
Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer
 , , ,
, , ,  and
and 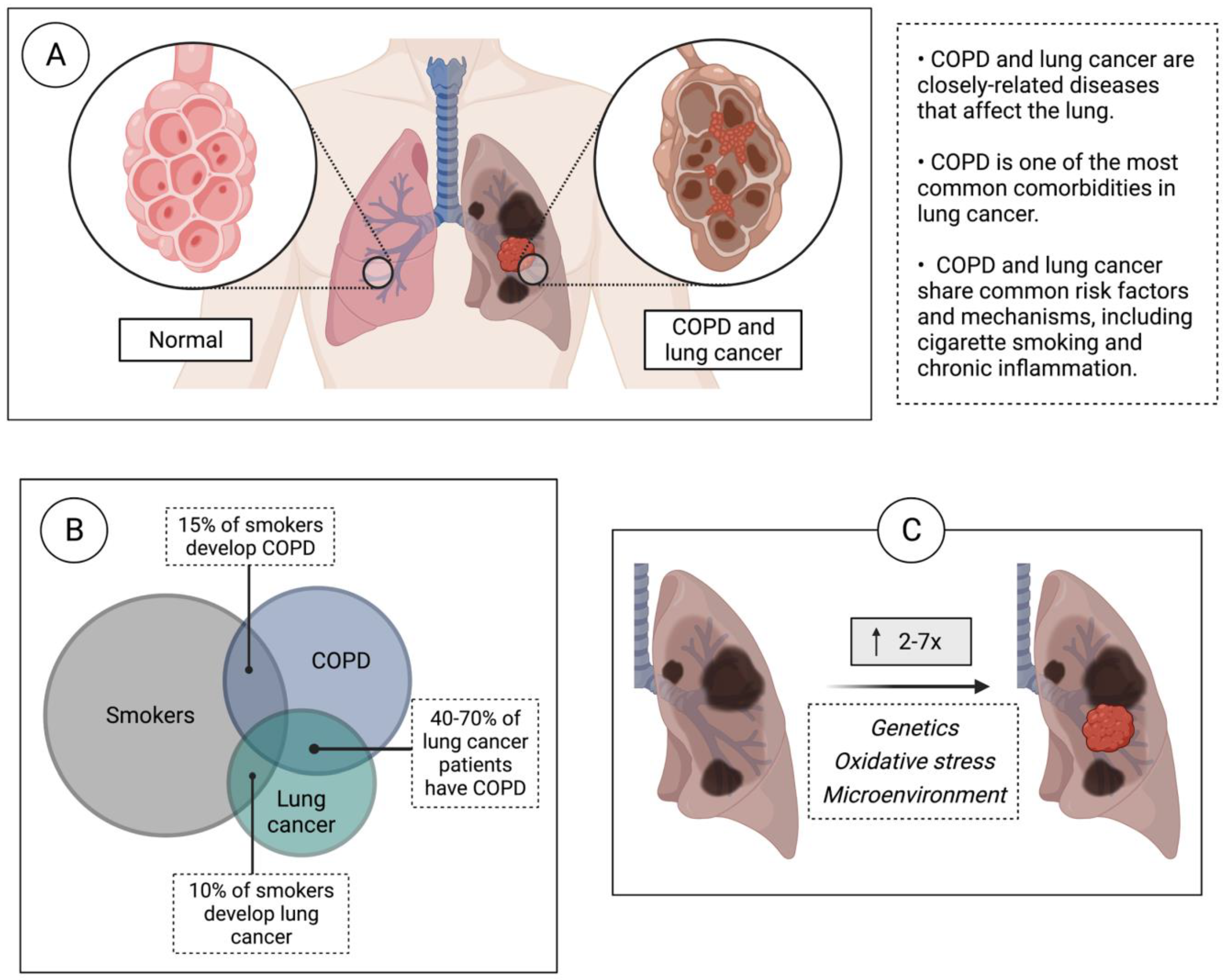{kind=link}
{kind=link}
{kind=link}
{kind=link}
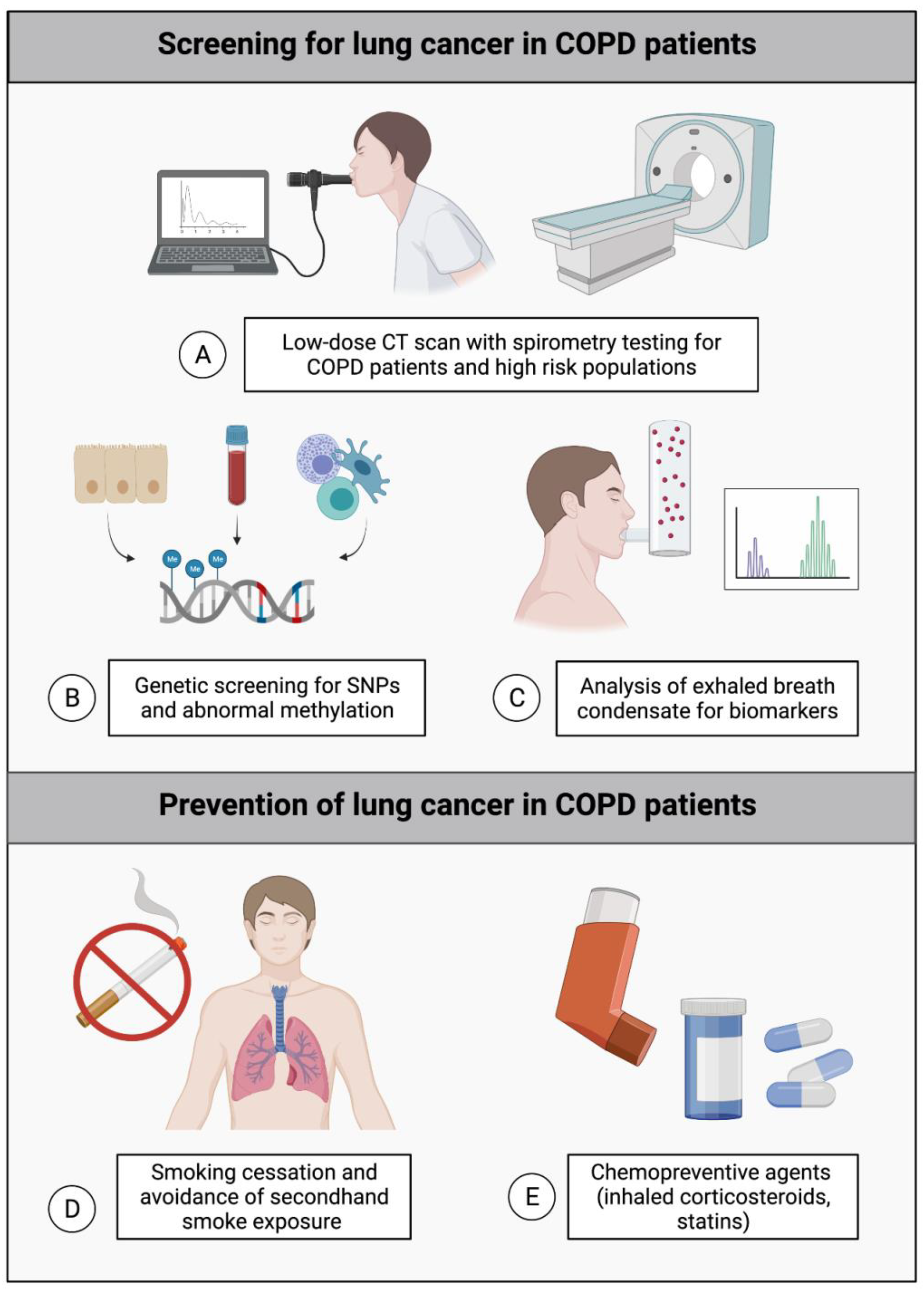{kind=link}
Abstract
1. Introduction
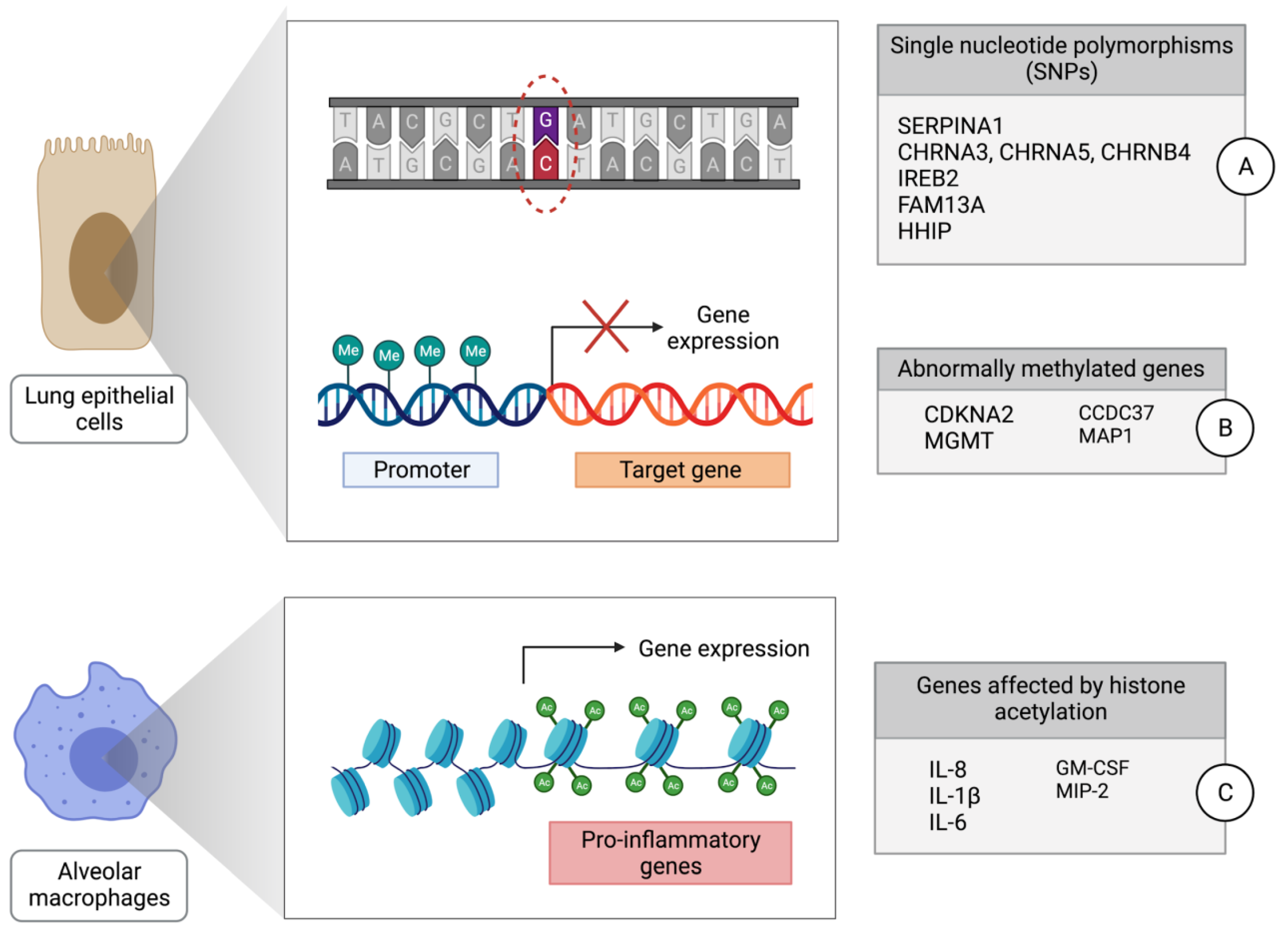
2. Genetic Mechanisms of Lung Cancer Development in Individuals with COPD
3. Epigenetic Mechanisms of Lung Cancer Development in Individuals with COPD
4. Alterations of Non-coding RNAs in COPD and Lung Cancer
4.1. MicroRNAs
4.2. Long Non-Coding RNAs
4.3. Small Nucleolar RNAs
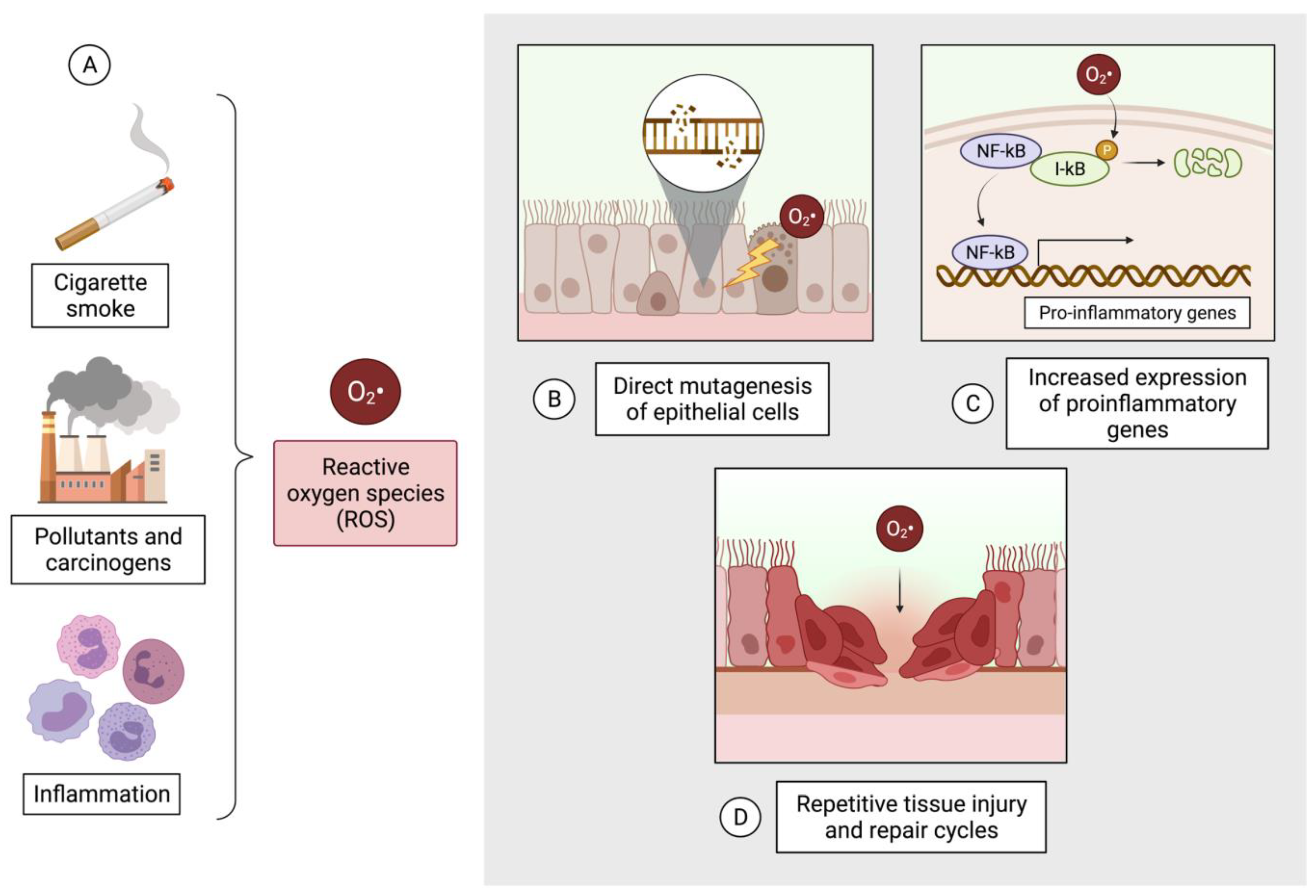
5. Oxidative Stress Contributes to Pathogenesis in COPD and Lung Cancer
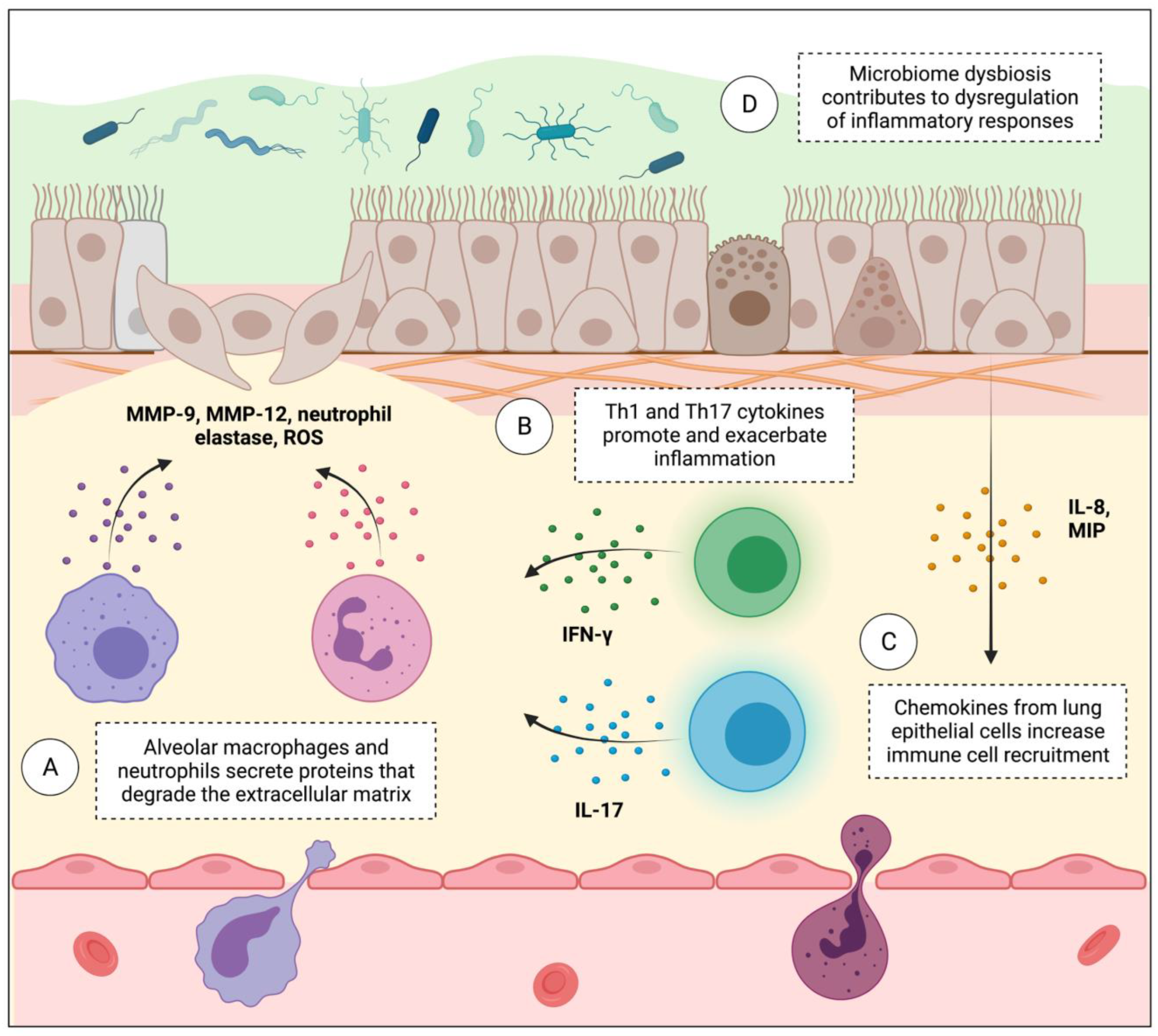
6. Lung Immune Microenvironment Dysregulation Contributes to Both COPD and Lung Cancer Progression
7. The Lung Microbiome in COPD and Lung Cancer
8. Sex Differences in COPD and Lung Cancer Risk
9. Implications for Lung Cancer Screening and Prevention
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COPD | Chronic Obstructive Pulmonary Disease |
| CT | Computed tomography |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| FEV | Forced expiratory volume |
| GWAS | Genome-wide association studies |
| ICS | Inhaled corticosteroids |
| lncRNA | Long non-coding RNA |
| miRNA | MicroRNA |
| MMP | Matrix metalloproteinase |
| NE | Neutrophil elastase |
| ncRNA | Non-coding RNA |
| NLR | Neutrophil to lymphocyte ratio |
| NSCLC | Non-small cell lung cancer |
| PRS | Polygenic risk score |
| ROS | Reactive oxygen species |
| SCLC | Small cell lung cancer |
| SNP | Single nucleotide polymorphism |
| USPSTF | United States Preventive Services Task Force |
References
- Zhang, T.; Joubert, P.; Ansari-Pour, N.; Zhao, W.; Hoang, P.H.; Lokanga, R.; Moye, A.L.; Rosenbaum, J.; Gonzalez-Perez, A.; Martínez-Jiménez, F.; et al. Genomic and Evolutionary Classification of Lung Cancer in Never Smokers. Nat. Genet. 2021, 53, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Adib, E.; Nassar, A.H.; Abou Alaiwi, S.; Groha, S.; Akl, E.W.; Sholl, L.M.; Michael, K.S.; Awad, M.M.; Jänne, P.A.; Gusev, A.; et al. Variation in Targetable Genomic Alterations in Non-Small Cell Lung Cancer by Genetic Ancestry, Sex, Smoking History, and Histology. Genome Med. 2022, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Dou, S.; Dong, W.; Xie, M.; Cui, L.; Zheng, C.; Xiao, W. Impact of COPD on Prognosis of Lung Cancer: From a Perspective on Disease Heterogeneity. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 3767–3776. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xie, M.; Dou, S.; Cui, L.; Zheng, C.; Xiao, W. The Link between Chronic Obstructive Pulmonary Disease Phenotypes and Histological Subtypes of Lung Cancer: A Case-Control Study. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; Yu, X.; Shafer, A.; Wain, J.C.; Christiani, D.C. The Impact of Coexisting COPD on Survival of Patients with Early-Stage Non-Small Cell Lung Cancer Undergoing Surgical Resection. Chest 2014, 145, 346–353. [Google Scholar] [CrossRef]
- Qiang, G.; Liang, C.; Xiao, F.; Yu, Q.; Wen, H.; Song, Z.; Tian, Y.; Shi, B.; Guo, Y.; Liu, D. Impact of Chronic Obstructive Pulmonary Disease on Postoperative Recurrence in Patients with Resected Non-Small-Cell Lung Cancer. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 43–49. [Google Scholar] [CrossRef]
- Huang, R.; Wei, Y.; Hung, R.J.; Liu, G.; Su, L.; Zhang, R.; Zong, X.; Zhang, Z.-F.; Morgenstern, H.; Brüske, I.; et al. Associated Links Among Smoking, Chronic Obstructive Pulmonary Disease, and Small Cell Lung Cancer: A Pooled Analysis in the International Lung Cancer Consortium. EBioMedicine 2015, 2, 1677–1685. [Google Scholar] [CrossRef]
- Congleton, J.; Muers, M.F. The Incidence of Airflow Obstruction in Bronchial Carcinoma, Its Relation to Breathlessness, and Response to Bronchodilator Therapy. Respir. Med. 1995, 89, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Hopkins, R.J.; Christmas, T.; Black, P.N.; Metcalf, P.; Gamble, G.D. COPD Prevalence Is Increased in Lung Cancer, Independent of Age, Sex and Smoking History. Eur. Respir. J. 2009, 34, 380–386. [Google Scholar] [CrossRef]
- Young, R.P.; Duan, F.; Chiles, C.; Hopkins, R.J.; Gamble, G.D.; Greco, E.M.; Gatsonis, C.; Aberle, D. Airflow Limitation and Histology Shift in the National Lung Screening Trial. The NLST-ACRIN Cohort Substudy. Am. J. Respir. Crit. Care Med. 2015, 192, 1060–1067. [Google Scholar] [CrossRef]
