
The Gene Expression of Proteins Involved in Intercellular Signaling and Neurodegeneration in the Substantia Nigra in a Mouse Subchronic Model of Parkinson’s Disease

 , and
, and 
Abstract
:1. Introduction
2. Results
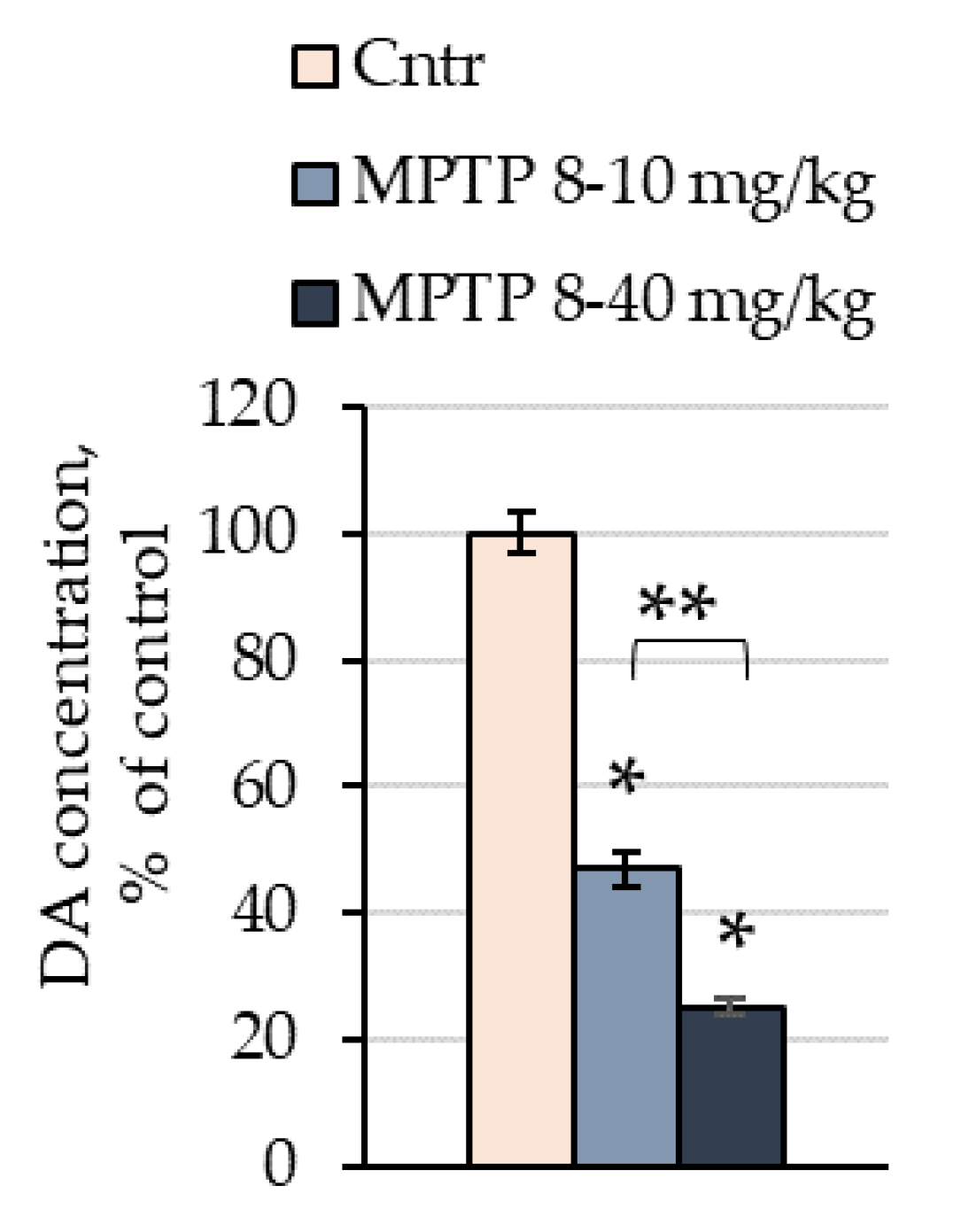
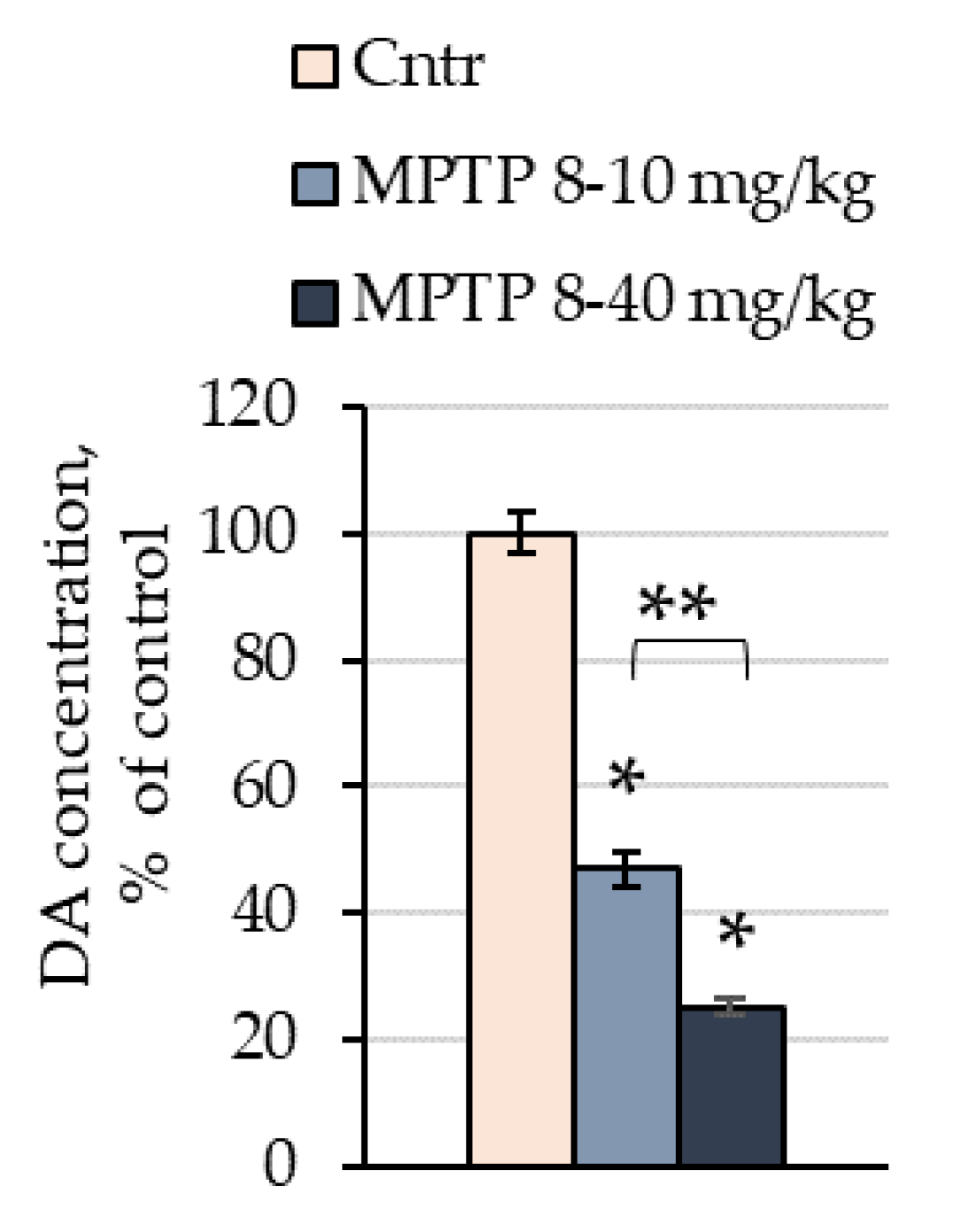
2.1. The Concentration of Dopamine in the Striata of Mice after the Administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
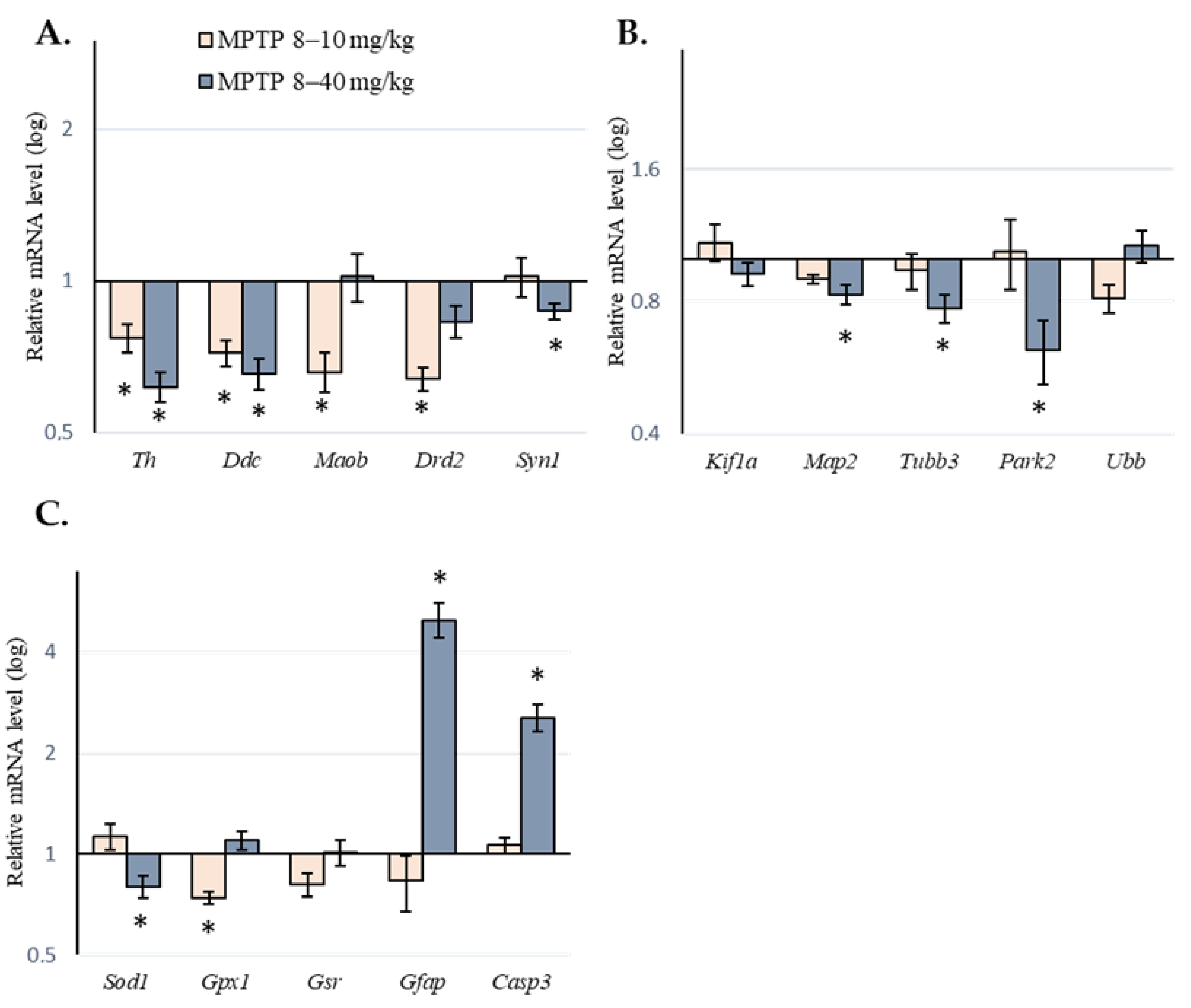
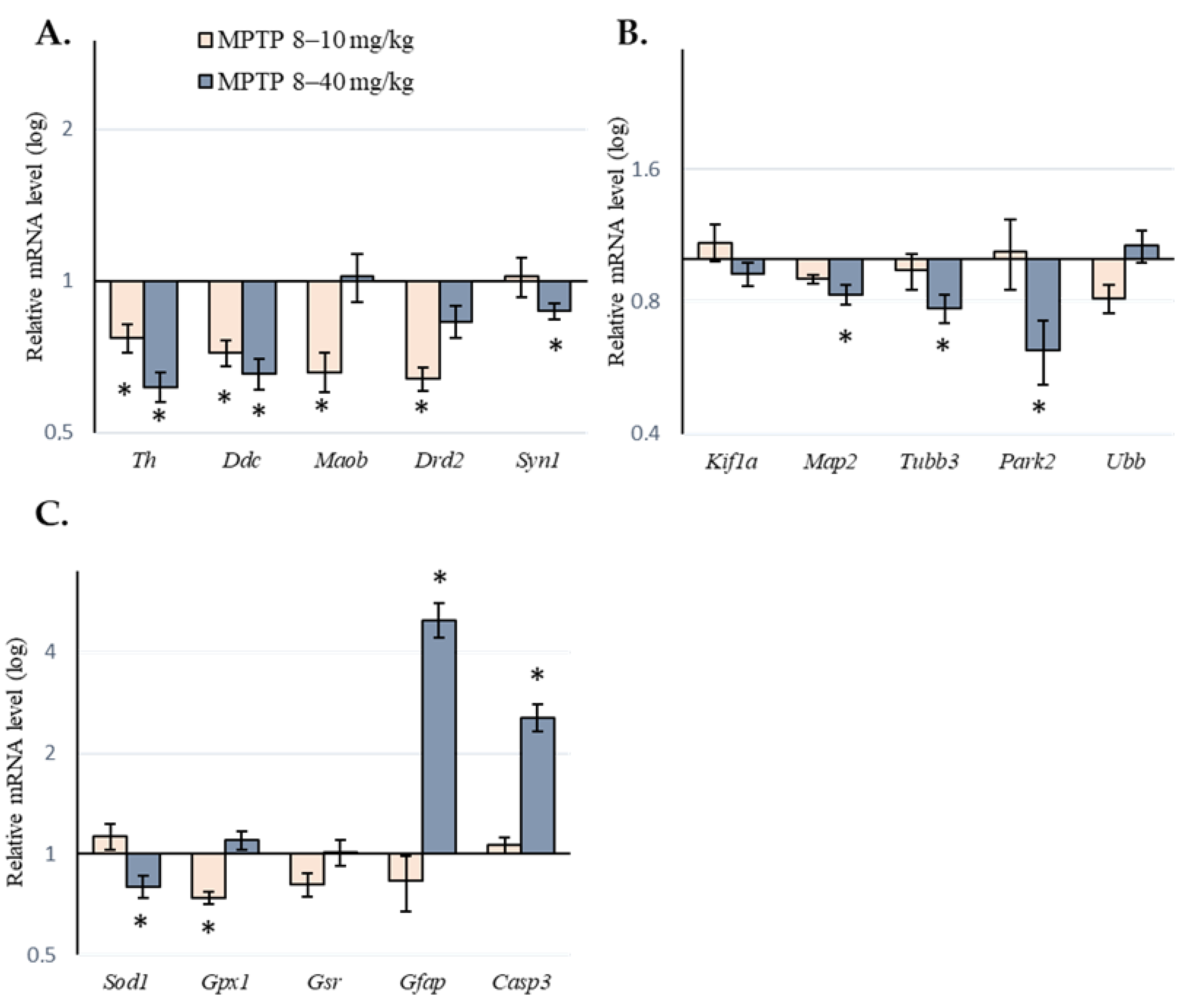
2.2. Expression of Genes of Interest in the Substantia Nigra of Mice following Administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
3. Discussion
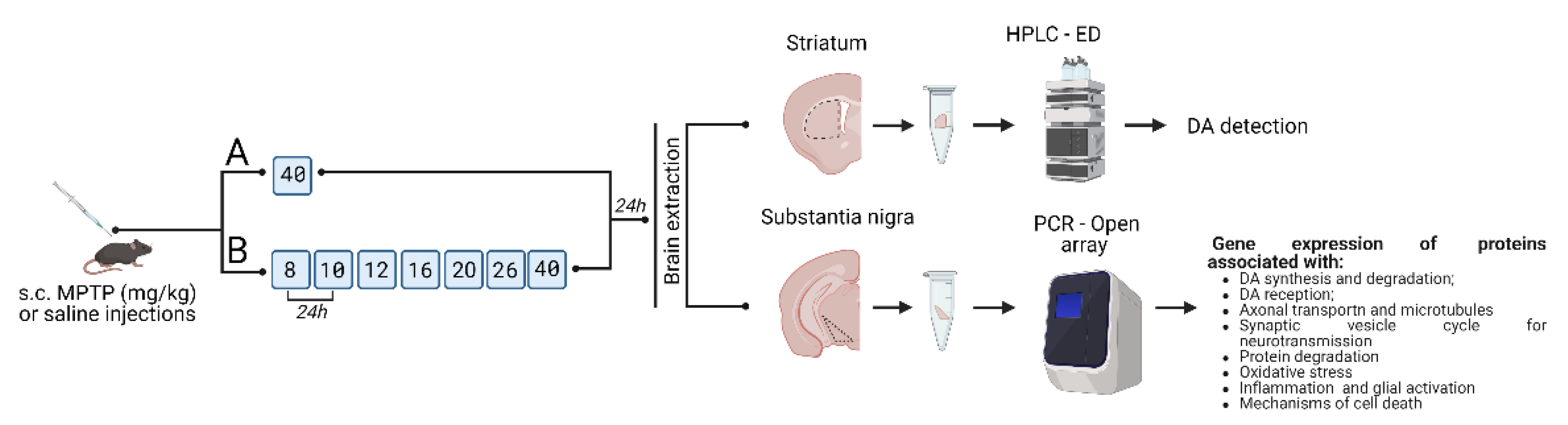
4. Materials and Methods
4.1. Animals
4.2. Sample Preparation for Analysis
4.3. Methods
4.3.1. High Performance Liquid Chromatography with Electrochemical Detection
4.3.2. Real-Time PCR
4.3.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DA | dopamine |
| DAergic | dopaminergic |
| MPTP | 1–methyl–4–phenyl–1,2,3,6–tetrahydropyridine |
| PD | Parkinson’s disease |
| SN | substantia nigra |
References
- Alves, G.; Forsaa, E.B.; Pedersen, K.F.; Dreetz Gjerstad, M.; Larsen, J.P. Epidemiology of Parkinson’s Disease. J. Neurol. 2008, 255 (Suppl. 5), 18–32. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s Disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D. Prodromal Parkinson’s Disease: The Decade Past, the Decade to Come. Mov. Disord. 2019, 34, 665–675. [Google Scholar] [CrossRef]
- Olanow, C.W. Rationale for Considering That Propargylamines Might Be Neuroprotective in Parkinson’s Disease. Neurology 2006, 66, S69–S79. [Google Scholar] [CrossRef]
- Ugrumov, M. Development of Early Diagnosis of Parkinson’s Disease: Illusion or Reality? CNS Neurosci. Ther. 2020, 26, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Kolacheva, A.; Bannikova, A.; Pavlova, E.; Bogdanov, V.; Ugrumov, M. Modeling of the Progressive Degradation of the Nigrostriatal Dopaminergic System in Mice to Study the Mechanisms of Neurodegeneration and Neuroplasticity in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 683. [Google Scholar] [CrossRef]
- Ichinose, H.; Ohye, T.; Fujita, K.; Pantucek, F.; Lange, K.; Riederer, P.; Nagatsu, T. Quantification of MRNA of Tyrosine Hydroxylase and Aromatic L-Amino Acid Decarboxylase in the Substantia Nigra in Parkinson’s Disease and Schizophrenia. J. Neural Transm. Park. Dis. Dement. Sect. 1994, 8, 149–158. [Google Scholar] [CrossRef]
- Harrington, K.A.; Augood, S.J.; Kingsbury, A.E.; Foster, O.J.; Emson, P.C. Dopamine Transporter (Dat) and Synaptic Vesicle Amine Transporter (VMAT2) Gene Expression in the Substantia Nigra of Control and Parkinson’s Disease. Mol. Brain Res. 1996, 36, 157–162. [Google Scholar] [CrossRef]
- Grünblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene Expression Profiling of Parkinsonian Substantia Nigra Pars Compacta; Alterations in Ubiquitin-Proteasome, Heat Shock Protein, Iron and Oxidative Stress Regulated Proteins, Cell Adhesion/Cellular Matrix and Vesicle Trafficking Genes. J. Neural Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef]
