
Inter-Regional Proteomic Profiling of the Human Brain Using an Optimized Protein Extraction Method from Formalin-Fixed Tissue to Identify Signaling Pathways



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Optimized Protocol to Evaluate the Proteome of Formalin-Fixed Human Brain Tissue
Quantitative Reproducibility of Formalin-Fixed Human Brain Tissue
2.2. Inter-Regional Analysis of the Human Brain Cortex
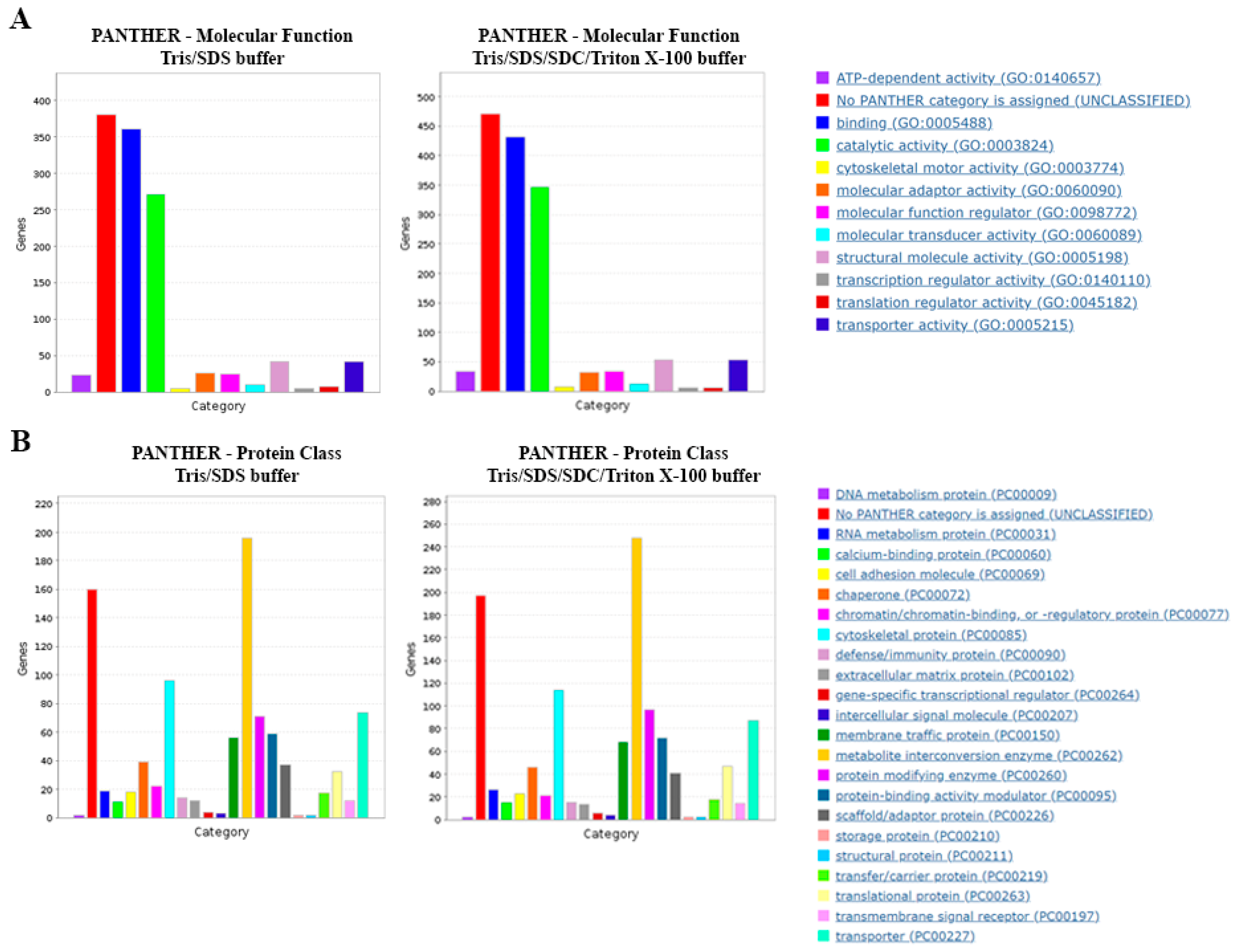
Gene Ontology Analysis
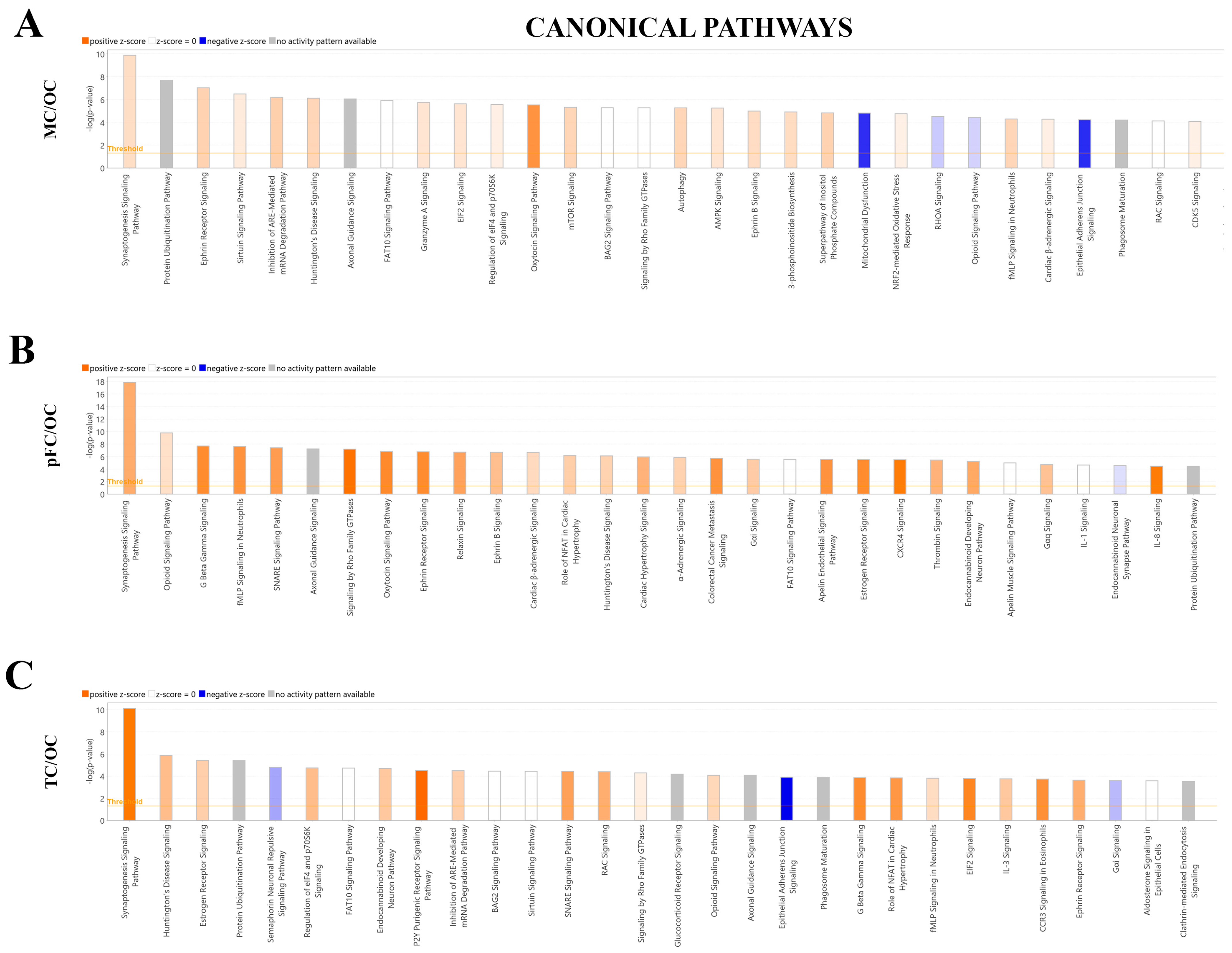
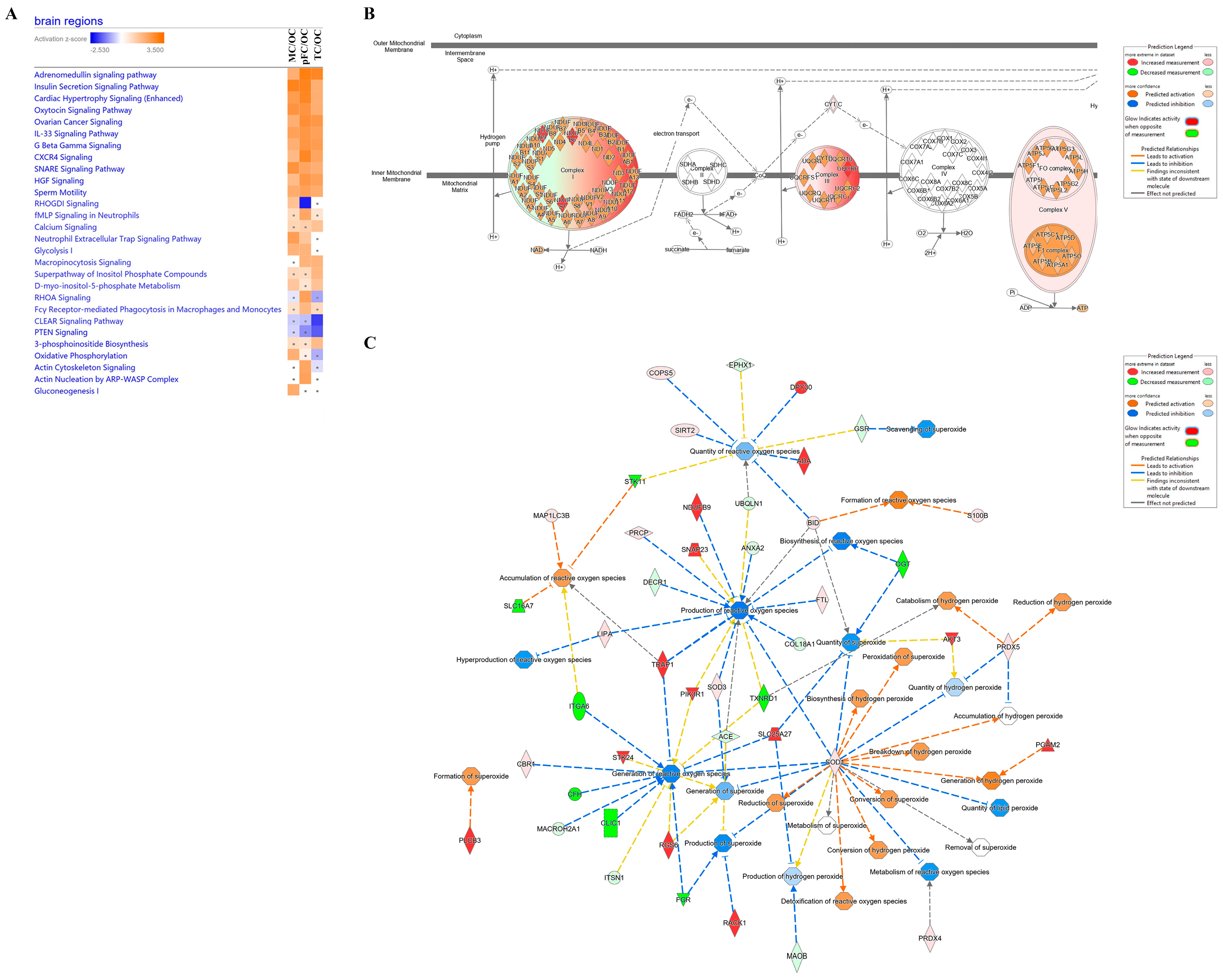
3. Discussion
4. Materials and Methods
4.1. Tissue Collection
4.2. Protein Extraction from Formalin-Fixed Tissue
4.3. Protocol Optimization
4.4. Protein Digestion of Brain Tissues
4.5. LC-MS/MS Analysis and MS Data Processing
4.6. Statistical and Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glasser, M.F.; Coalson, T.S.; Robinson, E.C.; Hacker, C.D.; Harwell, J.; Yacoub, E.; Ugurbil, K.; Andersson, J.; Beckmann, C.F.; Jenkinson, M.; et al. A multi-modal parcellation of human cerebral cortex. Nature 2016, 536, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.-C. Population-based tract-to-region connectome of the human brain and its hierarchical topology. Nat. Commun. 2022, 13, 4933. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Shao, C.; Zhang, Y.; Qiu, W.; Li, W.; Zhu, W.; Yang, Q.; Huang, Y.; Pan, L.; Dong, Y.; et al. A Global Multiregional Proteomic Map of the Human Cerebral Cortex. Genom. Proteom. Bioinform. 2022, 20, 614–632. [Google Scholar] [CrossRef] [PubMed]
- Carlyle, B.; Kitchen, R.R.; Kanyo, J.E.; Voss, E.Z.; Pletikos, M.; Sousa, A.M.M.; Lam, T.T.; Gerstein, M.B.; Sestan, N.; Nairn, A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017, 20, 1787–1795. [Google Scholar] [CrossRef]
- Grasby, K.L.; Jahanshad, N.; Painter, J.N.; Colodro-Conde, L.; Bralten, J.; Hibar, D.P.; Lind, P.A.; Pizzagalli, F.; Ching, C.R.K.; McMahon, M.A.B.; et al. The genetic architecture of the human cerebral cortex. Science 2020, 367, eaay6690. [Google Scholar] [CrossRef]