- Takiguchi, Y.; Sekine, I.; Iwasawa, S.; Kurimoto, R.; Tatsumi, K. Chronic Obstructive Pulmonary Disease as a Risk Factor for Lung Cancer. World J. Clin. Oncol. 2014, 5, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Adcock, I.M. The Relationship between COPD and Lung Cancer. Lung Cancer Amst. Neth. 2015, 90, 121–127. [Google Scholar] [CrossRef]
- Liu, J.; Lin, P.C.; Zhou, B.P. Inflammation Fuels Tumor Progress and Metastasis. Curr. Pharm. Des. 2015, 21, 3032–3040. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. Cancer: Inflaming Metastasis. Nature 2009, 457, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Inflammation: A Driving Force Speeds Cancer Metastasis. Cell Cycle Georget. Tex 2009, 8, 3267–3273. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Sun, L.; Wang, R.; Guo, Y.; Xie, C. Overexpression of Muscarinic Receptor 3 Promotes Metastasis and Predicts Poor Prognosis in Non-Small-Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2014, 9, 170–178. [Google Scholar] [CrossRef]
- Bekaert, S.; Fillet, M.; Detry, B.; Pichavant, M.; Marée, R.; Noel, A.; Rocks, N.; Cataldo, D. Inflammation-Generated Extracellular Matrix Fragments Drive Lung Metastasis. Cancer Growth Metastasis 2017, 10, 1179064417745539. [Google Scholar] [CrossRef]
- Jing, B.; Wang, T.; Sun, B.; Xu, J.; Xu, D.; Liao, Y.; Song, H.; Guo, W.; Li, K.; Hu, M.; et al. IL6/STAT3 Signaling Orchestrates Premetastatic Niche Formation and Immunosuppressive Traits in Lung. Cancer Res. 2020, 80, 784–797. [Google Scholar] [CrossRef]
- El Rayes, T.; Catena, R.; Lee, S.; Stawowczyk, M.; Joshi, N.; Fischbach, C.; Powell, C.A.; Dannenberg, A.J.; Altorki, N.K.; Gao, D.; et al. Lung Inflammation Promotes Metastasis through Neutrophil Protease-Mediated Degradation of Tsp-1. Proc. Natl. Acad. Sci. USA 2015, 112, 16000–16005. [Google Scholar] [CrossRef]
- Park, H.Y.; Kang, D.; Shin, S.H.; Yoo, K.-H.; Rhee, C.K.; Suh, G.Y.; Kim, H.; Shim, Y.M.; Guallar, E.; Cho, J.; et al. Chronic Obstructive Pulmonary Disease and Lung Cancer Incidence in Never Smokers: A Cohort Study. Thorax 2020, 75, 506–509. [Google Scholar] [CrossRef]
- Wasswa-Kintu, S.; Gan, W.Q.; Man, S.F.P.; Pare, P.D.; Sin, D.D. Relationship between Reduced Forced Expiratory Volume in One Second and the Risk of Lung Cancer: A Systematic Review and Meta-Analysis. Thorax 2005, 60, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Parris, B.A.; O’Farrell, H.E.; Fong, K.M.; Yang, I.A. Chronic Obstructive Pulmonary Disease (COPD) and Lung Cancer: Common Pathways for Pathogenesis. J. Thorac. Dis. 2019, 11, S2155–S2172. [Google Scholar] [CrossRef] [PubMed]
- Benusiglio, P.R.; Fallet, V.; Sanchis-Borja, M.; Coulet, F.; Cadranel, J. Lung Cancer is Also a Hereditary Disease. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2021, 30, 210045. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Kadota, T.; Matsuzaki, J.; Yoshida, Y.; Yamamoto, Y.; Nakagawa, K.; Takizawa, S.; Aoki, Y.; Nakamura, E.; Miura, J.; et al. A MiRNA-Based Diagnostic Model Predicts Resectable Lung Cancer in Humans with High Accuracy. Commun. Biol. 2020, 3, 134. [Google Scholar] [CrossRef] [PubMed]
- Varella-Garcia, M. Chromosomal and Genomic Changes in Lung Cancer. Cell Adhes. Migr. 2010, 4, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Hobbs, B.D.; Silverman, E.K. Genetics of Chronic Obstructive Pulmonary Disease: Understanding the Pathobiology and Heterogeneity of a Complex Disorder. Lancet Respir. Med. 2022, 10, 485–496. [Google Scholar] [CrossRef]
- Wells, A.D.; Woods, A.; Hilleman, D.E.; Malesker, M.A. Alpha-1 Antitrypsin Replacement in Patients With COPD. P T Peer-Rev. J. Formul. Manag. 2019, 44, 412–415. [Google Scholar]
- Lerman, I.; Hammes, S.R. Neutrophil Elastase in the Tumor Microenvironment. Steroids 2018, 133, 96–101. [Google Scholar] [CrossRef]
- Stewart, J.M.; Hall, M.E. Neuropeptide Processing in Pathophysiology. Agents Actions. Suppl. 1993, 42, 211–226. [Google Scholar] [CrossRef]
- Zhou, J.J.; Cho, M.H.; Castaldi, P.J.; Hersh, C.P.; Silverman, E.K.; Laird, N.M. Heritability of Chronic Obstructive Pulmonary Disease and Related Phenotypes in Smokers. Am. J. Respir. Crit. Care Med. 2013, 188, 941–947. [Google Scholar] [CrossRef]
- Moll, M.; Sakornsakolpat, P.; Shrine, N.; Hobbs, B.D.; DeMeo, D.L.; John, C.; Guyatt, A.L.; McGeachie, M.J.; Gharib, S.A.; Obeidat, M.; et al. Chronic Obstructive Pulmonary Disease and Related Phenotypes: Polygenic Risk Scores in Population-Based and Case-Control Cohorts. Lancet Respir. Med. 2020, 8, 696–708. [Google Scholar] [CrossRef]