- Hauser, M.A.; Li, Y.-J.; Xu, H.; Noureddine, M.A.; Shao, Y.S.; Gullans, S.R.; Scherzer, C.R.; Jensen, R.V.; McLaurin, A.C.; Gibson, J.R.; et al. Expression Profiling of Substantia Nigra in Parkinson Disease, Progressive Supranuclear Palsy, and Frontotemporal Dementia with Parkinsonism. Arch. Neurol. 2005, 62, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Noureddine, M.A.; Li, Y.-J.; van der Walt, J.M.; Walters, R.; Jewett, R.M.; Xu, H.; Wang, T.; Walter, J.W.; Scott, B.L.; Hulette, C.; et al. Genomic Convergence to Identify Candidate Genes for Parkinson Disease: SAGE Analysis of the Substantia Nigra. Mov. Disord. 2005, 20, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional Analysis of Multiple Brain Regions in Parkinson’s Disease Supports the Involvement of Specific Protein Processing, Energy Metabolism, and Signaling Pathways, and Suggests Novel Disease Mechanisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 137, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.B.; Duke, D.C.; Deprez, M.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Whole Genome Expression Profiling of the Medial and Lateral Substantia Nigra in Parkinson’s Disease. Neurogenetics 2006, 7, 1–11. [Google Scholar] [CrossRef]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.M.; Lockner, R.J.; Palaniappan, C.; Federoff, H.J. Robust Dysregulation of Gene Expression in Substantia Nigra and Striatum in Parkinson’s Disease. Neurobiol. Dis. 2006, 21, 305–313. [Google Scholar] [CrossRef]
- Duke, D.C.; Moran, L.B.; Pearce, R.K.B.; Graeber, M.B. The Medial and Lateral Substantia Nigra in Parkinson’s Disease: MRNA Profiles Associated with Higher Brain Tissue Vulnerability. Neurogenetics 2007, 8, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.B.; Croisier, E.; Duke, D.C.; Kalaitzakis, M.E.; Roncaroli, F.; Deprez, M.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Analysis of Alpha-Synuclein, Dopamine and Parkin Pathways in Neuropathologically Confirmed Parkinsonian Nigra. Acta Neuropathol. 2007, 113, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Bossers, K.; Meerhoff, G.; Balesar, R.; van Dongen, J.W.; Kruse, C.G.; Swaab, D.F.; Verhaagen, J. Analysis of Gene Expression in Parkinson’s Disease: Possible Involvement of Neurotrophic Support and Axon Guidance in Dopaminergic Cell Death. Brain Pathol. 2009, 19, 91–107. [Google Scholar] [CrossRef]
- Liu, H.; Wei, L.; Tao, Q.; Deng, H.; Ming, M.; Xu, P.; Le, W. Decreased NURR1 and PITX3 Gene Expression in Chinese Patients with Parkinson’s Disease. Eur. J. Neurol. 2012, 19, 870–875. [Google Scholar] [CrossRef]
- Glaab, E.; Schneider, R. Comparative Pathway and Network Analysis of Brain Transcriptome Changes during Adult Aging and in Parkinson’s Disease. Neurobiol. Dis. 2015, 74, 1–13. [Google Scholar] [CrossRef]
- Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L.T.; Krichevsky, A.M.; Andersen, S.L.; Stephens, R.M.; Benes, F.M.; Sonntag, K.C. Gene Expression Profiling of Substantia Nigra Dopamine Neurons: Further Insights into Parkinson’s Disease Pathology. Brain 2009, 132, 1795–1809. [Google Scholar] [CrossRef] [Green Version]
- Simunovic, F.; Yi, M.; Wang, Y.; Stephens, R.; Sonntag, K.C. Evidence for Gender-Specific Transcriptional Profiles of Nigral Dopamine Neurons in Parkinson Disease. PLoS ONE 2010, 5, e8856. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, A.; Antinori, P.; Licker, V.; Kövari, E.; Lobrinus, J.A.; Burkhard, P.R. Multiomic Analyses of Dopaminergic Neurons Isolated from Human Substantia Nigra in Parkinson’s Disease: A Descriptive and Exploratory Study. Cell. Mol. Neurobiol. 2022, 42, 2805–2818. [Google Scholar] [CrossRef] [PubMed]