- Biswas, D.; Shenoy, S.V.; Chetanya, C.; Lachén-Montes, M.; Barpanda, A.; Athithyan, A.P.; Ghosh, S.; Ausín, K.; Zelaya, M.V.; Fernández-Irigoyen, J.; et al. Deciphering the Interregional and Interhemisphere Proteome of the Human Brain in the Context of the Human Proteome Project. J. Proteome Res. 2021, 20, 5280–5293. [Google Scholar] [CrossRef]
- Toikumo, S.; Xu, H.; Gelernter, J.; Kember, R.L.; Kranzler, H.R. Integrating human brain proteomic data with genome-wide association study findings identifies novel brain proteins in substance use traits. Neuropsychopharmacology 2022, 47, 2292–2299. [Google Scholar] [CrossRef]
- Ping, L.; Kundinger, S.R.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. Sci. Data 2020, 7, 315. [Google Scholar] [CrossRef]
- Laszlo, Z.I.; Hindley, N.; Avila, A.S.; Kline, R.A.; Eaton, S.L.; Lamont, D.J.; Smith, C.; Spires-Jones, T.L.; Wishart, T.M.; Henstridge, C.M. Synaptic proteomics reveal distinct molecular signatures of cognitive change and C9ORF72 repeat expansion in the human ALS cortex. Acta Neuropathol. Commun. 2022, 10, 1–20. [Google Scholar] [CrossRef]
- O’Rourke, M.B.; Smith, C.C.; De La Monte, S.M.; Sutherland, G.; Padula, M.; La Monte, S.M. Higher Mass Accuracy MALDI-TOF/TOF Lipid Imaging of Human Brain Tissue in Alzheimer’s Disease. Curr. Protoc. Mol. Biol. 2018, 126, e86. [Google Scholar] [CrossRef]
- Wingo, T.S.; Duong, D.M.; Zhou, M.; Dammer, E.B.; Wu, H.; Cutler, D.J.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Integrating Next-Generation Genomic Sequencing and Mass Spectrometry To Estimate Allele-Specific Protein Abundance in Human Brain. J. Proteome Res. 2017, 16, 3336–3347. [Google Scholar] [CrossRef] [PubMed]
- Kandigian, S.E.; Ethier, E.C.; Kitchen, R.R.; Lam, T.T.; Arnold, S.E.; Carlyle, B.C. Proteomic characterization of post-mortem human brain tissue following ultracentrifugation-based subcellular fractionation. Brain Commun. 2022, 4, fcac103. [Google Scholar] [CrossRef]
- Davalieva, K.; Kiprijanovska, S.; Dimovski, A.; Rosoklija, G.; Dwork, A.J. Comparative evaluation of two methods for LC-MS/MS proteomic analysis of formalin fixed and paraffin embedded tissues. J. Proteom. 2021, 235, 104117. [Google Scholar] [CrossRef]