- Shrine, N.; Izquierdo, A.G.; Chen, J.; Packer, R.; Hall, R.J.; Guyatt, A.L.; Batini, C.; Thompson, R.J.; Pavuluri, C.; Malik, V.; et al. Multi-Ancestry Genome-Wide Association Study Improves Resolution of Genes, Pathways and Pleiotropy for Lung Function and Chronic Obstructive Pulmonary Disease. medRxiv 2022. [Google Scholar] [CrossRef]
- Hung, R.J.; McKay, J.D.; Gaborieau, V.; Boffetta, P.; Hashibe, M.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; et al. A Susceptibility Locus for Lung Cancer Maps to Nicotinic Acetylcholine Receptor Subunit Genes on 15q25. Nature 2008, 452, 633–637. [Google Scholar] [CrossRef]
- Joost, O.; Wilk, J.B.; Cupples, L.A.; Harmon, M.; Shearman, A.M.; Baldwin, C.T.; O’Connor, G.T.; Myers, R.H.; Gottlieb, D.J. Genetic Loci Influencing Lung Function: A Genome-Wide Scan in the Framingham Study. Am. J. Respir. Crit. Care Med. 2002, 165, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.A.; Holloway, J.W.; Fong, K.M. Genetic Susceptibility to Lung Cancer and Co-Morbidities. J. Thorac. Dis. 2013, 5 (Suppl. 5), S454–S462. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.J.; Duan, F.; Gamble, G.D.; Chiles, C.; Cavadino, A.; Billings, P.; Aberle, D.; Young, R.P. Chr15q25 Genetic Variant (Rs16969968) Independently Confers Risk of Lung Cancer, COPD and Smoking Intensity in a Prospective Study of High-Risk Smokers. Thorax 2021, 76, 272–280. [Google Scholar] [CrossRef]
- Jang, J.S.; Choi, Y.Y.; Lee, W.K.; Choi, J.E.; Cha, S.I.; Kim, Y.J.; Kim, C.H.; Kam, S.; Jung, T.H.; Park, J.Y. Telomere Length and the Risk of Lung Cancer. Cancer Sci. 2008, 99, 1385–1389. [Google Scholar] [CrossRef]
- Arimura-Omori, M.; Kiyohara, C.; Yanagihara, T.; Yamamoto, Y.; Ogata-Suetsugu, S.; Harada, E.; Hamada, N.; Tsuda, T.; Takata, S.; Shimabukuro, I.; et al. Association between Telomere-Related Polymorphisms and the Risk of IPF and COPD as a Precursor Lesion of Lung Cancer: Findings from the Fukuoka Tobacco-Related Lung Disease (FOLD) Registry. Asian Pac. J. Cancer Prev. APJCP 2020, 21, 667–673. [Google Scholar] [CrossRef]
- Furuie, H.; Arimura-Omori, M.; Hamada, N.; Yanagihara, T.; Kiyohara, C. The Association of Aging-Related Polymorphisms with Susceptibility to Lung Cancer: A Case-Control Study in a Japanese Population. Asian Pac. J. Cancer Prev. APJCP 2021, 22, 1279–1285. [Google Scholar] [CrossRef]
- DeMeo, D.L.; Mariani, T.; Bhattacharya, S.; Srisuma, S.; Lange, C.; Litonjua, A.; Bueno, R.; Pillai, S.G.; Lomas, D.A.; Sparrow, D.; et al. Integration of Genomic and Genetic Approaches Implicates IREB2 as a COPD Susceptibility Gene. Am. J. Hum. Genet. 2009, 85, 493–502. [Google Scholar] [CrossRef]
- Xu, J.; Shang, Y.; Cai, F.; Zhang, S.; Xiao, Z.; Wang, H.; Fan, Y.; Li, T.; Sheng, S.; Fu, Y.; et al. Correlation between Lung Cancer and the HHIP Polymorphisms of Chronic Obstructive Pulmonary Disease (COPD) in the Chinese Han Population. Genes Immun. 2019, 20, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Vucic, E.A.; Chari, R.; Thu, K.L.; Wilson, I.M.; Cotton, A.M.; Kennett, J.Y.; Zhang, M.; Lonergan, K.M.; Steiling, K.; Brown, C.J.; et al. DNA Methylation Is Globally Disrupted and Associated with Expression Changes in Chronic Obstructive Pulmonary Disease Small Airways. Am. J. Respir. Cell Mol. Biol. 2014, 50, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Y.; Breitling, L.P.; Brenner, H. Tobacco Smoking and Methylation of Genes Related to Lung Cancer Development. Oncotarget 2016, 7, 59017–59028. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Sotgiu, E.; Fois, A.G.; Zinellu, E.; Sotgia, S.; Ena, S.; Mangoni, A.A.; Carru, C.; Pirina, P. Blood Global DNA Methylation Is Decreased in Non-Severe Chronic Obstructive Pulmonary Disease (COPD) Patients. Pulm. Pharmacol. Ther. 2017, 46, 11–15. [Google Scholar] [CrossRef]
- Groth, E.E.; Weber, M.; Bahmer, T.; Pedersen, F.; Kirsten, A.; Börnigen, D.; Rabe, K.F.; Watz, H.; Ammerpohl, O.; Goldmann, T. Exploration of the Sputum Methylome and Omics Deconvolution by Quadratic Programming in Molecular Profiling of Asthma and COPD: The Road to Sputum Omics 2.0. Respir. Res. 2020, 21, 274. [Google Scholar] [CrossRef]
