

The Central Role of the NAD+ Molecule in the Development of Aging and the Prevention of Chronic Age-Related Diseases: Strategies for NAD+ Modulation

{kind=link}
{kind=link}
Abstract
:1. Introduction
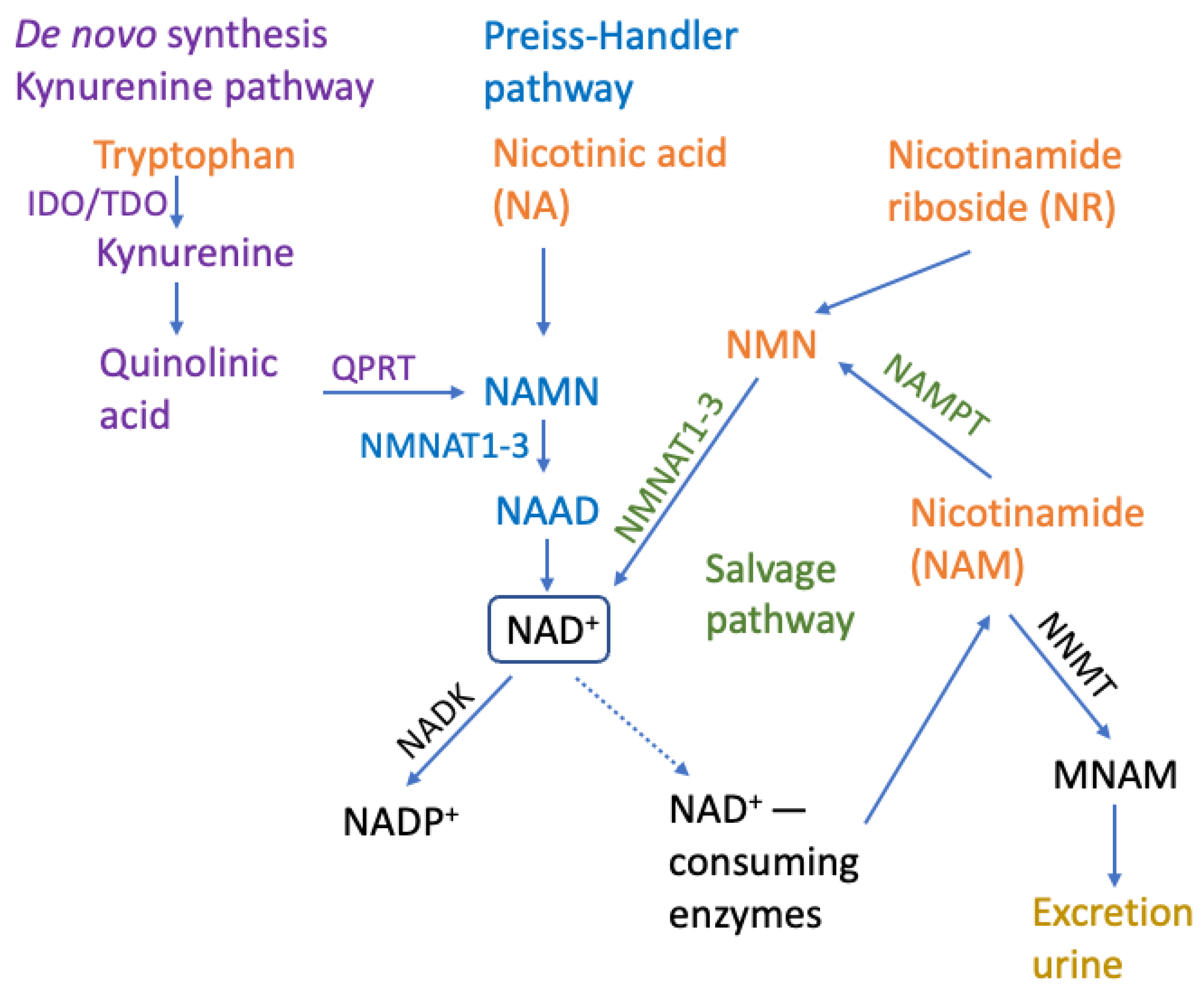
2. NAD+ Usage and Degradation
3. Consequences of Decreased NAD+ Levels
4. Methods to Increase NAD+ Levels
5. Beneficial Effects of NAD+ Boosting
6. The Effects of NAD+ on Cancer and Inflammation
7. Side Effects of NAD+ Precursors Observed in Human Trials
8. The Future Research Directions and Strategies
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hong, W.; Mo, F.; Zhang, Z.; Huang, M.; Wei, X. Nicotinamide Mononucleotide: A Promising Molecule for Therapy of Diverse Diseases by Targeting NAD+ Metabolism. Front. Cell Dev. Biol. 2020, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B. NAD+ in Cancer Prevention and Treatment: Pros and Cons. J. Clin. Exp. Oncol. 2016, 5, 4. [Google Scholar] [CrossRef]
- Imai, S.I.; Guarente, L. Ten years of NAD-dependent SIR2 family deacetylases: Implications for metabolic diseases. Trends Pharmacol. Sci. 2010, 31, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef]
- Lavery, G.G.; Oakey, L.A.; Fletcher, R.S.; Elhassan, Y.S.; Cartwright, D.M.; Doig, C.L.; Garten, A.; Thakker, A.; Maddocks, O.D.K.; Zhang, T.; et al. Metabolic tracing reveals novel adaptations to skeletal muscle cell energy production pathways in response to NAD + depletion. Wellcome Open Res. 2019, 3, 147. [Google Scholar] [CrossRef]
- Gallo, C.M.; Daniel, L.; Smith, J.; Smith, J.S. Nicotinamide Clearance by Pnc1 Directly Regulates Sir2-Mediated Silencing and Longevity. Mol. Cell. Biol. 2004, 24, 1301. [Google Scholar] [CrossRef]
- Sun, W.P.; Li, D.; Lun, Y.Z.; Gong, X.J.; Sun, S.X.; Guo, M.; Jing, L.X.; Zhang, L.B.; Xiao, F.C.; Zhou, S.S. Excess nicotinamide inhibits methylation-mediated degradation of catecholamines in normotensives and hypertensives. Hypertens. Res. 2012, 35, 180–185. [Google Scholar] [CrossRef]
- Ostrakhovitch, E.A.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Conze, D.; Brenner, C.; Kruger, C.L. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci. Rep. 2019, 9, 9772. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Sauve, A.A. Nicotinamide riboside, a trace nutrient in foods, is a Vitamin B3 with effects on energy metabolism and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Cambronne, X.; Kraus, W. Location, Location, Location: Compartmentalization of NAD + Synthesis and Functions in Mammalian Cells. Trends Biochem. Sci. 2020, 45, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.I. The NAD world: A new systemic regulatory network for metabolism and aaging-Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem. Biophys. 2009, 53, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.A.; Forouhar, F.; Tao, X.; Tong, L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin. Ther. Targets 2007, 11, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. Therapeutic potential of NAD+ for neurological diseases. Future Neurol. 2007, 2, 129–132. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Role of free radicals in aging and disease. Ann. N. Y. Acad. Sci. 1992, 673, 126–141. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef]
- Schultz, M.B.; Sinclair, D.A. Why NAD + Declines during Aging: It’s Destroyed. Cell Metab. 2016, 23, 965–966. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.S.; Andersen, C.; Daoud, M.; Anderson, K.A.; Laursen, J.S.; Chakladar, S.; Huynh, F.K.; Colaço, A.R.; Backos, D.S.; Fristrup, P.; et al. Investigating the Sensitivity of NAD+-dependent Sirtuin Deacylation Activities to NADH. J. Biol. Chem. 2016, 291, 7128–7141. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 2009, 394, 101–109. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef]