- Byrling, J.; Kristl, T.; Hu, D.; Pla, I.; Sanchez, A.; Sasor, A.; Andersson, R.; Marko-Varga, G.; Andersson, B. Mass spectrometry-based analysis of formalin-fixed, paraffin-embedded distal cholangiocarcinoma identifies stromal thrombospondin-2 as a potential prognostic marker. J. Transl. Med. 2020, 18, 343. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, S.C.; Bendou, H.; Blignaut, R.J.; Bell, L.; Rigby, J.; Christoffels, A.G. Evaluation of Protein Purification Techniques and Effects of Storage Duration on LC-MS/MS Analysis of Archived FFPE Human CRC Tissues. Pathol. Oncol. Res. 2021, 27, 622855. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Tian, S.; Bai, L.; Lv, Q.; Liu, J.; Liu, J.; Fang, Y.; Zhai, Y.; Shen, Q.; Rao, J.; et al. Comparative proteomic analysis of children FSGS FFPE tissues. BMC Pediatr. 2022, 22, 707. [Google Scholar] [CrossRef]
- Bao, X.; Wu, J.; Shuch, B.; LoRusso, P.; Bindra, R.S.; Li, J. Quantitative Profiling of Oncometabolites in Frozen and Formalin-Fixed Paraffin-Embedded Tissue Specimens by Liquid Chromatography Coupled with Tandem Mass Spectrometry. Sci. Rep. 2019, 9, 11238. [Google Scholar] [CrossRef]
- Cheng, F.; De Luca, A.; Hogan, A.L.; Rayner, S.L.; Davidson, J.M.; Watchon, M.; Stevens, C.H.; Muñoz, S.S.; Ooi, L.; Yerbury, J.J.; et al. Unbiased Label-Free Quantitative Proteomics of Cells Expressing Amyotrophic Lateral Sclerosis (ALS) Mutations in CCNF Reveals Activation of the Apoptosis Pathway: A Workflow to Screen Pathogenic Gene Mutations. Front. Mol. Neurosci. 2021, 14, 627740. [Google Scholar] [CrossRef]
- Li, Z.; Adams, R.M.; Chourey, K.; Hurst, G.B.; Hettich, R.L.; Pan, C. Systematic Comparison of Label-Free, Metabolic Labeling, and Isobaric Chemical Labeling for Quantitative Proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590. [Google Scholar] [CrossRef]
- Yokota, T.; Mishra, M.; Akatsu, H.; Tani, Y.; Miyauchi, T.; Yamamoto, T.; Kosaka, K.; Nagai, Y.; Sawada, T.; Heese, K. Brain site-specific gene expression analysis in Alzheimer’s disease patients. Eur. J. Clin. Investig. 2006, 36, 820–830. [Google Scholar] [CrossRef]