- Guzmán, L.; Depix, M.S.; Salinas, A.M.; Roldán, R.; Aguayo, F.; Silva, A.; Vinet, R. Analysis of Aberrant Methylation on Promoter Sequences of Tumor Suppressor Genes and Total DNA in Sputum Samples: A Promising Tool for Early Detection of COPD and Lung Cancer in Smokers. Diagn. Pathol. 2012, 7, 87. [Google Scholar] [CrossRef]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Liu, Y.; Weissfeld, J.L.; Siegfried, J.M.; Tesfaigzi, Y.; Belinsky, S.A. Epigenetic Repression of CCDC37 and MAP1B Links Chronic Obstructive Pulmonary Disease to Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 1181–1188. [Google Scholar] [CrossRef]
- Suzuki, M.; Wada, H.; Yoshino, M.; Tian, L.; Shigematsu, H.; Suzuki, H.; Alaa, M.; Tamura, H.; Fujiwara, T.; Nagato, K.; et al. Molecular Characterization of Chronic Obstructive Pulmonary Disease-Related Non-Small Cell Lung Cancer through Aberrant Methylation and Alterations of EGFR Signaling. Ann. Surg. Oncol. 2010, 17, 878–888. [Google Scholar] [CrossRef]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA Methylation Is Associated with Chronic Obstructive Pulmonary Disease and Lung Function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Sun, X.W.; Ding, Y.J.; Yan, Y.R.; Wang, Y.; Li, C.X.; Li, S.Q.; Zhang, L.; Song, H.J.; Li, H.P.; et al. SERPINA1 Methylation Levels Are Associated with Lung Cancer Development in Male Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2022, 17, 2117–2125. [Google Scholar] [CrossRef]
- Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. [Google Scholar] [CrossRef]
- Rahman, I.; Adcock, I.M. Oxidative Stress and Redox Regulation of Lung Inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Slaby, O.; Laga, R.; Sedlacek, O. Therapeutic Targeting of Non-Coding RNAs in Cancer. Biochem. J. 2017, 474, 4219–4251. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.-T.; Viereck, J.; Thum, T. Non-Coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Sato, T.; Liu, X.; Nelson, A.; Nakanishi, M.; Kanaji, N.; Wang, X.; Kim, M.; Li, Y.; Sun, J.; Michalski, J.; et al. Reduced MiR-146a Increases Prostaglandin E2in Chronic Obstructive Pulmonary Disease Fibroblasts. Am. J. Respir. Crit. Care Med. 2010, 182, 1020–1029. [Google Scholar] [CrossRef]
- Green, C.E.; Clarke, J.; Bicknell, R.; Turner, A.M. Pulmonary MicroRNA Changes Alter Angiogenesis in Chronic Obstructive Pulmonary Disease and Lung Cancer. Biomedicines 2021, 9, 830. [Google Scholar] [CrossRef]
- Cañas, J.A.; Rodrigo-Muñoz, J.M.; Sastre, B.; Gil-Martinez, M.; Redondo, N.; Del Pozo, V. MicroRNAs as Potential Regulators of Immune Response Networks in Asthma and Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11, 608666. [Google Scholar] [CrossRef]
- Zeng, Z.; He, S.; Lu, J.; Liu, C.; Lin, H.; Xu, C.; Xie, L.; Sun, S. MicroRNA-21 Aggravates Chronic Obstructive Pulmonary Disease by Promoting Autophagy. Exp. Lung Res. 2018, 44, 89–97. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative Stress Dependent MicroRNA-34a Activation via PI3Kα Reduces the Expression of Sirtuin-1 and Sirtuin-6 in Epithelial Cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Zampetaki, A.; Baker, J.R.; Birrell, M.A.; Starke, R.D.; Belvisi, M.G.; Donnelly, L.E.; Mayr, M.; Randi, A.M.; Barnes, P.J. Downregulation of MicroRNA-126 Augments DNA Damage Response in Cigarette Smokers and Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2018, 197, 665–668. [Google Scholar] [CrossRef]
- Mirra, D.; Cione, E.; Spaziano, G.; Esposito, R.; Sorgenti, M.; Granato, E.; Cerqua, I.; Muraca, L.; Iovino, P.; Gallelli, L.; et al. Circulating MicroRNAs Expression Profile in Lung Inflammation: A Preliminary Study. J. Clin. Med. 2022, 11, 5446. [Google Scholar] [CrossRef]
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.M.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56. [Google Scholar] [CrossRef]
- Cione, E.; Gallelli, L. Direct Detection of Circulating MicroRNAs Unveiled the Absence of MicroRNA-218-5p in Smoker Subjects. Am. J. Respir. Crit. Care Med. 2017, 196, 532. [Google Scholar] [CrossRef]
- Keller, A.; Fehlmann, T.; Ludwig, N.; Kahraman, M.; Laufer, T.; Backes, C.; Vogelmeier, C.; Diener, C.; Biertz, F.; Herr, C.; et al. Genome-Wide MicroRNA Expression Profiles in COPD: Early Predictors for Cancer Development. Genom. Proteom. Bioinform. 2018, 16, 162–171. [Google Scholar] [CrossRef]
- Molina-Pinelo, S.; Pastor, M.D.; Suarez, R.; Romero-Romero, B.; De la Peña, M.G.; Salinas, A.; García-Carbonero, R.; De Miguel, M.J.; Rodríguez-Panadero, F.; Carnero, A.; et al. MicroRNA Clusters: Dysregulation in Lung Adenocarcinoma and COPD. Eur. Respir. J. 2014, 43, 1740–1749. [Google Scholar] [CrossRef]