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression Analysis of Dopaminergic Neurons in Parkinson’s Disease and Aging Links Transcriptional Dysregulation of Energy Metabolism to Cell Death. Acta Neuropathol. 2011, 122, 75–86. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, I.; Keller-McGandy, C.; Bouzou, B.; Asteris, G.; Clark, T.W.; Frosch, M.P.; Standaert, D.G. Effects of Gender on Nigral Gene Expression and Parkinson Disease. Neurobiol. Dis. 2007, 26, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Tiklová, K.; Gillberg, L.; Volakakis, N.; Lundén-Miguel, H.; Dahl, L.; Serrano, G.E.; Adler, C.H.; Beach, T.G.; Perlmann, T. Disease Duration Influences Gene Expression in Neuromelanin-Positive Cells From Parkinson’s Disease Patients. Front. Mol. Neurosci. 2021, 14, 763777. [Google Scholar] [CrossRef]
- Shin, J.Y.; Park, H.J.; Ahn, Y.H.; Lee, P.H. Neuroprotective effect of L-dopa on dopaminergic neurons is comparable to pramipexol in MPTP-treated animal model of Parkinson’s disease: A direct comparison study. J. Neurochem. 2009, 111, 1042–1050. [Google Scholar] [CrossRef]
- Charbonnier-Beaupel, F.; Malerbi, M.; Alcacer, C.; Tahiri, K.; Carpentier, W.; Wang, C.; During, M.; Xu, D.; Worley, P.F.; Girault, J.-A.; et al. Gene Expression Analyses Identify Narp Contribution in the Development of L-DOPA-Induced Dyskinesia. J. Neurosci. 2015, 35, 96–111. [Google Scholar] [CrossRef]
- Kolacheva, A.; Alekperova, L.; Pavlova, E.; Bannikova, A.; Ugrumov, M.V. Changes in Tyrosine Hydroxylase Activity and Dopamine Synthesis in the Nigrostriatal System of Mice in an Acute Model of Parkinson’s Disease as a Manifestation of Neurodegeneration and Neuroplasticity. Brain Sci. 2022, 12, 779. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kiuchi, K.; Kojima, K.; Kondo, T.; Narabayashi, H.; Rausch, D.; Riederer, P.; Jellinger, K.; Nagatsu, T. Homospecific Activity (Activity per Enzyme Protein) of Tyrosine Hydroxylase Increases in Parkinsonian Brain. J. Neural Transm. 1988, 72, 77–82. [Google Scholar] [CrossRef]
- Nagatsu, T. Change of Tyrosine Hydroxylase in the Parkinsonian Brain and in the Brain of MPTP-Treated Mice as Revealed by Homospecific Activity. Neurochem. Res. 1990, 15, 425–429. [Google Scholar] [CrossRef]
- Kozina, E.A.; Khakimova, G.R.; Khaindrava, V.G.; Kucheryanu, V.G.; Vorobyeva, N.E.; Krasnov, A.N.; Georgieva, S.G.; Kerkerian-Le Goff, L.; Ugrumov, M.V. Tyrosine Hydroxylase Expression and Activity in Nigrostriatal Dopaminergic Neurons of MPTP-Treated Mice at the Presymptomatic and Symptomatic Stages of Parkinsonism. J. Neurol. Sci. 2014, 340, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in Axonal Transport Motor Proteins in Sporadic and Experimental Parkinson’s Disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.; Ratnakaran, N.; Koushika, S.P. Neurodegeneration and Microtubule Dynamics: Death by a Thousand Cuts. Front. Cell. Neurosci. 2015, 9, 343. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferro, P.; Burke, R.E. Retrograde Axonal Degeneration in Parkinson Disease. J. Park. Dis. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gcwensa, N.Z.; Russell, D.L.; Cowell, R.M.; Volpicelli-Daley, L.A. Molecular Mechanisms Underlying Synaptic and Axon Degeneration in Parkinson’s Disease. Front. Cell. Neurosci. 2021, 15, 626128. [Google Scholar] [CrossRef]