- Amé, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; De Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Gonzalez, R.; Mendoza-Alvarez, H. Dissection of ADP-ribose polymer synthesis into individual steps of initiation, elongation, and branching. Biochimie 1995, 77, 403–407. [Google Scholar] [CrossRef]
- Cakir-Kiefer, C.; Muller-Steffner, H.; Oppenheimer, N.; Schuber, F. Kinetic competence of the cADP-ribose-CD38 complex as an intermediate in the CD38/NAD+ glycohydrolase-catalysed reactions: Implication for CD38 signalling. Biochem. J. 2001, 358, 399–406. [Google Scholar] [CrossRef]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Nicotinamide Adenine Dinucleotide Metabolism and Neurodegeneration. Antioxid. Redox Signal. 2018, 28, 1652–1668. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD + Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Altmeyer, M.; Hottiger, M.O. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging 2009, 1, 458–469. [Google Scholar] [CrossRef]
- Kulikova, V.; Shabalin, K.; Nerinovski, K.; Dölle, C.; Niere, M.; Yakimov, A.; Redpath, P.; Khodorkovskiy, M.; Migaud, M.E.; Ziegler, M.; et al. Generation, release, and uptake of the NAD precursor nicotinic acid riboside by human cells. J. Biol. Chem. 2015, 290, 27124–27137. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I. NAD+ as the Link between Oxidative Stress, Inflammation, Caloric Restriction, Exercise, DNA Repair, Longevity, and Health Span. Rejuvenation Res. 2016, 19, 406–413. [Google Scholar] [CrossRef]
- Johnson, S.; Imai, S. NAD+ biosynthesis, aging, and disease. F1000Research 2018, 7, 132. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Lee, H.; Min, K. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.I.; Guarente, L. It takes two to tango: Nad+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta Proteins Proteomics 2016, 1864, 1787–1800. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- De Grey, A.D.N.J. A mechanism proposed to explain the rise in oxidative stress during aging. J. Anti. Aging. Med. 1998, 1, 53–66. [Google Scholar] [CrossRef]
- Brewer, G.J. Epigenetic oxidative redox shift (EORS) theory of aging unifies the free radical and insulin signaling theories. Exp. Gerontol. 2010, 45, 173–179. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Poljsak, B.; Kovac, V.; Dahmane, R.; Levec, T.; Starc, A. Cancer Etiology: A Metabolic Disease Originating from Life’s Major Evolutionary Transition? Hindawi Limited: London, UK, 2019; Volume 2019. [Google Scholar]
- Ying, W. NAD+ and NADH in cellular functions and cell death. Front. Biosci. 2006, 11, 3129–3148. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Yoshino, J. Adipose tissue NAD+ biology in obesity and insulin resistance: From mechanism to therapy. BioEssays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Mills, K.F.; Satoh, A.; Imai, S.I. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 2008, 7, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Yamagishi, S. Advanced Glycation End Products and Insulin Resistance. Curr. Pharm. Des. 2008, 14, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.; Malik, A.; Butt, T.T.; Ashraf, M.A.B.; Rasool, R.; Zahid, A.; Waquar, S.; Asif, M.; Zaheer, A.; Jabbar, A.; et al. Implications of advanced oxidation protein products (AOPPs), advanced glycation end products (AGEs) and other biomarkers in the development of cardiovascular diseases. Saudi J. Biol. Sci. 2019, 26, 334–339. [Google Scholar] [CrossRef]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z.; Luca, M. Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes. Oxid. Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, H.; Grueter, C.; Collins, A.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, N.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef]