- Fathinavid, A.; Ghobadi, M.Z.; Najafi, A.; Masoudi-Nejad, A. Identification of Common MicroRNA between COPD and Non-Small Cell Lung Cancer through Pathway Enrichment Analysis. BMC Genom. Data 2021, 22, 41. [Google Scholar] [CrossRef]
- Mateu-Jimenez, M.; Curull, V.; Rodríguez-Fuster, A.; Aguiló, R.; Sánchez-Font, A.; Pijuan, L.; Gea, J.; Barreiro, E. Profile of Epigenetic Mechanisms in Lung Tumors of Patients with Underlying Chronic Respiratory Conditions. Clin. Epigenet. 2018, 10, 7. [Google Scholar] [CrossRef]
- Ni, K.; Wang, D.; Xu, H.; Mei, F.; Wu, C.; Liu, Z.; Zhou, B. MiR-21 Promotes Non-Small Cell Lung Cancer Cells Growth by Regulating Fatty Acid Metabolism. Cancer Cell Int. 2019, 19, 219. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, J.; Tao, Y.; Guo, M.; Ya, Z.; Chen, C.; Qin, N.; Zheng, J.; Luo, J.; Xu, L. MicroRNA-21: A Promising Biomarker for the Prognosis and Diagnosis of Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 16, 2777–2782. [Google Scholar] [CrossRef]
- Xue, X.; Liu, Y.; Wang, Y.; Meng, M.; Wang, K.; Zang, X.; Zhao, S.; Sun, X.; Cui, L.; Pan, L.; et al. MiR-21 and MiR-155 Promote Non-Small Cell Lung Cancer Progression by Downregulating SOCS1, SOCS6, and PTEN. Oncotarget 2016, 7, 84508–84519. [Google Scholar] [CrossRef]
- Pop-Bica, C.; Pintea, S.; Magdo, L.; Cojocneanu, R.; Gulei, D.; Ferracin, M.; Berindan-Neagoe, I. The Clinical Utility of MiR-21 and Let-7 in Non-Small Cell Lung Cancer (NSCLC). A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 516850. [Google Scholar] [CrossRef]
- Kim, R.Y.; Sunkara, K.P.; Bracke, K.R.; Jarnicki, A.G.; Donovan, C.; Hsu, A.C.; Ieni, A.; Beckett, E.L.; Galvão, I.; Wijnant, S.; et al. A MicroRNA-21-Mediated SATB1/S100A9/NF-ΚB Axis Promotes Chronic Obstructive Pulmonary Disease Pathogenesis. Sci. Transl. Med. 2021, 13, eaav7223. [Google Scholar] [CrossRef]
- Cannataro, R.; Caroleo, M.C.; Fazio, A.; La Torre, C.; Plastina, P.; Gallelli, L.; Lauria, G.; Cione, E. Ketogenic Diet and MicroRNAs Linked to Antioxidant Biochemical Homeostasis. Antioxid. Basel Switz. 2019, 8. [Google Scholar] [CrossRef]
- Groot, M.; Zhang, D.; Jin, Y. Long Non-Coding RNA Review and Implications in Lung Diseases. JSM Bioinforma. Genom. Preteom. 2018, 3, 1033. [Google Scholar]
- Lai, X.; Zhong, J.; Zhang, A.; Zhang, B.; Zhu, T.; Liao, R. Focus on Long Non-Coding RNA MALAT1: Insights into Acute and Chronic Lung Diseases. Front. Genet. 2022, 13, 1003964. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, S.; Zhao, M.; Chen, F. LncRNA MALAT1 Accelerates Non-Small Cell Lung Cancer Progression via Regulating MiR-185-5p/MDM4 Axis. Cancer Med. 2020, 9, 9138–9149. [Google Scholar] [CrossRef] [PubMed]
- Rong, F.; Liu, L.; Zou, C.; Zeng, J.; Xu, Y. MALAT1 Promotes Cell Tumorigenicity Through Regulating MiR-515-5p/EEF2 Axis in Non-Small Cell Lung Cancer. Cancer Manag. Res. 2020, 12, 7691–7701. [Google Scholar] [CrossRef]
- Sun, L.; Xu, A.; Li, M.; Xia, X.; Li, P.; Han, R.; Fei, G.; Zhou, S.; Wang, R. Effect of Methylation Status of LncRNA-MALAT1 and MicroRNA-146a on Pulmonary Function and Expression Level of COX2 in Patients With Chronic Obstructive Pulmonary Disease. Front. Cell Dev. Biol. 2021, 9, 667624. [Google Scholar] [CrossRef]
- Liu, S.; Liu, M.; Dong, L. The Clinical Value of LncRNA MALAT1 and Its Targets MiR-125b, MiR-133, MiR-146a, and MiR-203 for Predicting Disease Progression in Chronic Obstructive Pulmonary Disease Patients. J. Clin. Lab. Anal. 2020, 34, e23410. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, L. New Insights into Long Non-Coding RNA MALAT1 in Cancer and Metastasis. Cancers 2019, 11, 216. [Google Scholar] [CrossRef]
- Mei, J.; Zhang, Y.; Lu, S.; Wang, J. Long Non-Coding RNA NNT-AS1 Regulates Proliferation, Apoptosis, Inflammation and Airway Remodeling of Chronic Obstructive Pulmonary Disease via Targeting MiR-582-5p/FBXO11 Axis. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 129, 110326. [Google Scholar] [CrossRef]
- Wei, X.; Xie, F.; Zhou, X.; Wu, Y.; Yan, H.; Liu, T.; Huang, J.; Wang, F.; Zhou, F.; Zhang, L. Role of Pyroptosis in Inflammation and Cancer. Cell. Mol. Immunol. 2022, 19, 971–992. [Google Scholar] [CrossRef]
- Mo, R.; Li, J.; Chen, Y.; Ding, Y. LncRNA GAS5 Promotes Pyroptosis in COPD by Functioning as a CeRNA to Regulate the MiR-223-3p/NLRP3 Axis. Mol. Med. Rep. 2022, 26, 219. [Google Scholar] [CrossRef]