- Kawashima, Y.; Kodera, Y.; Singh, A.; Matsumoto, M.; Matsumoto, H. Efficient extraction of proteins from formalin-fixed paraffin-embedded tissues requires higher concentration of tris(hydroxymethyl)aminomethane. Clin. Proteom. 2014, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Weißer, J.; Biniossek, M.L.; Kuehs, M.; Mayer, B.; Drendel, V.; Timme, S.; Shahinian, H.; Küsters, S.; Wellner, U.F.; et al. Impact of routinely employed procedures for tissue processing on the proteomic analysis of formalin-fixed paraffin-embedded tissue. Proteom.-Clin. Appl. 2014, 8, 796–804. [Google Scholar] [CrossRef]
- Jiang, X.; Jiang, X.; Feng, S.; Tian, R.; Ye, M.; Zou, H. Development of Efficient Protein Extraction Methods for Shotgun Proteome Analysis of Formalin-Fixed Tissues. J. Proteome Res. 2007, 6, 1038–1047. [Google Scholar] [CrossRef]
- Shi, S.-R.; Liu, C.; Balgley, B.M.; Lee, C.; Taylor, C.R. Protein Extraction from Formalin-fixed, Paraffin-embedded Tissue Sections: Quality Evaluation by Mass Spectrometry. J. Histochem. Cytochem. 2006, 54, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Sprung, R.W.; Brock, J.W.C.; Tanksley, J.P.; Li, M.; Washington, M.K.; Slebos, R.J.C.; Liebler, D.C. Equivalence of Protein Inventories Obtained from Formalin-fixed Paraffin-embedded and Frozen Tissue in Multidimensional Liquid Chromatography-Tandem Mass Spectrometry Shotgun Proteomic Analysis. Mol. Cell. Proteom. 2009, 8, 1988. [Google Scholar] [CrossRef]
- Nirmalan, N.J.; Hughes, C.; Peng, J.; McKenna, T.; Langridge, J.; Cairns, D.A.; Harnden, P.; Selby, P.J.; Banks, R.E. Initial Development and Validation of a Novel Extraction Method for Quantitative Mining of the Formalin-Fixed, Paraffin-Embedded Tissue Proteome for Biomarker Investigations. J. Proteome Res. 2010, 10, 896–906. [Google Scholar] [CrossRef]
- Scicchitano, M.S.; Dalmas, D.A.; Boyce, R.W.; Thomas, H.C.; Frazier, K.S. Protein Extraction of Formalin-fixed, Paraffin-embedded Tissue Enables Robust Proteomic Profiles by Mass Spectrometry. J. Histochem. Cytochem. 