; Makin, R.; Yang, H.; Mavlyutov, T.A.; Gelfand, B.; Cebulla, C.M.; Guo, L.W. TMEM97 Ablation Aggravates Oxidant-Induced Retinal Degeneration. Cell Signal. 2021, 86, 110078. [Google Scholar] [CrossRef]
- Ostenfeld, M.S.; Høyer-Hansen, M.; Bastholm, L.; Fehrenbacher, N.; Olsen, O.D.; Groth-Pedersen, L.; Puustinen, P.; Kirkegaard-Sørensen, T.; Nylandsted, J.; Farkas, T.; et al. Anti-Cancer Agent Siramesine Is a Lysosomotropic Detergent That Induces Cytoprotective Autophagosome Accumulation. Autophagy 2008, 4, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.R.; Regen, S.L. The Structural Role of Cholesterol in Cell Membranes: From Condensed Bilayers to Lipid Rafts. Acc. Chem. Res. 2014, 47, 3512–3521. [Google Scholar] [CrossRef] [PubMed]
- Gylys, K.H.; Fein, J.A.; Tan, A.M.; Cole, G.M. Apolipoprotein E Enhances Uptake of Soluble but Not Aggregated Amyloid-β Protein into Synaptic Terminals. J. Neurochem. 2003, 84, 1442–1451. [Google Scholar] [CrossRef]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of ApoE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 10, 14. [Google Scholar] [CrossRef]
- Wisniewski, T.; Drummond, E. APOE-Amyloid Interaction: Therapeutic Targets. Neurobiol. Dis. 2020, 138, 104784. [Google Scholar] [CrossRef]
- Smith, L.M.; Strittmatter, S.M. Binding Sites for Amyloid-β Oligomers and Synaptic Toxicity. Cold Spring Harb. Perspect. Med. 2017, 7, a024075. [Google Scholar] [CrossRef]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic Glutamate Receptor 5 Is a Coreceptor for Alzheimer Aβ Oligomer Bound to Cellular Prion Protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef]
- Spurrier, J.; Nicholson, L.; Fang, X.T.; Stoner, A.J.; Toyonaga, T.; Holden, D.; Sieger, T.R.; Laird, W.; Allnutt, M.A.; Chiasseu, M.; et al. Reversal of Synapse Loss in Alzheimer Mouse Models by Targeting MGluR5 to Prevent Synaptic Tagging by C1Q. Sci. Transl. Med. 2022, 14, eabi8593. [Google Scholar] [CrossRef]
- Salazar, S.V.; Gallardo, C.; Kaufman, A.C.; Herber, C.S.; Haas, L.T.; Robinson, S.; Manson, J.C.; Lee, M.K.; Strittmatter, S.M. Conditional Deletion of Prnp Rescues Behavioral and Synaptic Deficits after Disease Onset in Transgenic Alzheimer’s Disease. J. Neurosci. 2017, 37, 9207–9221. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally Secreted Oligomers of Amyloid β Protein Potently Inhibit Hippocampal Long-Term Potentiation in Vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular Prion Protein Mediates Impairment of Synaptic Plasticity by Amyloid-β Oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Chung, E.; Ji, Y.; Sun, Y.; Kascsak, R.J.; Kascsak, R.B.; Mehta, P.D.; Strittmatter, S.M.; Wisniewski, T. Anti-PrPC Monoclonal Antibody Infusion as a Novel Treatment for Cognitive Deficits in an Alzheimer’s Disease Model Mouse. BMC Neurosci. 2010, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic Degeneration in Alzheimer Disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar] [CrossRef]
- Carson, R.E.; Naganawa, M.; Toyonaga, T.; Koohsari, S.; Yang, Y.; Chen, M.K.; Matuskey, D.; Finnema, S.J. Imaging of Synaptic Density in Neurodegenerative Disorders. J. Nucl. Med. 2022, 63, 60S–67S. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, S.D.; Ruthazer, E.S.; Kennedy, T.E. Guiding Synaptic Plasticity: Novel Roles for Netrin-1 in Synaptic Plasticity and Memory Formation in the Adult Brain. J. Physiol. 2020, 599, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.W.; Glasgow, S.D.; Trigiani, L.J.; Chitsaz, D.; Rymar, V.; Sadikot, A.; Ruthazer, E.S.; Hamel, E.; Kennedy, T.E. Spatial Memory Formation Requires Netrin-1 Expression by Neurons in the Adult Mammalian Brain. Learn. Mem. 2019, 26, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.A. Progesterone Receptor Membrane Component 1: An Integrative Review. J. Steroid Biochem. Mol. Biol. 2007, 105, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Huganir, R.L.; Nicoll, R.A. AMPARs and Synaptic Plasticity: The Last 25 Years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Nakane, T.; Koike, I.; Yamamoto, T.; Sugiura, Y.; Harada, E.; Sugase, K.; Shimamura, T.; Ohmura, M.; Muraoka, K.; et al. Haem-Dependent Dimerization of PGRMC1/Sigma-2 Receptor Facilitates Cancer Proliferation and Chemoresistance. Nat. Commun. 