- Mourksi, N.-E.-H.; Morin, C.; Fenouil, T.; Diaz, J.-J.; Marcel, V. SnoRNAs Offer Novel Insight and Promising Perspectives for Lung Cancer Understanding and Management. Cells 2020, 9, 541. [Google Scholar] [CrossRef]
- Liao, J.; Yu, L.; Mei, Y.; Guarnera, M.; Shen, J.; Li, R.; Liu, Z.; Jiang, F. Small Nucleolar RNA Signatures as Biomarkers for Non-Small-Cell Lung Cancer. Mol. Cancer 2010, 9, 198. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical Causes of Cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Aoshiba, K.; Zhou, F.; Tsuji, T.; Nagai, A. DNA Damage as a Molecular Link in the Pathogenesis of COPD in Smokers. Eur. Respir. J. 2012, 39, 1368–1376. [Google Scholar] [CrossRef]
- Caramori, G.; Adcock, I.M.; Casolari, P.; Ito, K.; Jazrawi, E.; Tsaprouni, L.; Villetti, G.; Civelli, M.; Carnini, C.; Chung, K.F.; et al. Unbalanced Oxidant-Induced DNA Damage and Repair in COPD: A Link towards Lung Cancer. Thorax 2011, 66, 521–527. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling Hypoxia and Free Radicals Regulate Angiogenesis and Radiotherapy Response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Covey, T.M.; Edes, K.; Coombs, G.S.; Virshup, D.M.; Fitzpatrick, F.A. Alkylation of the Tumor Suppressor PTEN Activates Akt and β-Catenin Signaling: A Mechanism Linking Inflammation and Oxidative Stress with Cancer. PLoS ONE 2010, 5, e13545. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Liu, M.; Li, J.; Xu, D.; Li, J. Cigarette Smoke Extract Amplifies NADPH Oxidase-Dependent ROS Production to Inactivate PTEN by Oxidation in BEAS-2B Cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 150, 112050. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Potdar, P.; Gairola, C.G.; Aggarwal, B.B. Curcumin (Diferuloylmethane) down-Regulates Cigarette Smoke-Induced NF-KappaB Activation through Inhibition of IkappaBalpha Kinase in Human Lung Epithelial Cells: Correlation with Suppression of COX-2, MMP-9 and Cyclin D1. Carcinogenesis 2003, 24, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-M.; Wu, T.-C.; Wang, Y.-C.; Cheng, Y.-W.; Sheu, G.-T.; Chen, C.-Y.; Lee, H. Activation of NF-ΚB by SOD2 Promotes the Aggressiveness of Lung Adenocarcinoma by Modulating NKX2-1-Mediated IKKβ Expression. Carcinogenesis 2013, 34, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiró, T.; Serrano, A.; Cortijo, J. Epithelial to Mesenchymal Transition Is Increased in Patients with COPD and Induced by Cigarette Smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef]
- Gohy, S.T.; Hupin, C.; Fregimilicka, C.; Detry, B.R.; Bouzin, C.; Gaide Chevronay, H.; Lecocq, M.; Weynand, B.; Ladjemi, M.Z.; Pierreux, C.E.; et al. Imprinting of the COPD Airway Epithelium for Dedifferentiation and Mesenchymal Transition. Eur. Respir. J. 2015, 45, 1258–1272. [Google Scholar] [CrossRef]
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef]
- Taucher, E.; Mykoliuk, I.; Lindenmann, J.; Smolle-Juettner, F.-M. Implications of the Immune Landscape in COPD and Lung Cancer: Smoking Versus Other Causes. Front. Immunol. 2022, 13, 846605. [Google Scholar] [CrossRef]
- Sekine, Y.; Hata, A.; Koh, E.; Hiroshima, K. Lung Carcinogenesis from Chronic Obstructive Pulmonary Disease: Characteristics of Lung Cancer from COPD and Contribution of Signal Transducers and Lung Stem Cells in the Inflammatory Microenvironment. Gen. Thorac. Cardiovasc. Surg. 2014, 62, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Willemse, B.W.M.; ten Hacken, N.H.T.; Rutgers, B.; Lesman-Leegte, I.G.A.T.; Postma, D.S.; Timens, W. Effect of 1-Year Smoking Cessation on Airway Inflammation in COPD and Asymptomatic Smokers. Eur. Respir. J. 2005, 26, 835–845. [Google Scholar] [CrossRef]
- King, P.T. Inflammation in Chronic Obstructive Pulmonary Disease and Its Role in Cardiovascular Disease and Lung Cancer. Clin. Transl. Med. 2015, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Bozinovski, S.; Vlahos, R.; Anthony, D.; McQualter, J.; Anderson, G.; Irving, L.; Steinfort, D. COPD and Squamous Cell Lung Cancer: Aberrant Inflammation and Immunity Is the Common Link. Br. J. Pharmacol. 2016, 173, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Wang, Y.-H.; Liu, Y.-M.; Ma, L.-X. Prognostic Significance of the Neutrophil to Lymphocyte Ratio in Patients with Non-Small Cell Lung Cancer: A Systemic Review and Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 3098–3106. [Google Scholar]