2009, 57, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Romano, A.; Stanca, E.; La Pesa, V.; Aloisi, L.; De Domenico, S.; Franck, J.; Cicalini, I.; Giudetti, A.; Storelli, E.; et al. Proteomic expression profile of injured rat peripheral nerves revealed biological networks and processes associated with nerve regeneration. J. Cell. Physiol. 2018, 233, 6207–6223. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Sasaki, H.; Terasaki, T. Establishment and validation of highly accurate formalin-fixed paraffin-embedded quantitative proteomics by heat-compatible pressure cycling technology using phase-transfer surfactant and SWATH-MS. Sci. Rep. 2020, 10, 11271. [Google Scholar] [CrossRef]
- Schoffman, H.; Levin, Y.; Itzhaki-Alfia, A.; Tselekovits, L.; Gonen, L.; Vainer, G.W.; Hout-Siloni, G.; Barshack, I.; Cohen, Z.R.; Margalit, N.; et al. Comparison of matched formalin-fixed paraffin embedded and fresh frozen meningioma tissue reveals bias in proteomic profiles. Proteomics 2022, 22, 2200085. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.S.; Ozcan, S.; Ramsey, J.M.; Wang, Z.; Ma’Ayan, A.; Rustogi, N.; Gottschalk, M.G.; Webster, M.J.; Weickert, C.S.; Buxbaum, J.D.; et al. Temporal proteomic profiling of postnatal human cortical development. Transl. Psychiatry 2018, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Körner, S.; Böselt, S.; Thau, N.; Rath, K.J.; Dengler, R.; Petri, S. Differential Sirtuin Expression Patterns in Amyotrophic Lateral Sclerosis (ALS) Postmortem Tissue: Neuroprotective or Neurotoxic Properties of Sirtuins in ALS? Neurodegener. Dis. 2012, 11, 141–152. [Google Scholar] [CrossRef]
- Sharma, A.; Mahur, P.; Muthukumaran, J.; Singh, A.K.; Jain, M. Shedding light on structure, function and regulation of human sirtuins: A comprehensive review. 3 Biotech 2022, 13, 29. [Google Scholar] [CrossRef]
- Canty, A.J.; Greferath, U.; Turnley, A.M.; Murphy, M. Eph tyrosine kinase receptor EphA4 is required for the topographic mapping of the corticospinal tract. Proc. Natl. Acad. Sci. USA 2006, 103, 15629–15634. [Google Scholar] [CrossRef]