2016, 7, 11030. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Yamamoto, T.; Kajimura, M.; Sugiura, Y.; Koike, I.; Ohmura, M.; Nakamura, T.; Tokumoto, Y.; Tsugawa, H.; Handa, H.; et al. Cystathionine Beta-Synthase and PGRMC1 as CO Sensors. Free Radic. Biol. Med. 2016, 99, 333–344. [Google Scholar] [CrossRef]
- Zhuo, M.; Small, S.A.; Kandel, E.R.; Hawkins, R.D. Nitric Oxide and Carbon Monoxide Produce Activity-Dependent Long-Term Synaptic Enhancement in Hippocampus. Science 1993, 260, 1946–1950. [Google Scholar] [CrossRef]
- Yi, B.; Sahn, J.J.; Ardestani, P.M.; Evans, A.K.; Scott, L.; Chan, J.Z.; Iyer, S.; Crisp, A.; Zuniga, G.; Pierce-Shimomura, J.; et al. Small Molecule Modulator of Sigma 2 Receptor Is Neuroprotective and Reduces Cognitive Deficits and Neuro-Inflammation in Experimental Models of Alzheimer’s Disease. J. Neurochem. 2017, 140, 561–575. [Google Scholar] [CrossRef]
- Mondal, S.; Hegarty, E.; Sahn, J.J.; Scott, L.L.; Gökçe, S.K.; Martin, C.; Ghorashian, N.; Satarasinghe, P.N.; Iyer, S.; Sae-Lee, W.; et al. High-Content Microfluidic Screening Platform Used To Identify σ2R/Tmem97 Binding Ligands That Reduce Age-Dependent Neurodegeneration in C. Elegans SC_APP Model. ACS Chem. Neurosci. 2018, 9, 1014–1026. [Google Scholar] [CrossRef]
- Terada, K.; Migita, K.; Matsushima, Y.; Sugimoto, Y.; Kamei, C.; Matsumoto, T.; Mori, M.; Matsunaga, K.; Takata, J.; Karube, Y. Cholinesterase Inhibitor Rivastigmine Enhances Nerve Growth Factor-Induced Neurite Outgrowth in PC12 Cells via Sigma-1 and Sigma-2 Receptors. PLoS ONE 2018, 13, e0209250. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic Targeting by Alzheimer’s-Related Amyloid β Oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Selkoe, D.J. Preventing Alzheimer’s Disease. Science 2012, 337, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Kim, D.-H. Alzheimer’s Disease Genes and Autophagy. Brain Res. 2016, 1649, 201–209. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Haung Yu, W.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A Novel β-Amyloid Peptide-Generating Pathway Activated in Alzheimer’s Disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The Autophagy-Related Protein Beclin 1 Shows Reduced Expression in Early Alzheimer Disease and Regulates Amyloid β Accumulation in Mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty Autolysosome Acidification in Alzheimer’s Disease Mouse Models Induces Autophagic Build-up of Aβ in Neurons, Yielding Senile Plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef]
- Koffie, R.M.; Hashimoto, T.; Tai, H.-C.; Kay, K.R.; Serrano-Pozo, A.; Joyner, D.; Hou, S.; Kopeikina, K.J.; Frosch, M.P.; Lee, V.M.; et al. Apolipoprotein E4 Effects in Alzheimer’s Disease Are Mediated by Synaptotoxic Oligomeric Amyloid-β. Brain 2012, 135, 2155–2168. [Google Scholar] [CrossRef]
- Lue, L.-F.; Kuo, Y.-M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble Amyloid β Peptide Concentration as a Predictor of Synaptic Change in Alzheimer’s Disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Koffie, R.M.; Wegmann, S.; Henstridge, C.M.; Herrmann, A.G.; Colom-Cadena, M.; Lleo, A.; Kay, K.R.; Vaught, M.; Soberman, R.; et al. Non-Fibrillar Oligomeric Amyloid-β within Synapses. J. Alzheimer’s Dis. 2016, 53, 787–800. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-Beta Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Limegrover, C.S.; LeVine, H.; Izzo, N.J.; Yurko, R.; Mozzoni, K.; Rehak, C.; Sadlek, K.; Safferstein, H.; Catalano, S.M. Alzheimer’s Protection Effect of A673T Mutation May Be Driven by Lower Aβ Oligomer Binding Affinity. J. Neurochem. 2020, 157, 1316–1330. [Google Scholar] [CrossRef] [PubMed]