- Pirinen, E.; Auranen, M.; Khan, N.A.; Brilhante, V.; Urho, N.; Pessia, A.; Hakkarainen, A.; Kuula, J.; Heinonen, U.; Schmidt, M.S.; et al. Niacin Cures Systemic NAD+ Deficiency and Improves Muscle Performance in Adult-Onset Mitochondrial Myopathy. Cell Metab. 2020, 31, 1078–1090.e5. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Rechsteiner, M.; Hillyard, D.; Olivera, B.M. Turnover of nicotinamide adenine dinucleotide in cultures of human cells. J. Cell. Physiol. 1976, 88, 207–217. [Google Scholar] [CrossRef]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD+. Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef]
- Ryu, K.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.; Kraus, W. Metabolic regulation of transcription through compartmentalized NAD + biosynthesis. Science 2018, 360, eaan5780. [Google Scholar] [CrossRef]
- Sallin, O.; Reymond, L.; Gondrand, C.; Raith, F.; Koch, B.; Johnsson, K. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. eLife 2018, 7, e32638. [Google Scholar] [CrossRef]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ Metabolism in Health and Disease; Elsevier Ltd.: Amsterdam, The Netherlands, 2007; Volume 32, pp. 12–19. [Google Scholar]
- Stein, L.R.; Imai, S.I. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ homeostasis in man. Cell. Mol. Life Sci. 2004, 61, 19–34. [Google Scholar] [CrossRef]
- Aksoy, P.; Escande, C.; White, T.A.; Thompson, M.; Soares, S.; Benech, J.C.; Chini, E.N. Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006, 349, 353–359. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.M.A.M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.S.M.A.M.S.; Brenner, C. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.W.W.; Loro, E.; Liu, L.; Davila, A.; Chellappa, K.; Silverman, I.M.M.; Quinn, W.J.J.; Gosai, S.J.J.; Tichy, E.D.D.; Davis, J.G.G.; et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016, 24, 269–282. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Partial reversal of skeletal muscle aging by restoration of normal NAD + levels. Rejuvenation Res. 2014, 17, 62–69. [Google Scholar] [CrossRef]
- Wu, L.E.; Sinclair, D.A. Restoring stem cells-all you need is NAD+. Cell Res. 2016, 26, 971–972. [Google Scholar] [CrossRef]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef]
- Nóbrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Brown, K.D.; Maqsood, S.; Huang, J.Y.; Pan, Y.; Harkcom, W.; Li, W.; Sauve, A.; Verdin, E.; Jaffrey, S.R. Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014, 20, 1059–1068. [Google Scholar] [CrossRef]
- Poljsak, B.; Kovač, V.; Milisav, I. Healthy Lifestyle Recommendations: Do the Beneficial Effects Originate from NAD+Amount at the Cellular Level? Oxid. Med. Cell. Longev. 2020, 2020, 8819627. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Yiasemides, E.; Sivapirabu, G.; Halliday, G.M.; Park, J.; Damian, D.L. Oral nicotinamide protects against ultraviolet radiation-induced immunosuppression in humans. Carcinogenesis 2009, 30, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Barbosa, M.T.P.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Energy metabolism, altered proteins, sirtuins and ageing: Converging mechanisms? Biogerontology 2008, 9, 49–55. [Google Scholar] [CrossRef]