- Ren, Y.R.; Zhao, J.; Feng, J. Parkin Binds to Alpha/Beta Tubulin and Increases Their Ubiquitination and Degradation. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 3316–3324. [Google Scholar] [CrossRef]
- Kuzuhara, S.; Mori, H.; Izumiyama, N.; Yoshimura, M.; Ihara, Y. Lewy Bodies Are Ubiquitinated. A Light and Electron Microscopic Immunocytochemical Study. Acta Neuropathol. 1988, 75, 345–353. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Shashidharan, P.; Perl, D.P.; Jenner, P.; Olanow, C.W. Aggresome-Related Biogenesis of Lewy Bodies. Eur. J. Neurosci. 2002, 16, 2136–2148. [Google Scholar] [CrossRef]
- Kopito, R.R. Aggresomes, Inclusion Bodies and Protein Aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone Suppression of Alpha-Synuclein Toxicity in a Drosophila Model for Parkinson’s Disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative Damage Linked to Neurodegeneration by Selective Alpha-Synuclein Nitration in Synucleinopathy Lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Goto, K.; Mori, H.; Mizuno, Y. Histochemical Detection of Apoptosis in Parkinson’s Disease. J. Neurol. Sci. 1996, 137, 120–123. [Google Scholar] [CrossRef]
- Kingsbury, A.E.; Mardsen, C.D.; Foster, O.J. DNA Fragmentation in Human Substantia Nigra: Apoptosis or Perimortem Effect? Mov. Disord. 1998, 13, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Kosel, S.; Egensperger, R.; von Eitzen, U.; Mehraein, P.; Graeber, M.B. On the Question of Apoptosis in the Parkinsonian Substantia Nigra. Acta Neuropathol. 1997, 93, 105–108. [Google Scholar] [CrossRef]
- Banati, R.B.; Daniel, S.E.; Blunt, S.B. Glial Pathology but Absence of Apoptotic Nigral Neurons in Long-Standing Parkinson’s Disease. Mov. Disord. 1998, 13, 221–227. [Google Scholar] [CrossRef]
- Graeber, M.B.; Grasbon-Frodl, E.; Abell-Aleff, P.; Kösel, S. Nigral Neurons Are Likely to Die of a Mechanism Other than Classical Apoptosis in Parkinson’s Disease. Park. Relat. Disord. 1999, 5, 187–192. [Google Scholar] [CrossRef]
- Jellinger, K.A. Cell Death Mechanisms in Parkinson’s Disease. J. Neural Transm. 2000, 107, 1–29. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Novikova, L.; Garris, B.L.; Garris, D.R.; Lau, Y.-S. Early Signs of Neuronal Apoptosis in the Substantia Nigra Pars Compacta of the Progressive Neurodegenerative Mouse 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine/Probenecid Model of Parkinson’s Disease. Neuroscience 2006, 140, 67–76. [Google Scholar] [CrossRef]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A Vulnerability Factor and Final Effector in Apoptotic Death of Dopaminergic Neurons in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turmel, H.; Hartmann, A.; Parain, K.; Douhou, A.; Srinivasan, A.; Agid, Y.; Hirsch, E.C. Caspase-3 Activation in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP)-Treated Mice. Mov. Disord. 2001, 16, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Ohnuki, T.; Nakamura, A.; Okuyama, S.; Nakamura, S. Gene expression profiling in progressively MPTP-lesioned macaques reveals molecular pathways associated with sporadic Parkinson’s disease. Brain Res. 2010, 1346, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.T.; Matigian, N.A.; Chalk, A.M.; Anderson, M.J.; Silburn, P.A.; Mackay-Sim, A.; Wells, C.A.; Mellick, G.D. A Cross-Study Transcriptional Analysis of Parkinson’s Disease. PLoS ONE 2009, 4, e4955. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.P. The Role of D2-Autoreceptors in Regulating Dopamine Neuron Activity and Transmission. Neuroscience 2014, 282, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kang, X.; Zhou, L.; Chai, Z.; Wu, Q.; Huang, R.; Xu, H.; Hu, M.; Sun, X.; Sun, S.; et al. Synaptotagmin-11 Is a Critical Mediator of Parkin-Linked Neurotoxicity and Parkinson’s Disease-like Pathology. Nat. Commun. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates, Compact, 2nd ed.; Academic Press: Cambridge, UK, 2001. [Google Scholar]
- Ugrumov, M.V.; Khaindrava, V.G.; Kozina, E.A.; Kucheryanu, V.G.; Bocharov, E.V.; Kryzhanovsky, G.N.; Kudrin, V.S.; Narkevich, V.B.; Klodt, P.M.; Rayevsky, K.S.; et al. Modeling of Presymptomatic and Symptomatic Stages of Parkinsonism in Mice. Neuroscience 2011, 181, 175–188. [Google Scholar] [CrossRef]
- Bengtsson, M.; Ståhlberg, A.; Rorsman, P.; Kubista, M. Gene Expression Profiling in Single Cells from the Pancreatic Islets of Langerhans Reveals Lognormal Distribution of MRNA Levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Function | Scheme of Subcutaneous Administration of MPTP, mg/kg | |||
|---|---|---|---|---|---|---|
| 8–10 | 8–40 | |||||
| Fold Change | p | Fold Change | p | |||
| Dopamine synthesis and degradation | ||||||
| Th | Tyrosine hydroxylase | DA synthesis | 0.77 * | 0.01 | 0.64 * | 0.01 |
| Ddc | Aromatic L-amino acid decarboxylase | DA synthesis | 0.72 * | 0.01 | 0.69 * | 0.01 |
| Maoa | Monoamine oxidase A | DA degradation | 1.18 | 0.37 | 1.06 | 0.99 |
| Maob | Monoamine oxidase B | DA degradation | 0.66 * | 0.01 | 1.02 | 0.98 |
| Comt | Catechol-O-methyltransferase | DA degradation | 0.87 | 0.99 | 1.05 | 0.99 |
| Dopamine reception | ||||||
| Drd2 | Dopamine receptor 2 | DA reception | 0.64 * | 0.01 | 0.83 | 0.37 |
| Axonal transport and microtubules | ||||||
| Kif1a | Kinesin family member 1A | Axonal transport | 1.09 | 0.95 | 1.00 | 0.99 |
| Kif1b | Kinesin family member 1B | Axonal transport | 0.98 | 0.99 | 1.17 | 0.99 |
| Kif5a | Kinesin family member 5A | Axonal transport | 0.89 | 0.88 | 1.14 | 0.62 |
| Dync1h1 | Dynein cytoplasmic 1 heavy chain 1 | Axonal transport | 1.17 | 0.69 | 0.90 | 0.97 |
| Dynll1 | Dynein light chain | Axonal transport | 0.85 | 0.72 | 1.07 | 0.98 |
| Dctn1 | Dynactin 1 | Axonal transport | 1.09 | 0.87 | 1.04 | 0.99 |
| Map2 | Microtubule-associated protein 2 | Axonal transport | 0.90 | 0.61 | 0.83 * | 0.04 |
| Mapt | Microtubule-associated protein tau | Axonal transport | 1.15 | 0.36 | 0.95 | 0.98 |
| Mark2 | MAP/microtubule affinity regulating kinase 2 | Axonal transport | 0.97 | 0.99 | 1.49 | 0.13 |
| Tubb3 | β-tubulin | Axonal transport | 0.94 | 0.94 | 0.77 * | 0.02 |
| Tuba1a | α-tubulin | Axonal transport | 0.98 | 0.90 | 0.74 * | 0.04 |
| Synaptic vesicle cycle of neurotransmission | ||||||
| Snca | α-Synuclein | Neurotransmission | 0.78 | 0.18 | 0.93 | 0.90 |
| Syn1 | Synapsin 1, SNARE-complex | Vesicular cycle | 1.02 | 0.98 | 0.70 * | 0.01 |
| Stx1a | Syntaxin 1A | Vesicular cycle | 0.76 | 0.96 | 1.17 | 0.94 |
| Syt1 | Synaptotagmin I | Vesicular cycle | 0.89 | 0.75 | 0.79 | 0.12 |
| Syt11 | Synaptotagmin 11 | Endocytosis | 0.72 * | 0.02 | 0.87 | 0.05 |
| Rab5a | RAB5A | Endocytosis | 0.89 | 0.49 | 0.92 | 0.60 |
| Rab7 | RAB7 | Endocytosis | 0.93 | 0.98 | 1.03 | 0.98 |
| Nsf | N-Ethylmaleimide sensitive fusion protein | Vesicular cycle | 0.92 | 0.96 | 0.96 | 0.98 |
| Dnm1 | Dynamin 1-like protein | Vesicular cycle | 0.81 | 0.48 | 1.40 | 0.13 |
| Protein degradation | ||||||
| Park2 | Parkin | Ubiquitinylation | 1.04 | 0.90 | 0.62 * | 0.01 |
| Uba3 | Ubiquitin-like modifier activating enzyme 3 | Ubiquitinylation | 0.82 | 0.49 | 1.21 | 0.72 |
| Usp47 | Ubiquitin specific peptidase 47 | Ubiquitinylation | 0.79 | 0.79 | 1.62 | 0.98 |
| Ubb | Ubiquitin B | Ubiquitinylation | 0.81 | 0.16 | 1.07 | 0.71 |
| Ube2n | Ubiquitin-conjugating enzyme E2N | Ubiquitinylation | 0.83 | 0.49 | 0.92 | 0.72 |
| Psmb4 | Proteasome 20S Subunit Beta 4 | Protein degradation | 0.93 | 0.95 | 0.88 | 0.45 |
| Psmc3 | Proteasome 26S subunit ATPase 3 | Protein degradation | 1.07 | 0.89 | 0.76 * | 0.02 |
| Psmd4 | Proteasome 26S subunit non-ATPase 4 | Protein degradation | 0.87 | 0.99 | 1.04 | 0.83 |
| Oxidative stress | ||||||
| Sod1 | Superoxide dismutase 1 | Antioxidant system | 1.13 | 0.60 | 0.80 * | 0.05 |
| Gpx1 | Glutathione peroxidase 1 | Antioxidant system | 0.74 * | 0.04 | 1.10 | 0.72 |
| Gsr | Glutathione reductase | Antioxidant system | 0.81 | 0.07 | 1.01 | 0.87 |
| Txnrd1 | Thioredoxin reductase 1 | Antioxidant system | 0.80 | 052 | 1.09 | 0.99 |
| Nos1 | Nitric oxide synthase 1, neuronal | Antioxidant system | 0.61 | 0.72 | 1.02 | 0.99 |
| Prdx1 | Peroxiredoxin 1 | Antioxidant system | 0.81 | 0.65 | 0.93 | 0.74 |
| Nfe2l2 | Nuclear factor erythroid 2-related factor 2 | Regulation of antioxidant system | 1.14 | 0.92 | 1.03 | 1.00 |
| Keap1 | Kelch-like ECH-associated protein 1 | Regulation of antioxidant system | 1.31 | 0.45 | 0.60 * | 0.04 |
| Sigmar1 | Sigma-1 receptor | Chaperone protein | 0.97 | 0.99 | 1.09 | 0.88 |
| Inflammation and glial activation | ||||||
| Gfap | Glial fibrillary acidic protein | Glial activation | 0.83 | 0.99 | 4.99 * | 0.01 |
| Akt1 | Protein kinase B alpha | Inflammatory signaling pathway | 0.83 | 0.48 | 0.81 * | 0.02 |
| Tgfb1 | Transforming growth factor, beta 1 | Anti-inflammatory cytokine | 0.75 | 0.65 | 1.35 | 0.65 |
| Cell death | ||||||
| Casp3 | Caspase 3 | Apoptosis | 1.06 | 0.99 | 2.55 * | 0.01 |
| Parp1 | Poly [ADP-ribose] polymerase 1 | Apoptosis | 1.01 | 0.96 | 1.72 * | 0.02 |
| Aifm1 | Apoptosis inducing factor mitochondria associated 1 | Apoptosis | 1.08 | 0.99 | 1.62 * | 0.04 |
| Bcl2l11 | BCL2 like 11 | Apoptosis | 0.82 | 0.8 | 1.38 | 0.33 |
| Map3k5 | Mitogen-activated protein kinase kinase kinase 5 | Apoptotic signaling | 0.64 | 0.89 | 0.95 | 0.65 |
| Cib1 | Calcium and integrin binding 1 | Apoptotic signaling | 0.94 | 0.96 | 2.55 * | 0.01 |
| Bax | Bax protein | Apoptosis | 0.90 | 0.94 | 1.17 | 0.60 |
| Trp53 | Transformation related protein 53 | Apoptosis | 1.04 | 0.99 | 0.10 | 0.99 |
| Fos | FBJ osteosarcoma oncogene | Apoptosis | 0.91 | 0.99 | 1.44 | 0.07 |
| Vps35 | Vacuolar protein sorting-associated protein 35 | Autophagy | 0.87 | 0.88 | 1.05 | 0.99 |
| Mapk8 | Mitogen-activated protein kinase 8 | Autophagy | 1.03 | 0.95 | 0.85 | 0.95 |
| Lamp2 | Lysosomal-associated membrane protein 2 | Autophagy | 0.83 | 0.59 | 0.87 | 0.70 |