- Grundman, M.; Morgan, R.; Lickliter, J.D.; Schneider, L.S.; DeKosky, S.; Izzo, N.J.; Guttendorf, R.; Higgin, M.; Pribyl, J.; Mozzoni, K.; et al. A Phase 1 Clinical Trial of the Sigma-2 Receptor Complex Allosteric Antagonist CT1812, a Novel Therapeutic Candidate for Alzheimer’s Disease. Alzheimer’s Dement. 2019, 5, 20–26. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.L.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy Modulates SNCA/α-Synuclein Release, Thereby Generating a Hostile Microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Gao, P.; Arzberger, T.; Höllerhage, M.; Herms, J.; Höglinger, G.; Koeglsperger, T. Alpha-Synuclein Defects Autophagy by Impairing SNAP29-Mediated Autophagosome-Lysosome Fusion. Cell Death Dis. 2021, 12, 854. [Google Scholar] [CrossRef]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014.e8. [Google Scholar] [CrossRef]
- Colebc, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid Droplet Binding and Oligomerization Properties of the Parkinson’s Disease Protein α-Synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant Alpha-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-Produced α-Synuclein Oligomers Are Targeted to, and Impair, the 26S Proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef]
- Petrou, M.; Dwamena, B.A.; Foerster, B.R.; Maceachern, M.P.; Bohnen, N.I.; Müller, M.L.; Albin, R.L.; Frey, K.A. Amyloid Deposition in Parkinson’s Disease and Cognitive Impairment: A Systematic Review. Mov. Disord. 2015, 30, 928–935. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A Large Genome-Wide Association Study of Age-Related Macular Degeneration Highlights Contributions of Rare and Common Variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef]
- Yan, Q.; Ding, Y.; Liu, Y.; Sun, T.; Fritsche, L.G.; Clemons, T.; Ratnapriya, R.; Klein, M.L.; Cook, R.J.; Liu, Y.; et al. Genome-Wide Analysis of Disease Progression in Age-Related Macular Degeneration. Hum. Mol. Genet. 2018, 27, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Hamby, M.E. Targeting the Sigma-2 Receptor for Dry Age-Related Macular Degeneration (AMD). Available online: https://cogrx.com/news/events/fifth-iss2r/ (accessed on 4 December 2021).
- Mengel, E.; Klünemann, H.H.; Lourenço, C.M.; Hendriksz, C.J.; Sedel, F.; Walterfang, M.; Kolb, S.A. Niemann-Pick Disease Type C Symptomatology: An Expert-Based Clinical Description. Orphanet J. Rare Dis. 2013, 8, 166. [Google Scholar] [CrossRef]
- Stampfer, M.; Theiss, S.; Amraoui, Y.; Jiang, X.; Keller, S.; Ory, D.S.; Mengel, E.; Fischer, C.; Runz, H. Niemann-Pick Disease Type C Clinical Database: Cognitive and Coordination Deficits Are Early Disease Indicators. Orphanet J. Rare Dis. 2013, 8, 35. [Google Scholar] [CrossRef]
- Patterson, M.C.; Mengel, E.; Wijburg, F.A.; Muller, A.; Schwierin, B.; Drevon, H.; Vanier, M.T.; Pineda, M. Disease and Patient Characteristics in NP-C Patients: Findings from an International Disease Registry. Orphanet J. Rare Dis. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Havla, J.; Moser, M.; Sztatecsny, C.; Lotz-Havla, A.S.; Maier, E.M.; Hizli, B.; Schinner, R.; Kümpfel, T.; Strupp, M.; Bremova-Ertl, T.; et al. Retinal Axonal Degeneration in Niemann–Pick Type C Disease. J. Neurol. 2020, 267, 2070–2082. [Google Scholar] [CrossRef]
- Keefe, R.S.; Harvey, P.; Khan, A.; Saoud, J.B.; Staner, C.; Davidson, M.; Luthringer, R. Cognitive Effects of MIN-101 in Patients With Schizophrenia and Negative Symptoms. J. Clin. Psychiatry 2018, 79, 17m11753. [Google Scholar] [CrossRef] [PubMed]
- Schilling, G.; Klevytska, A.; Tebbenkamp, A.T.N.; Juenemann, K.; Cooper, J.; Gonzales, V.; Slunt, H.; Poirer, M.; Ross, C.A.; Borchelt, D.R. Characterization of Huntingtin Pathologic Fragments in Human Huntington Disease, Transgenic Mice, and Cell Models. J. Neuropathol. Exp. Neurol. 2007, 66, 313–320. [Google Scholar] [CrossRef]