- Morris, K.C.; Lin, H.W.; Thompson, J.W.; Perez-Pinzon, M.A. Pathways for ischemic cytoprotection: Role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J. Cereb. Blood Flow Metab. 2011, 31, 1003–1019. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, 10. [Google Scholar] [CrossRef]
- Curtin, N. PARP inhibitors for anticancer therapy. Biochem. Soc. Trans. 2014, 42, 82–88. [Google Scholar] [CrossRef]
- Prolla, T.A.; Denu, J.M. NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab. 2014, 19, 178–180. [Google Scholar] [CrossRef] [Green Version]
- Dollerup, O.; Chubanava, S.; Agerholm, M.; Søndergård, S.; Altıntaş, A.; Møller, A.; Høyer, K.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Lavery, G.; et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 2020, 598, 731–754. [Google Scholar] [CrossRef]
- Remie, C.M.E.; Roumans, K.H.M.; Moonen, M.P.B.; Connell, N.J.; Havekes, B.; Mevenkamp, J.; Lindeboom, L.; Wit, D.; Van De Weijer, T.; Arts, S.; et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am. J. Clin. Nutr. 2020, 112, 413–426. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-Tolerated and elevates NAD+ in healthy middle-Aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Kallio, K.; Hätönen, K.; Lehto, M.; Salomaa, V.; Männistö, S.; Pussinen, P. Endotoxemia, nutrition, and cardiometabolic disorders. Acta Diabetol. 2015, 52, 395–404. [Google Scholar] [CrossRef]
- Lee, C.; Song, E.; Yoo, C.; Kwak, Y.; Han, M. Lipopolysaccharide induces CD38 expression and solubilization in J774 macrophage cells. Mol. Cells 2012, 34, 573–576. [Google Scholar] [CrossRef]
- Kim, K.A.; Jeong, J.J.; Yoo, S.Y.; Kim, D.H. Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC Microbiol. 2016, 16, 9. [Google Scholar] [CrossRef]
- Ghosh, S.; Lertwattanarak, R.; Garduño Jde, J.; Galeana, J.; Li, J.; Zamarripa, F.; Lancaster, J.; Mohan, S.; Hussey, S.; Musi, N. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2015, 70, 232–246. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Grozio, A.; Sociali, G.; Sturla, I.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef]
- Kellenberger, E.; Kuhn, I.; Schuber, F.; Muller-Steffner, H. Flavonoids as inhibitors of human CD38. Bioorg. Med. Chem. Lett. 2011, 21, 3939–3942. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef]
- Haffner, C.D.; Becherer, J.D.; Boros, E.E.; Cadilla, R.; Carpenter, T.; Cowan, D.; Deaton, D.N.; Guo, Y.; Harrington, W.; Henke, B.R.; et al. Discovery, synthesis, and biological evaluation of thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J. Med. Chem. 2015, 58, 3548–3571. [Google Scholar] [CrossRef]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Ferracin, M.; Ghimenti, C.; Massaia, M.; Horenstein, A.; Malavasi, F. Anti-CD38 antibody therapy: Windows of opportunity yielded by the functional characteristics of the target molecule. Mol. Med. 2013, 19, 99–108. [Google Scholar] [CrossRef]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef]
- Gardell, S.J.; Hopf, M.; Khan, A.; Dispagna, M.; Hampton Sessions, E.; Falter, R.; Kapoor, N.; Brooks, J.; Culver, J.; Petucci, C.; et al. Boosting NAD+ with a small molecule that activates NAMPT. Nat. Commun. 2019, 10, 3421. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhu, T.; Zhou, P.; Xu, H.; Meng, X.; Ding, T.; Nan, F.; Sun, G.; Sun, X. Notoginseng Leaf Triterpenes Ameliorates OGD/R-Induced Neuronal Injury via SIRT1/2/3-Foxo3a-MnSOD/PGC-1 α Signaling Pathways Mediated by the NAMPT-NAD Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 7308386. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhu, T.; Zhou, P.; Xu, H.; Meng, X.; Ding, T.; Nan, F.; Sun, G.; Sun, X. Notoginseng leaf triterpenes ameliorates mitochondrial oxidative injury via the NAMPT-SIRT1/2/3 signaling pathways in cerebral ischemic model rats. J. Ginseng Res. 2020. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Son, M.; Kerek, E.; Cromwell, C.R.; Wingert, B.M.; Wu, K.; Jovel, J.; Camacho, C.J.; Hubbard, B.P.; Wu, J. Tripeptide IRW Upregulates NAMPT Protein Levels in Cells and Obese C57BL/6J Mice. J. Agric. Food Chem. 2021, 69, 1555–1566. [Google Scholar] [CrossRef]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose Restriction Inhibits Skeletal Myoblast Differentiation by Activating SIRT1 through AMPK-Mediated Regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef]
- Yang, Y.; Cimen, H.; Han, M.J.; Shi, T.; Deng, J.H.; Koc, H.; Palacios, O.M.; Montier, L.; Bai, Y.; Tong, Q.; et al. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J. Biol. Chem. 2010, 285, 7417–7429. [Google Scholar] [CrossRef] [Green Version]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Costford, S.R.; Bajpeyi, S.; Pasarica, M.; Albarado, D.C.; Thomas, S.C.; Xie, H.; Church, T.S.; Jubrias, S.A.; Conley, K.E.; Smith, S.R. Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E117. [Google Scholar] [CrossRef] [PubMed]
- Van Haren, M.J.; Zhang, Y.; Thijssen, V.; Buijs, N.; Gao, Y.; Mateuszuk, L.; Fedak, F.A.; Kij, A.; Campagna, R.; Sartini, D.; et al. Macrocyclic peptides as allosteric inhibitors of nicotinamide N-methyltransferase (NNMT). RSC Chem. Biol. 2021, 2, 1546–1555. [Google Scholar] [CrossRef]
- Lee, H.Y.; Suciu, R.M.; Horning, B.D.; Vinogradova, E.V.; Ulanovskaya, O.A.; Cravatt, B.F. Covalent inhibitors of nicotinamide N-methyltransferase (NNMT) provide evidence for target engagement challenges in situ. Bioorg. Med. Chem. Lett. 2018, 28, 2682–2687. [Google Scholar] [CrossRef] [PubMed]
- Policarpo, R.L.; Decultot, L.; May, E.; Kuzmič, P.; Carlson, S.; Huang, D.; Chu, V.; Wright, B.A.; Dhakshinamoorthy, S.; Kannt, A.; et al. High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). J. Med. Chem. 2019, 62, 9837–9873. [Google Scholar] [CrossRef]
- Gao, Y.; Van Haren, M.J.; Moret, E.E.; Rood, J.J.M.; Sartini, D.; Salvucci, A.; Emanuelli, M.; Craveur, P.; Babault, N.; Jin, J.; et al. Bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) with enhanced activity. J. Med. Chem. 2019, 62, 6597–6614. [Google Scholar] [CrossRef]
- Gao, Y.; Van Haren, M.J.; Buijs, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W.; et al. Potent Inhibition of Nicotinamide N-Methyltransferase by Alkene-Linked Bisubstrate Mimics Bearing Electron Deficient Aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef]
- van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; de Castilla, P.E.M.; et al. Esterase-Sensitive Prodrugs of a Potent Bisubstrate Inhibitor of Nicotinamide N-Methyltransferase (NNMT) Display Cellular Activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef]
- Lu, X.M.; Long, H. Nicotinamide N-methyltransferase as a potential marker for cancer. Neoplasma 2018, 65, 656–663. [Google Scholar] [CrossRef]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.Y.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboums, R.M. Human Liver Nicotinamide N-Methyltransferase. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; De Oliveira, R.M.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Brandauer, J.; Vienberg, S.G.; Andersen, M.A.; Ringholm, S.; Risis, S.; Larsen, P.S.; Kristensen, J.M.; Frøsig, C.; Leick, L.; Fentz, J.; et al. AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J. Physiol. 2013, 591, 5207–5220. [Google Scholar] [CrossRef] [PubMed]