- Zhang, L.; Cheng, Z.; Liu, W.; Wu, K. Expression of Interleukin (IL)-10, IL-17A and IL-22 in Serum and Sputum of Stable Chronic Obstructive Pulmonary Disease Patients. COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 459–465. [Google Scholar] [CrossRef]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; Hunt, P.; Han, W.; McLoed, A.G.; Saxon, J.A.; Tanjore, H.; Gulleman, P.M.; Young, L.R.; et al. Chronic NF-ΚB Activation Links COPD and Lung Cancer through Generation of an Immunosuppressive Microenvironment in the Lungs. Oncotarget 2016, 7, 5470–5482. [Google Scholar] [CrossRef]
- Scrimini, S.; Pons, J.; Agustí, A.; Clemente, A.; Sallán, M.C.; Bauçà, J.M.; Soriano, J.B.; Cosio, B.G.; Lopez, M.; Crespi, C.; et al. Expansion of Myeloid-Derived Suppressor Cells in Chronic Obstructive Pulmonary Disease and Lung Cancer: Potential Link between Inflammation and Cancer. Cancer Immunol. Immunother. CII 2015, 64, 1261–1270. [Google Scholar] [CrossRef]
- Natalini, J.G.; Singh, S.; Segal, L.N. The Dynamic Lung Microbiome in Health and Disease. Nat. Rev. Microbiol. 2022, 1–14. [Google Scholar] [CrossRef]
- Mao, Q.; Jiang, F.; Yin, R.; Wang, J.; Xia, W.; Dong, G.; Ma, W.; Yang, Y.; Xu, L.; Hu, J. Interplay between the Lung Microbiome and Lung Cancer. Cancer Lett. 2018, 415, 40–48. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The Respiratory Tract Microbiome and Lung Inflammation: A Two-Way Street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef]
- He, J.-Q.; Chen, Q.; Wu, S.-J.; Wang, D.-Q.; Zhang, S.-Y.; Zhang, S.-Z.; Chen, R.-L.; Wang, J.-F.; Wang, Z.; Yu, C.-H. Potential Implications of the Lung Microbiota in Patients with Chronic Obstruction Pulmonary Disease and Non-Small Cell Lung Cancer. Front. Cell. Infect. Microbiol. 2022, 12, 937864. [Google Scholar] [CrossRef]
- Sze, M.A.; Dimitriu, P.A.; Hayashi, S.; Elliott, W.M.; McDonough, J.E.; Gosselink, J.V.; Cooper, J.; Sin, D.D.; Mohn, W.W.; Hogg, J.C. The Lung Tissue Microbiome in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1073–1080. [Google Scholar] [CrossRef]
- Marshall, E.A.; Filho, F.S.L.; Sin, D.D.; Lam, S.; Leung, J.M.; Lam, W.L. Distinct Bronchial Microbiome Precedes Clinical Diagnosis of Lung Cancer. Mol. Cancer 2022, 21, 68. [Google Scholar] [CrossRef]
- Mur, L.A.; Huws, S.A.; Cameron, S.J.; Lewis, P.D.; Lewis, K.E. Lung Cancer: A New Frontier for Microbiome Research and Clinical Translation. Ecancermedicalscience 2018, 12, 866. [Google Scholar] [CrossRef]
- Cameron, S.J.S.; Lewis, K.E.; Huws, S.A.; Hegarty, M.J.; Lewis, P.D.; Pachebat, J.A.; Mur, L.A.J. A Pilot Study Using Metagenomic Sequencing of the Sputum Microbiome Suggests Potential Bacterial Biomarkers for Lung Cancer. PLoS ONE 2017, 12, e0177062. [Google Scholar] [CrossRef]
- Greathouse, K.L.; White, J.R.; Vargas, A.J.; Bliskovsky, V.V.; Beck, J.A.; von Muhlinen, N.; Polley, E.C.; Bowman, E.D.; Khan, M.A.; Robles, A.I.; et al. Interaction between the Microbiome and TP53 in Human Lung Cancer. Genome Biol. 2018, 19, 123. [Google Scholar] [CrossRef]
- Liang, H.-Y.; Li, X.-L.; Yu, X.-S.; Guan, P.; Yin, Z.-H.; He, Q.-C.; Zhou, B.-S. Facts and Fiction of the Relationship between Preexisting Tuberculosis and Lung Cancer Risk: A Systematic Review. Int. J. Cancer 2009, 125, 2936–2944. [Google Scholar] [CrossRef]
- Cummins, J.; Tangney, M. Bacteria and Tumours: Causative Agents or Opportunistic Inhabitants? Infect. Agent. Cancer 2013, 8, 11. [Google Scholar] [CrossRef]
- Barnes, P.J. Sex Differences in Chronic Obstructive Pulmonary Disease Mechanisms. Am. J. Respir. Crit. Care Med. 2016, 193, 813–814. [Google Scholar] [CrossRef]
- Prescott, E.; Bjerg, A.M.; Andersen, P.K.; Lange, P.; Vestbo, J. Gender Difference in Smoking Effects on Lung Function and Risk of Hospitalization for COPD: Results from a Danish Longitudinal Population Study. Eur. Respir. J. 1997, 10, 822–827. [Google Scholar] [CrossRef]
- Shukla, S.D.; Shastri, M.D.; Jha, N.K.; Gupta, G.; Chellappan, D.K.; Bagade, T.; Dua, K. Female Gender as a Risk Factor for Developing COPD. EXCLI J. 2021, 20, 1290–1293. [Google Scholar] [CrossRef]