- Mende-Mueller, L.M.; Toneff, T.; Hwang, S.R.; Chesselet, M.F.; Hook, V.Y.H. Tissue-Specific Proteolysis of Huntingtin (Htt) in Human Brain: Evidence of Enhanced Levels of N- and C-Terminal Htt Fragments in Huntington’s Disease Striatum. J. Neurosci. 2001, 21, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, K.A.; Mann, D.M.A. The Selective Vulnerability of Nerve Cells in Huntington’s Disease. Neuropathol. Appl. Neurobiol. 2001, 27, 1–21. [Google Scholar] [CrossRef]
- Blass, B.E.; Rogers, J.P. The Sigma-2 (σ-2) Receptor: A Review of Recent Patent Applications: 2013–2018. Expert Opin. Ther. Pat. 2018, 28, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Brimson, J.M.; Brimson, S.; Chomchoei, C.; Tencomnao, T. Using Sigma-Ligands as Part of a Multi-Receptor Approach to Target Diseases of the Brain. Expert Opin. Ther. Targets 2020, 24, 1009–1028. [Google Scholar] [CrossRef]
- Kargbo, R.B. Sigma-1 and Sigma-2 Receptor Modulators as Potential Therapeutics for Alzheimer’s Disease. ACS Med. Chem. Lett. 2021, 12, 178–179. [Google Scholar] [CrossRef]
- Cahill, M.A. Quo Vadis PGRMC? Grand-Scale Biology in Human Health and Disease. Front. Biosci.-Landmark 2022, 27, 318. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The Clinical Promise of Biomarkers of Synapse Damage or Loss in Alzheimer’s Disease. Alzheimers. Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Niso, M.; Berardi, F. Sigma-2 Receptor: Past, Present and Perspectives on Multiple Therapeutic Exploitations. Future Med. Chem. 2018, 10, 1997–2018. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions of S2R | Molecules Involved | Disease Relevance | Sections in Text |
|---|---|---|---|
| Blocks amyloid-β oligomers from binding neuronal synapses | TMEM97, PGRMC1 oligomer receptor | Alzheimer’s disease | 5.4, 6.1–4 |
| Blocks α-synuclein oligomers from binding neuronal synapses | TMEM97 | Dementia with Lewy bodies Parkinson’s disease | 6.5–6 |
| Mediates synaptoprotection | TMEM97, PGRMC1 mGluR5, oligomer receptor | Alzheimer’s disease Parkinson’s disease Dementia with Lewy bodies | 6.1–3 |
| Regulates autophagy | TMEM97, PGRMC1 LAMP2A, MAP1LC3B | Dry AMD Dementia with Lewy bodies Parkinson’s disease | 5.4, 6.7 6.5–6 |
| Regulates cholesterol homeostasis | TMEM97, PGRMC1, LDLR, Apo-E, NPC1 | Alzheimer’s disease Niemann–Pick disease type C | 5.4, 6.1 5.4, 6.8 |
| Regulates membrane trafficking | TMEM97, PGRMC1, LDLR | Alzheimer’s disease Parkinson’s disease Dementia with Lewy bodies | 6.1–4 6.5–6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lizama, B.N.; Kahle, J.; Catalano, S.M.; Caggiano, A.O.; Grundman, M.; Hamby, M.E. Sigma-2 Receptors—From Basic Biology to Therapeutic Target: A Focus on Age-Related Degenerative Diseases. Int. J. Mol. Sci. 2023, 24, 6251. https://doi.org/10.3390/ijms24076251
Lizama BN, Kahle J, Catalano SM, Caggiano AO, Grundman M, Hamby ME. Sigma-2 Receptors—From Basic Biology to Therapeutic Target: A Focus on Age-Related Degenerative Diseases. International Journal of Molecular Sciences. 2023; 24(7):6251. https://doi.org/10.3390/ijms24076251
Chicago/Turabian StyleLizama, Britney N., Jennifer Kahle, Susan M. Catalano, Anthony O. Caggiano, Michael Grundman, and Mary E. Hamby. 2023. "Sigma-2 Receptors—From Basic Biology to Therapeutic Target: A Focus on Age-Related Degenerative Diseases" International Journal of Molecular Sciences 24, no. 7: 6251. https://doi.org/10.3390/ijms24076251
APA StyleLizama, B. N., Kahle, J., Catalano, S. M., Caggiano, A. O., Grundman, M., & Hamby, M. E. (2023). Sigma-2 Receptors—From Basic Biology to Therapeutic Target: A Focus on Age-Related Degenerative Diseases. International Journal of Molecular Sciences, 24(7), 6251. https://doi.org/10.3390/ijms24076251