| Atg16l1 | Autophagy related 16-like 1 (S. cerevisiae) | Autophagy | 0.87 | 0.91 | 0.87 | 0.77 |
| Atg5 | Autophagy related 5 | Autophagy | 0.77 | 0.65 | 1.00 | 0.99 |
| Ctsb | Cathepsin B | Necrosis | 0.85 | 0.52 | 1.31 * | 0.01 |
| Capn1 | Calpain 1 | Necrosis | 1.16 | 0.78 | 1.19 | 0.98 |
| Eif2ak3 | Endoplasmic reticulum (ER) to nucleus signaling 2 | ER stress | 0.86 | 0.92 | 1.53 | 0.31 |
| Atf6 | Activating transcription factor 6 | ER stress | 1.18 | 0.78 | 1.02 | 0.99 |
| Gene | Protein | Function | CSSM | SN PD | Articles |
|---|---|---|---|---|---|
| Dopamine synthesis and degradation | |||||
| Th | Tyrosine hydroxylase | DA synthesis | ↓ | ↓ | [7] |
| Ddc | Aromatic L-amino acid decarboxylase | DA synthesis | ↓ | ↓ | [7,14] |
| Maoa | Monoamine oxidase A | DA degradation | → | ↑ | [15] |
| Axonal transport and microtubules | |||||
| Dynll1 | Dynein light chain | Axonal transport | → | ↓ | [14,17] |
| Mapt | Tau protein | Axonal transport | → | ↓ | [15,19] |
| Map2 | Microtubule-associated protein 2 | Axonal transport | ↓ | ↓/→ | [14,16] |
| Synaptic vesicle cycle of neurotransmission | |||||
| Snca | α-Synuclein | Neurotransmission | → | ↓/→ | [14,16,17] |
| Syt11 | Synaptotagmin 11 | Endocytosis | → | ↓ | [11] |
| Syt1 | Synaptotagmin 1 | Vesicular cycle | → | ↓ | [10,12,13,17] |
| Nsf | N-Ethylmaleimide sensitive fusion protein | Vesicular cycle | → | ↓ | [12,13,14,17] |
| Protein degradation | |||||
| Park2 | Parkin | Ubiquitinylation | ↓ | → | [16] |
| Ube2n | Ubiquitin-conjugating enzyme E2N | Ubiquitinylation | → | ↓ | [15] |
| Ubb | Ubiquitin B | Ubiquitinylation | → | ↓ | [11,15] |
| Psmc3 | Proteasome 26S subunit ATPase 3 | Protein degradation | ↓ | ↓ | [15] |
| Psmd4 | Proteasome 26S subunit non-ATPase 4 | Protein degradation | → | ↑ | [15] |
| Inflammation and glial activation | |||||
| Gfap | Glial fibrillary acidic protein | Glial activation | ↑ | ↓ | [11] |
| Cell death | |||||
| Casp3 | Caspase 3 | Apoptosis | ↑ | ↑ | [16] |
| Vps35 | VPS35 retromer complex component | Autophagy | → | ↓ | [11] |
| Mapk8 | Mitogen-activated protein kinase 8 | Autophagy | → | → | [16] |
| Ctsb | Cathepsin B | Necrosis | ↑ | ↓ | [11] |
| Gene | Protein | Function | CSSM | NMPC | Articles |
|---|---|---|---|---|---|
| Dopamine reception | |||||
| Drd2 | Dopamine receptor 2 | DA reception | → | ↑/→ | [21,22,23] |
| Axonal transport and microtubules | |||||
| Kif1a | Kinesin family member 1A | Axonal transport | → | ↓/→ | [20,22] |
| Kif5a | Kinesin family member 5A | Axonal transport | → | ↓/→ | [22,24] |
| Mark2 | MAP/microtubule affinity regulating kinase 2 | Axonal transport | → | ↑/↓ | [23,24] |
| Synaptic vesicle cycle of neurotransmission | |||||
| Syt1 | Synaptotagmin 1 | Vesicular cycle | → | ↓ | [20,21,22,23,24] |
| Gene | Protein | Protein Function | Subchronic Model Stages: | |
|---|---|---|---|---|
| PSSM | CSSM | |||
| Dopamine synthesis and degradation, reception | ||||
| Th | Tyrosine hydroxylase | DA synthesis | ↓ | ↓ |
| Ddc | Aromatic L-amino acid decarboxylase | DA synthesis | ↓ | ↓ |
| Maob | Monoamine oxidase B | DA degradation | ↓ | → |
| Dopamine reception | ||||
| Drd2 | Dopamine receptor 2 | DA reception | ↓ | → |
| Axonal transport and microtubules | ||||
| Map2 | Microtubule-associated protein 2 | Axonal transport | → | ↓ |