- Joshi, A.R.; Muke, I.; Bobylev, I.; Lehmann, H.C. ROCK inhibition improves axonal regeneration in a preclinical model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2019, 527, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Tönges, L.; Günther, R.; Suhr, M.; Jansen, J.; Balck, A.; Saal, K.-A.; Barski, E.; Nientied, T.; Götz, A.A.; Koch, J.-C.; et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 2013, 62, 217–232. [Google Scholar] [CrossRef]
- Vaishnavi, S.N.; Vlassenko, A.G.; Rundle, M.M.; Snyder, A.Z.; Mintun, M.A.; Raichle, M.E. Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 17757–17762. [Google Scholar] [CrossRef]
- Gur, R.C.; Mozley, L.H.; Mozley, P.D.; Resnick, S.M.; Karp, J.S.; Alavi, A.; Arnold, S.E.; Gur, R.E. Sex Differences in Regional Cerebral Glucose Metabolism During a Resting State. Science 1995, 267, 528–531. [Google Scholar] [CrossRef]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’Andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chayer, C.; Freedman, M. Frontal lobe functions. Curr. Neurol. Neurosci. Rep. 2001, 1, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Prefrontal Cortical Opioids and Dysregulated Motivation: A Network Hypothesis. Trends Neurosci. 2016, 39, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Seney, M.L.; Kim, S.-M.; Glausier, J.R.; Hildebrand, M.A.; Xue, X.; Zong, W.; Wang, J.; Shelton, M.A.; Phan, B.N.; Srinivasan, C.; et al. Transcriptional Alterations in Dorsolateral Prefrontal Cortex and Nucleus Accumbens Implicate Neuroinflammation and Synaptic Remodeling in Opioid Use Disorder. Biol. Psychiatry 2021, 90, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.M.; Wong, C.T.; Li, H.; Crawford, D.A. Prostaglandin E2 facilitates subcellular translocation of the EP4 receptor in neuroectodermal NE-4C stem cells. Biochem. Biophys. Rep. 2016, 7, 173–179. [Google Scholar] [CrossRef]
- Rai-Bhogal, R.; Wong, C.; Kissoondoyal, A.; Davidson, J.; Li, H.; Crawford, D.A. Maternal exposure to prostaglandin E2 modifies expression of Wnt genes in mouse brain—An autism connection. Biochem. Biophys. Rep. 2018, 14, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.M.; Wong, C.T.; Rai-Bhogal, R.; Li, H.; Crawford, D.A. Prostaglandin E2 elevates calcium in differentiated neuroectodermal stem cells. Mol. Cell. Neurosci. 2016, 74, 71–77. [Google Scholar] [CrossRef]