- de Guia, R.M.; Agerholm, M.; Nielsen, T.S.; Consitt, L.A.; Søgaard, D.; Helge, J.W.; Larsen, S.; Brandauer, J.; Houmard, J.A.; Treebak, J.T. Aerobic and resistance exercise training reverses age-dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiol. Rep. 2019, 7, e14139. [Google Scholar] [CrossRef]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S. Emerging therapeutic roles for NAD + metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 5, 25. [Google Scholar] [CrossRef]
- Wu, L.E.; Gomes, A.P.; Sinclair, D.A. Geroncogenesis: Metabolic changes during aging as a driver of tumorigenesis. Cancer Cell 2014, 25, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Gomes, A.L.; Yilmaz, M.; Graña, O.; Bakiri, L.; Ruppen, I.; Ximénez-Embún, P.; Sheshappanavar, V.; Rodriguez-Justo, M.; Pisano, D.G.; et al. Inhibition of De Novo NAD+ Synthesis by Oncogenic URI Causes Liver Tumorigenesis through DNA Damage. Cancer Cell 2014, 26, 826–839. [Google Scholar] [CrossRef]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investig. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [Green Version]
- Matasic, D.S.; Brenner, C.; London, B. Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease. Am. J. Physiol. Hear. Circ. Physiol. 2018, 314, H839–H852. [Google Scholar] [CrossRef] [PubMed]
- Mericskay, M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch. Cardiovasc. Dis. 2016, 109, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.S.; Abraham, D.M.; Hershberger, K.A.; Bhatt, D.P.; Mao, L.; Cui, H.; Liu, J.; Liu, X.; Muehlbauer, M.J.; Grimsrud, P.A.; et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2017, 2, e93885. [Google Scholar] [CrossRef] [PubMed]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; ichiro Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Csiszar, A.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Kiss, T.; Farkas, E.; Baur, J.A.; Ungvari, Z. Role of endothelial NAD + deficiency in age-related vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1253–H1266. [Google Scholar] [CrossRef]
- Ying, W. NAD+ and NADH in brain functions, brain diseases and brain aging. Front. Biosci. 2007, 12, 1863–1888. [Google Scholar] [CrossRef]
- Hikosaka, K.; Yaku, K.; Okabe, K.; Nakagawa, T. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr. Neurosci. 2021, 24, 371–383. [Google Scholar] [CrossRef]
- Klaidman, L.; Morales, M.; Kem, S.; Yang, J.; Chang, M.L.; Adams, J.D. Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology 2003, 69, 150–157. [Google Scholar] [CrossRef]
- Ugur, S.; Ulu, R.; Dogukan, A.; Gurel, A.; Yigit, I.P.; Gozel, N.; Aygen, B.; Ilhan, N. The renoprotective effect of curcumin in cisplatin-induced nephrotoxicity. Ren. Fail. 2015, 37, 332–336. [Google Scholar] [CrossRef]
- Zhuo, L.; Fu, B.; Bai, X.; Zhang, B.; Wu, L.; Cui, J.; Cui, S.; Wei, R.; Chen, X.; Cai, G. NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell. Physiol. Biochem. 2011, 27, 681–690. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Damian, D.L. Nicotinamide and the skin. Australas. J. Dermatol. 2014, 55, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Jukarainen, S.; Heinonen, S.; Rämö, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity is associated with low nad+/sirt pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Drew, J.E.; Farquharson, A.J.; Horgan, G.W.; Williams, L.M. Tissue-specific regulation of sirtuin and nicotinamide adenine dinucleotide biosynthetic pathways identified in C57Bl/6 mice in response to high-fat feeding. J. Nutr. Biochem. 2016, 37, 20–29. [Google Scholar] [CrossRef]
- Nielsen, K.N.; Peics, J.; Ma, T.; Karavaeva, I.; Dall, M.; Chubanava, S.; Basse, A.L.; Dmytriyeva, O.; Treebak, J.T.; Gerhart-Hines, Z. NAMPT-mediated NAD + biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol. Metab. 2018, 11, 178–188. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430. [Google Scholar] [CrossRef]