| Tubb3 | β-tubulin | Axonal transport | → | ↓ |
| Tuba1a | α-tubulin | Axonal transport | → | ↓ |
| Synaptic vesicle cycle of neurotransmission | ||||
| Syn1 | Synapsin 1 | Vesicular cycle | → | ↓ |
| Syt11 | Synaptotagmin 11 | Endocytosis | ↓ | → |
| Protein degradation | ||||
| Park2 | Parkin | Ubiquitinylation | → | ↓ |
| Psmc3 | Proteasome 26S subunit ATPase 3 | Protein degradation | → | ↓ |
| Oxidative stress | ||||
| Gpx1 | Glutathione peroxidase 1 | Antioxidant system | ↓ | → |
| Keap1 | Kelch-like ECH-associated protein 1 | Regulation of antioxidant system | → | ↓ |
| Inflammation and glial activation | ||||
| Gfap | Glial fibrillary acidic protein | Glial activation | → | ↑ |
| Akt1 | Protein kinase B alpha | Inflammatory signaling pathway | → | ↓ |
| Cell death | ||||
| Casp3 | Caspase 3 | Apoptosis | → | ↑ |
| Parp1 | Poly [ADP-ribose] polymerase 1 | Apoptosis | → | ↑ |
| Aifm1 | Apoptosis inducing factor mitochondria associated 1 | Apoptosis | → | ↑ |
| Cib1 | Calcium and integrin binding 1 | Apoptotic signaling | → | ↑ |
| Ctsb | Cathepsin B | Necrosis | → | ↑ |
| Cluster of Genes | Genes |
|---|---|
| Dopamine synthesis and degradation | Th, Ddc, Dbh, Pnmt, Maoa, Maob, and Comt |
| Dopamine reception | Drd1–Drd5 |
| Axonal transport and microtubules | Kif1a, Kif1b, Kif5a, Kif2c, Dync1h1, Dynll1, Dctn1, Mapt, Map2, Mark2, Tubb3, and Tuba1a |
| Synaptic vesicle cycle of neurotransmission | Snca, Syn1, Stx1a, Syt1, Syt11, Rab5a, Rab7, Nsf, Dnm1l, and Vps35 |
| Oxidative stress | Sod1, Gpx1, Gsr, Txnrd1, Nos1, Prdx1, Nfe2l2, Agtr2, Keap1, and Sigmar1 |
| Protein degradation | Park2, Ube2n, Uba3, Psmb4, Psmc3, Psmd4, Usp47, Ubb, Cacna1d, and Trpm2 |
| Inflammation and glial activation | Calb1, Ifng, Tgfb1, Akt1, Cnr1, Ptgs2, Traf1, and Cxcl11 |
| Cell death | Casp1, Casp3, Parp1, Aifm1, Bcl2l11, Map3k5, Cib1, Trp53, Bax, Fos, Mapk8, Lamp2, Atg16l1, Atg5, Tnf, Ctsb, Ern2, Eif2ak3, and Atf6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolacheva, A.; Pavlova, E.; Bannikova, A.; Bogdanov, V.; Troshev, D.; Ugrumov, M. The Gene Expression of Proteins Involved in Intercellular Signaling and Neurodegeneration in the Substantia Nigra in a Mouse Subchronic Model of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 3027. https://doi.org/10.3390/ijms24033027
Kolacheva A, Pavlova E, Bannikova A, Bogdanov V, Troshev D, Ugrumov M. The Gene Expression of Proteins Involved in Intercellular Signaling and Neurodegeneration in the Substantia Nigra in a Mouse Subchronic Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(3):3027. https://doi.org/10.3390/ijms24033027
Chicago/Turabian StyleKolacheva, Anna, Ekaterina Pavlova, Alyona Bannikova, Vsevolod Bogdanov, Dmitry Troshev, and Michael Ugrumov. 2023. "The Gene Expression of Proteins Involved in Intercellular Signaling and Neurodegeneration in the Substantia Nigra in a Mouse Subchronic Model of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 3: 3027. https://doi.org/10.3390/ijms24033027
APA StyleKolacheva, A., Pavlova, E., Bannikova, A., Bogdanov, V., Troshev, D., & Ugrumov, M. (2023). The Gene Expression of Proteins Involved in Intercellular Signaling and Neurodegeneration in the Substantia Nigra in a Mouse Subchronic Model of Parkinson’s Disease. International Journal of Molecular Sciences, 24(3), 3027. https://doi.org/10.3390/ijms24033027