- De Keijzer, S.; Meddens, M.B.M.; Torensma, R.; Cambi, A. The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle. Int. J. Mol. Sci. 2013, 14, 6542–6555. [Google Scholar] [CrossRef]
- Sathe, G.; Albert, M.; Darrow, J.; Saito, A.; Troncoso, J.; Pandey, A.; Moghekar, A. Quantitative proteomic analysis of the frontal cortex in Alzheimer’s disease. J. Neurochem. 2020, 156, 988–1002. [Google Scholar] [CrossRef]
- Etheridge, N.; Lewohl, J.M.; Mayfield, R.D.; Harris, R.A.; Dodd, P.R. Synaptic proteome changes in the superior frontal gyrus and occipital cortex of the alcoholic brain. Proteom.-Clin. Appl. 2009, 3, 730–742. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidson, J.M.; Rayner, S.L.; Liu, S.; Cheng, F.; Di Ieva, A.; Chung, R.S.; Lee, A. Inter-Regional Proteomic Profiling of the Human Brain Using an Optimized Protein Extraction Method from Formalin-Fixed Tissue to Identify Signaling Pathways. Int. J. Mol. Sci. 2023, 24, 4283. https://doi.org/10.3390/ijms24054283
Davidson JM, Rayner SL, Liu S, Cheng F, Di Ieva A, Chung RS, Lee A. Inter-Regional Proteomic Profiling of the Human Brain Using an Optimized Protein Extraction Method from Formalin-Fixed Tissue to Identify Signaling Pathways. International Journal of Molecular Sciences. 2023; 24(5):4283. https://doi.org/10.3390/ijms24054283
Chicago/Turabian StyleDavidson, Jennilee M., Stephanie L. Rayner, Sidong Liu, Flora Cheng, Antonio Di Ieva, Roger S. Chung, and Albert Lee. 2023. "Inter-Regional Proteomic Profiling of the Human Brain Using an Optimized Protein Extraction Method from Formalin-Fixed Tissue to Identify Signaling Pathways" International Journal of Molecular Sciences 24, no. 5: 4283. https://doi.org/10.3390/ijms24054283
APA StyleDavidson, J. M., Rayner, S. L., Liu, S., Cheng, F., Di Ieva, A., Chung, R. S., & Lee, A. (2023). Inter-Regional Proteomic Profiling of the Human Brain Using an Optimized Protein Extraction Method from Formalin-Fixed Tissue to Identify Signaling Pathways. International Journal of Molecular Sciences, 24(5), 4283. https://doi.org/10.3390/ijms24054283