- North, B.J.; Rosenberg, M.A.; Jeganathan, K.B.; Hafner, A.V.; Michan, S.; Dai, J.; Baker, D.J.; Cen, Y.; Wu, L.E.; Sauve, A.A.; et al. SIRT 2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014, 33, 1438–1453. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Gowda, G.A.N.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I. Vitamin B3 forms as precursors to NAD+: Are they safe? Trends Food Sci. Technol. 2018, 79, 198–203. [Google Scholar] [CrossRef]
- Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.S.; Kobor, M.S.; Horvath, S. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef]
- Tummala, K.S.; Djouder, N. Oncogene-induced NAD(+) depletion in tumorigenesis. Oncoscience 2015, 2, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.L.; Dame, A.J.; Pyrek, J.S.; Jacobson, M.K. Evaluating the role of niacin in human carcinogenesis. Biochimie 1995, 77, 394–398. [Google Scholar] [CrossRef]
- Smith, J.S. Human Sir2 and the “silencing” of p53 activity. Trends Cell Biol. 2002, 12, 404–406. [Google Scholar] [CrossRef]
- Tucci, P. Caloric restriction: Is mammalian life extension linked to p53? Aging 2012, 4, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.R.; Sheng, Y.; Pero, R.W.; Chaplin, D.J.; Horsman, M.R. DNA damage and repair in tumour and non-tumour tissues of mice induced by nicotinamide. Br. J. Cancer 1996, 74, 368. [Google Scholar] [CrossRef]
- Yang, T.; Sauve, A.A. NAD metabolism and sirtuins: Metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 2006, 8, E632. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. Interference between PARPs and SIRT1: A novel approach to healthy ageing? Aging 2011, 3, 543–547. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.T.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef]
- Wang, F.; Nguyen, M.; Qin, F.X.F.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, D.W.; Park, J.H.; Kim, S.J.; Lee, C.H.; Yong, J.I.; Ryu, E.J.; Cho, S.B.; Yeo, H.J.; Hyeon, J.; et al. PEP-1-SIRT2 inhibits inflammatory response and oxidative stress-induced cell death via expression of antioxidant enzymes in murine macrophages. Free Radic. Biol. Med. 2013, 63, 432–445. [Google Scholar] [CrossRef]
- Rawling, J.M.; Jackson, T.M.; Driscoll, E.R.; Kirkland, J.B. Dietary niacin deficiency lowers tissue poly(ADP-ribose) and NAD+ concentrations in Fischer-344 rats. J. Nutr. 1994, 124, 1597–1603. [Google Scholar] [CrossRef]
- Wright, S.C.; Wei, Q.S.; Kinder, D.H.; Larrick, J.W. Biochemical pathways of apoptosis: Nicotinamide adenine dinucleotide-deficient cells are resistant to tumor necrosis factor or ultraviolet light activation of the 24-kD apoptotic protease and DNA fragmentation. J. Exp. Med. 1996, 183, 463–471. [Google Scholar] [CrossRef]
- Roth, M.; Chen, W.Y. Sorting out functions of sirtuins in cancer. Oncogene 2014, 33, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, W.Y. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer 2013, 4, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. How does SIRT1 affect metabolism, senescence and cancer? Nat. Rev. Cancer 2009, 9, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Milisav, I. The Role of Antioxidants in Cancer, Friends or Foes? Curr. Pharm. Des. 2019, 24, 5234–5244. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an Endothelial NAD+-H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89. [Google Scholar] [CrossRef]
- Liu, T.F.; Brown, C.M.; El Gazzar, M.; McPhail, L.; Millet, P.; Rao, A.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. Fueling the flame: Bioenergy couples metabolism and inflammation. J. Leukoc. Biol. 2012, 92, 499. [Google Scholar] [CrossRef]
- Li, L.; Chen, Z.; Fu, W.; Cai, S.; Zeng, Z. Emerging evidence concerning the role of sirtuins in sepsis. Crit. Care Res. Pract. 2018, 2018, 8. [Google Scholar] [CrossRef]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD + decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Poljšak, B.; Kovač, V.; Milisav, I. Current Uncertainties and Future Challenges Regarding NAD+ Boosting Strategies. Antioxidants 2022, 11, 1637. [Google Scholar] [CrossRef]
- Braidy, N.; Liu, Y. NAD+ therapy in age-related degenerative disorders: A benefit/risk analysis. Exp. Gerontol. 2020, 132, 110831. [Google Scholar]
- Dragovic, J.; Kim, S.H.; Brown, S.L.; Kim, J.H. Nicotinamide pharmacokinetics in patients. Radiother. Oncol. 1995, 36, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.T.; Gillmor, H.A.; McLean, A.E.M.; Bingley, P.J.; Gale, E.A.M. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Kourtzidis, I.A.; Stoupas, A.T.; Gioris, I.S.; Veskoukis, A.S.; Margaritelis, N.V.; Tsantarliotou, M.; Taitzoglou, I.; Vrabas, I.S.; Paschalis, V.; Kyparos, A.; et al. The NAD+ precursor nicotinamide riboside decreases exercise performance in rats. J. Int. Soc. Sports Nutr. 2016, 13, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.; Bartoli, W.P.; Eddy, D.E.; Horn, M.K. Physiological and performance responses to nicotinic-acid ingestion during exercise. Med. Sci. Sports Exerc. 1995, 27, 1057–1062. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, Y.; Wang, D.; Zhang, X.; Hao, X.; Hu, M. Nicotinamide mononucleotide supplementation enhances aerobic capacity in amateur runners: A randomized, double-blind study. J. Int. Soc. Sports Nutr. 2021, 18, 54. [Google Scholar] [CrossRef]
- Igarashi, M.; Nakagawa-Nagahama, Y.; Miura, M.; Kashiwabara, K.; Yaku, K.; Sawada, M.; Sekine, R.; Fukamizu, Y.; Sato, T.; Sakurai, T.; et al. Chronic nicotinamide mononucleotide supplementation elevates blood nicotinamide adenine dinucleotide levels and alters muscle function in healthy older men. NPJ Aging 2022, 8, 1–11. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J.; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I. Restoring NAD(+) Levels with NAD(+) intermediates, the second law of thermodynamics, and aging delay. Rejuvenation Res. 2018, 21, 506–509. [Google Scholar] [CrossRef]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD+/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 2020, 12, 11325–11336. [Google Scholar] [CrossRef]
- Khaidizar, F.D.; Bessho, Y.; Nakahata, Y. Nicotinamide phosphoribosyltransferase as a key molecule of the aging/senescence process. Int. J. Mol. Sci. 2021, 22, 3709. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poljšak, B.; Kovač, V.; Špalj, S.; Milisav, I. The Central Role of the NAD+ Molecule in the Development of Aging and the Prevention of Chronic Age-Related Diseases: Strategies for NAD+ Modulation. Int. J. Mol. Sci. 2023, 24, 2959. https://doi.org/10.3390/ijms24032959
Poljšak B, Kovač V, Špalj S, Milisav I. The Central Role of the NAD+ Molecule in the Development of Aging and the Prevention of Chronic Age-Related Diseases: Strategies for NAD+ Modulation. International Journal of Molecular Sciences. 2023; 24(3):2959. https://doi.org/10.3390/ijms24032959
Chicago/Turabian StylePoljšak, Borut, Vito Kovač, Stjepan Špalj, and Irina Milisav. 2023. "The Central Role of the NAD+ Molecule in the Development of Aging and the Prevention of Chronic Age-Related Diseases: Strategies for NAD+ Modulation" International Journal of Molecular Sciences 24, no. 3: 2959. https://doi.org/10.3390/ijms24032959