- Ben-Zaken Cohen, S.; Paré, P.D.; Man, S.F.P.; Sin, D.D. The Growing Burden of Chronic Obstructive Pulmonary Disease and Lung Cancer in Women: Examining Sex Differences in Cigarette Smoke Metabolism. Am. J. Respir. Crit. Care Med. 2007, 176, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Meireles, S.I.; Esteves, G.H.; Hirata, R.J.; Peri, S.; Devarajan, K.; Slifker, M.; Mosier, S.L.; Peng, J.; Vadhanam, M.V.; Hurst, H.E.; et al. Early Changes in Gene Expression Induced by Tobacco Smoke: Evidence for the Importance of Estrogen within Lung Tissue. Cancer Prev. Res. 2010, 3, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Mollerup, S.; Ryberg, D.; Hewer, A.; Phillips, D.H.; Haugen, A. Sex Differences in Lung CYP1A1 Expression and DNA Adduct Levels among Lung Cancer Patients. Cancer Res. 1999, 59, 3317–3320. [Google Scholar]
- Van Winkle, L.S.; Gunderson, A.D.; Shimizu, J.A.; Baker, G.L.; Brown, C.D. Gender Differences in Naphthalene Metabolism and Naphthalene-Induced Acute Lung Injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1122–L1134. [Google Scholar] [CrossRef]
- Soriano, J.B.; Kendrick, P.J.; Paulson, K.R.; Gupta, V.; Abrams, E.M.; Adedoyin, R.A.; Adhikari, T.B.; Advani, S.M.; Agrawal, A.; Ahmadian, E.; et al. Prevalence and Attributable Health Burden of Chronic Respiratory Diseases, 1990-2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Ramírez-Venegas, A.; Sansores, R.H.; Pérez-Padilla, R.; Regalado, J.; Velázquez, A.; Sánchez, C.; Mayar, M.E. Survival of Patients with Chronic Obstructive Pulmonary Disease Due to Biomass Smoke and Tobacco. Am. J. Respir. Crit. Care Med. 2006, 173, 393–397. [Google Scholar] [CrossRef]
- Kiyohara, C.; Ohno, Y. Sex Differences in Lung Cancer Susceptibility: A Review. Gend. Med. 2010, 7, 381–401. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Calle, E.E. Tobacco Use and Cancer: An Epidemiologic Perspective for Geneticists. Oncogene 2002, 21, 7307–7325. [Google Scholar] [CrossRef]
- Zang, E.A.; Wynder, E.L. Differences in Lung Cancer Risk between Men and Women: Examination of the Evidence. J. Natl. Cancer Inst. 1996, 88, 183–192. [Google Scholar] [CrossRef]
- Henschke, C.I.; Yip, R.; Miettinen, O.S. Women’s Susceptibility to Tobacco Carcinogens and Survival after Diagnosis of Lung Cancer. J. Am. Med. Assoc. 2006, 296, 180–184. [Google Scholar] [CrossRef]
- Bain, C.; Feskanich, D.; Speizer, F.E.; Thun, M.; Hertzmark, E.; Rosner, B.A.; Colditz, G.A. Lung Cancer Rates in Men and Women with Comparable Histories of Smoking. J. Natl. Cancer Inst. 2004, 96, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Marang-van de Mheen, P.J.; Smith, G.D.; Hart, C.L.; Hole, D.J. Are Women More Sensitive to Smoking than Men? Findings from the Renfrew and Paisley Study. Int. J. Epidemiol. 2001, 30, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Ragavan, M.V.; Patel, M.I. Understanding Sex Disparities in Lung Cancer Incidence: Are Women More at Risk? Lung Cancer Manag. 2020, 9, LMT34. [Google Scholar] [CrossRef] [PubMed]
- Ragavan, M.; Patel, M.I. The Evolving Landscape of Sex-Based Differences in Lung Cancer: A Distinct Disease in Women. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2022, 31, 210100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Midthun, D.E.; Wampfler, J.A.; Deng, B.; Stoddard, S.M.; Zhang, S.; Yang, P. Trends in the Proportion of Patients with Lung Cancer Meeting Screening Criteria. J. Am. Med. Assoc. 2015, 313, 853–855. [Google Scholar] [CrossRef]
- Colson, Y.L.; Shepard, J.-A.O.; Lennes, I.T. New USPSTF Guidelines for Lung Cancer Screening: Better but Not Enough. JAMA Surg. 2021, 156, 513–514. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Klinge, D.M.; Dekker, J.D.; Smith, M.W.; Bocklage, T.J.; Gilliland, F.D.; Crowell, R.E.; Karp, D.D.; Stidley, C.A.; Picchi, M.A. Gene Promoter Methylation in Plasma and Sputum Increases with Lung Cancer Risk. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6505–6511. [Google Scholar] [CrossRef]
- Schotten, L.M.; Darwiche, K.; Seweryn, M.; Yildiz, V.; Kneuertz, P.J.; Eberhardt, W.E.E.; Eisenmann, S.; Welter, S.; Sisson, B.E.; Pietrzak, M.; et al. DNA Methylation of PTGER4 in Peripheral Blood Plasma Helps to Distinguish between Lung Cancer, Benign Pulmonary Nodules and Chronic Obstructive Pulmonary Disease Patients. Eur. J. Cancer Oxf. Engl. 2021, 147, 142–150. [Google Scholar] [CrossRef]
- Borrill, Z.L.; Roy, K.; Singh, D. Exhaled Breath Condensate Biomarkers in COPD. Eur. Respir. J. 2008, 32, 472–486. [Google Scholar] [CrossRef]
- Miao, T.-W.; Du, L.-Y.; Xiao, W.; Mao, B.; Wang, Y.; Fu, J.-J. Identification of Survival-Associated Gene Signature in Lung Cancer Coexisting With COPD. Front. