

Unveiling the Potentiality of Shikonin Derivatives Inhibiting SARS-CoV-2 Main Protease by Molecular Dynamic Simulation Studies

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
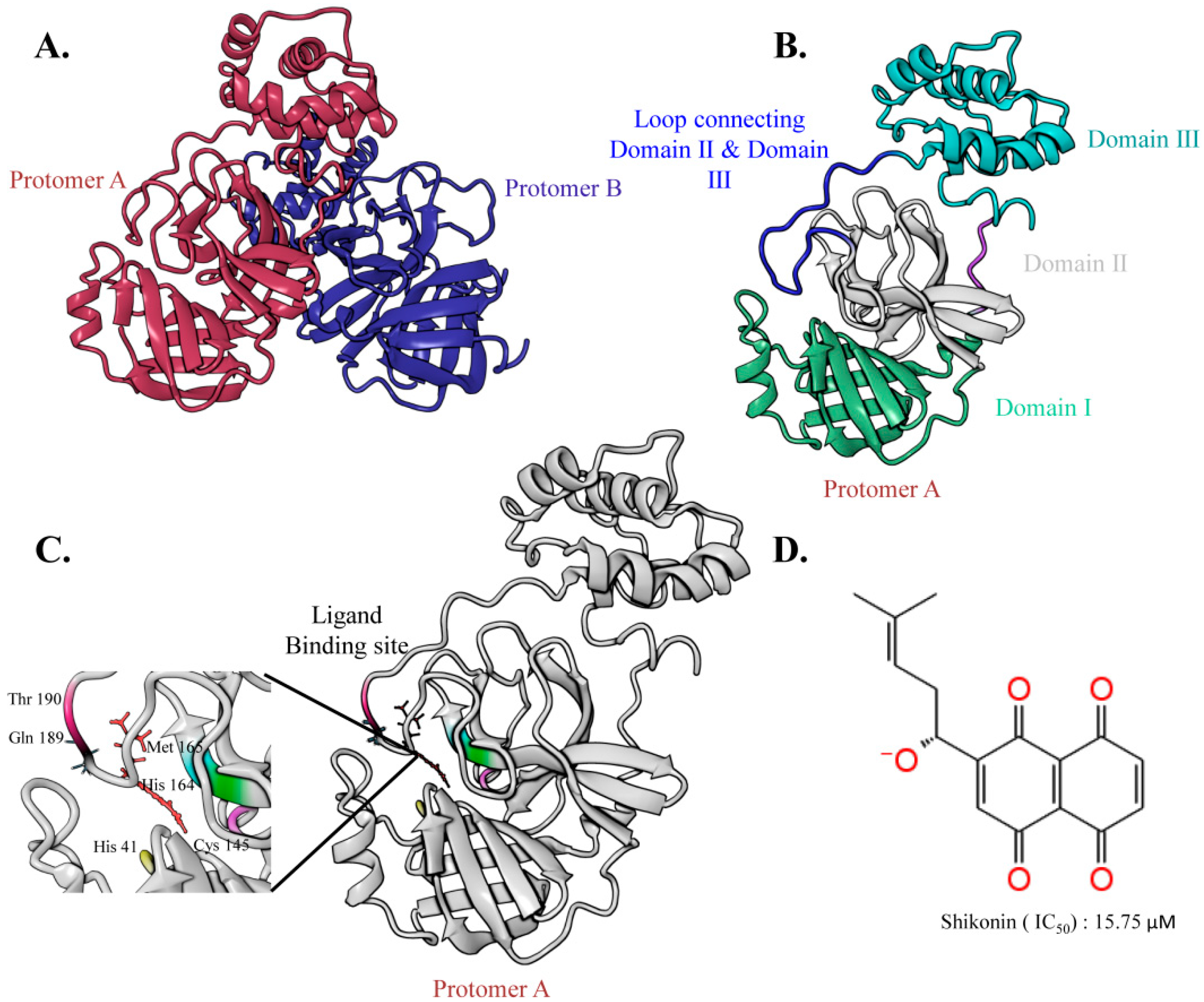
2.1. The Overall Architecture of the SARS-CoV-2 Main Protease
2.2. Molecular Docking
2.3. Drug-Likeness Prediction Studies
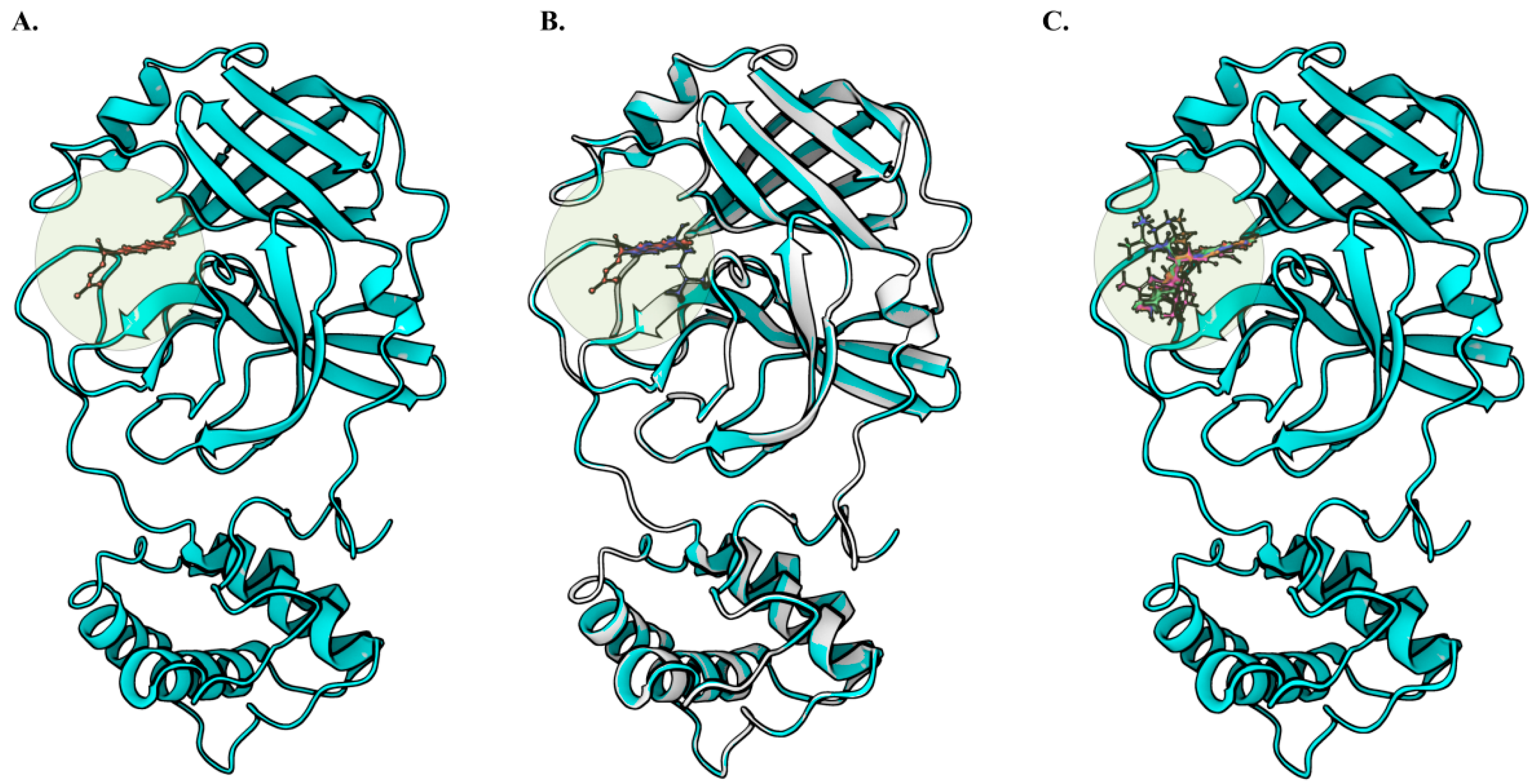
2.4. Molecular Docking Analysis
2.5. Molecular Dynamics Simulations
2.5.1. Effect of Ligands on Conformational Stability
2.5.2. Conformational Dynamics of Protein–Ligand Binding
2.5.3. Ligand-Induced Changes on Protein Dynamics
3. Discussion
4. Materials and Methods
4.1. Ligand Preparation
4.2. Protein Preparation and Grid Generation
4.3. Molecular Docking Simulation
4.4. Prime MM-GBSA for Affinity Prediction
4.5. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Leibowitz, J.L. Coronavirus pathogenesis. Adv. Virus Res. 2011, 81, 85–164. [Google Scholar] [PubMed]
- De Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.M.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (mers-cov): Announcement of the coronavirus study group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I.; et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics ap-proach. Biochim. Et Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Chitranshi, N.; Gupta, V.K.; Rajput, R.; Godinex, A.; Pushpita, K.; Shen, T.; Mirzaei, M.; You, Y.; Basavarajappa, D.; Gupta, V.; et al. Evolving geographic diversity in SARS-CoV2 and in silico analysis of replicating enzyme 3CLpro targeting repurposed drug candidates. J. Transl. Med. 2020, 18, 278. [Google Scholar] [CrossRef]
- Prajapat, M.; Sarma, P.; Shekar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattachacharyya, A.; Kumar, H.; Bansal, S.; et al. Drug targets for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56–65. [Google Scholar]
- Al-Qaaneh, A.M.; Alshammari, T.; Aldahhan, R.; Aldossary, H.; Alkhalifah, Z.A.; Borgio, J.F. Genome composition and genetic characterization of SARS-CoV-2. Saudi J. Biol. Sci. 2021, 28, 1978–1989. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID. Cells 2021, 10, 821. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Bideau, M.L.; Duflot, I.; Jardot, P.; Rolland, C.; Boxberger, M.; Wurtz, N.; Rolain, J.M.; Colson, P.; Scola, B.L.; et al. In vitro testing of combined hydroxychloroquine and azithromycin on SARS-CoV-2 shows synergistic effect. Microb. Pathog. 2020, 145, 104228. [Google Scholar] [CrossRef] [PubMed]
- Meo, S.A.; Bukhari, I.A.; Akram, J.; Meo, A.S.; Klonoff, D.C. COVID-19 vaccines: Comparison of biological, pharmacological characteristics and adverse effects of Pfizer/BioNTech and Moderna Vaccines. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1663–1669. [Google Scholar]
- Farooqi, T.; Malik, J.A.; Mulla, A.H.; Al Hagbani, T.; Almansour, K.; Ubaid, M.A.; Alghamdi, S.; Anwar, S. An overview of SARS-COV-2 epidemiology, mutant variants, vaccines, and management strategies. J. Infect. Public Health 2021, 14, 1299–1312. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Gorbalenya, A.E.; Snijder, E.J. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Heusipp, G.; Siddell, S.G. Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J. Virol. 1997, 71, 3992–3997. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Yilmaz, N.K.; Thompson, P.R.; Schiffer, C.A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, X.; Zhang, Y.; Zhong, F.; Lin, C.; Cormick, P.J.M.; Jiang, F.; Luo, J.; Zhou, H.; Wang, Q.; et al. Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci. Bull. 2021, 66, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Boulos, J.C.; Rahama, M.; Hegazy, M.-E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Lu, L.; Qin, A.; Huang, H.; Zhou, P.; Zhang, C.; Liu, N.; Li, S.; Wen, G.; Zhang, C.; Dong, W.; et al. Shikonin extracted from medicinal Chinese herbs exerts anti-inflammatory effect via proteasome inhibition. Eur. J. Pharmacol. 2011, 658, 242–247. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, H.; Qiu, H.; Lin, H.; Yu, L.; Zhu, W.; Qi, J.; Yang, R.; Pang, Y.; Wang, X.; et al. Antiviral activity of a synthesized shikonin ester against influenza A (H1N1) virus and insights into its mechanism. Biomed. Pharmacother. 2017, 93, 636–645. [Google Scholar] [CrossRef]
- Wang, F.; Yao, X.; Zhang, Y.; Tang, J. Synthesis, biological function and evaluation of Shikonin in cancer therapy. Fitoterapia 2019, 134, 329–339. [Google Scholar] [CrossRef]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Anand, K.; Palm, J.G.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Abu-Saleh, A.A.A.; Awad, I.E.; Yadav, A.; Poirier, R.A. Discovery of potent inhibitors for SARS-CoV-2’s main protease by ligand-based/structure-based virtual screening, MD simulations, and binding energy calculations. Phys. Chem. Chem. Phys. 2020, 22, 23099–23106. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Kang, S.G. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 M(pro) via combinatorial molecular simulation calculations. Life Sci. 2020, 257, 118080. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and de-velopment settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Press, S. QikProp 3.4 User Manual; LLC: New York, NY, USA, 2011. [Google Scholar]
- Dash, R.; Choi, H.J.; Moon, I.S. Mechanistic insights into the deleterious roles of Nasu-Hakola disease associated TREM2 variants. Sci. Rep. 2020, 10, 3663. [Google Scholar] [CrossRef]
- Dash, R.; Ali, C.; Rana, L.; Munni, Y.A.; Barua, L.; Jahan, I.; Haque, M.F.; Hannan, A.; Moon, I.S. Computational SNP Analysis and Molecular Simulation Revealed the Most Deleterious Missense Variants in the NBD1 Domain of Human ABCA1 Transporter. Int. J. Mol. Sci. 2020, 21, 7606. [Google Scholar] [CrossRef]
- Wang, R.; Yin, R.; Zhou, W.; Xu, D.; Li, S. Shikonin and its derivatives: A patent review. Expert Opin. Ther. Pat. 2012, 22, 977–997. [Google Scholar] [CrossRef]
- Zhao, Q.; Kretschmer, N.; Bauer, R.; Efferth, T. Shikonin and its derivatives inhibit the epidermal growth factor receptor signaling and synergistically kill glioblastoma cells in combination with erlotinib. Int. J. Cancer 2015, 137, 1446–1456. [Google Scholar] [CrossRef]
- Han, H.; Sun, W.; Feng, L.; Wen, Z.; Yang, M.; Ma, Y.; Fu, J.; Ma, X.; Xu, X.; Wang, Z.; et al. Differential relieving effects of shikonin and its derivatives on inflammation and mucosal barrier damage caused by ulcerative colitis. PeerJ 2021, 9, e10675. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Bhachoo, J.; Beuming, T. Investigating Protein–Peptide Interactions Using the Schrödinger Computational Suite Modeling Pep-Tide-Protein Interactions. Model. Pept.-Protein Interact. 2017, 1561, 235–254. [Google Scholar] [CrossRef]
- Li, J.; Zhou, X.; Zhang, Y.; Zhong, F.; Lin, C.; Cormick, P.J.M.; Jiang, F.; Luo, J.; Zhou, H.; Wang, Q.; et al. Crystal structure of SARS-CoV-2 main protease in complex with a Chinese herb inhibitor shikonin. bioRxiv 2020. [Google Scholar] [CrossRef]
- Banerjee, K.; Gupta, U.; Gupta, S.; Wadhwa, G.; Gabrani, R.; Sharma, S.K.; Jain, C.K. Molecular docking of glucosamine-6-phosphate synthase in Rhizopus oryzae. Bioinformation 2011, 7, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Vijayakumar, B.; Umamaheswari, A.; Puratchikody, A.; Velmurugan, D. Selection of an improved HDAC8 inhibitor through structure-based drug design. Bioinformation 2011, 7, 134–141. [Google Scholar] [CrossRef]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Kassir, Y.; Kupiec, M.; Shalom, A.; Simchen, G. Cloning and mapping of CDC40, a Saccharomyces cerevisiae gene with a role in DNA repair. Curr. Genet. 1985, 9, 253–257. [Google Scholar] [CrossRef]
- Li, J.; Ngan, A.H.; Gumbsch, P. Atomistic modeling of mechanical behavior. Acta Mater. 2003, 51, 5711–5742. [Google Scholar] [CrossRef]
- Reddy, S.; Reddy, K.T.; Kumari, V.V.; Basha, S.H. Molecular docking and dynamic simulation studies evidenced plausible immunotherapeutic anticancer property by Withaferin A targeting indoleamine 2,3-dioxygenase. J. Biomol. Struct. Dyn. 2015, 33, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Basha, S.H.; Bethapudi, P.; Rambabu, F.M. Anti-angiogenesis property by quercetin compound targeting VEGFR2 elucidated in a computational approach. Eur. J. Biotechnol. Biosci. 2014, 2, 30–46. [Google Scholar]
- Zhang, Y.; Gao, H.; Hu, X.; Wang, Q.; Zhong, F.; Zhou, X.; Lin, C.; Yang, Y.; Wei, J.; Du, W.; et al. Structure-based discovery and structural basis of a novel broad-spectrum natural product against the main protease of coronavirus. J. Virol. 2022, 96, e0125321. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, R.; Habiba, S.U.; Dash, R.; Seo, Y.; Woo, J. Unveiling the Potentiality of Shikonin Derivatives Inhibiting SARS-CoV-2 Main Protease by Molecular Dynamic Simulation Studies. Int. J. Mol. Sci. 2023, 24, 3100. https://doi.org/10.3390/ijms24043100
Das R, Habiba SU, Dash R, Seo Y, Woo J. Unveiling the Potentiality of Shikonin Derivatives Inhibiting SARS-CoV-2 Main Protease by Molecular Dynamic Simulation Studies. International Journal of Molecular Sciences. 2023; 24(4):3100. https://doi.org/10.3390/ijms24043100
Chicago/Turabian StyleDas, Raju, Sarmin Ummey Habiba, Raju Dash, Yohan Seo, and Joohan Woo. 2023. "Unveiling the Potentiality of Shikonin Derivatives Inhibiting SARS-CoV-2 Main Protease by Molecular Dynamic Simulation Studies" International Journal of Molecular Sciences 24, no. 4: 3100. https://doi.org/10.3390/ijms24043100
APA StyleDas, R., Habiba, S. U., Dash, R., Seo, Y., & Woo, J. (2023). Unveiling the Potentiality of Shikonin Derivatives Inhibiting SARS-CoV-2 Main Protease by Molecular Dynamic Simulation Studies. International Journal of Molecular Sciences, 24(4), 3100. https://doi.org/10.3390/ijms24043100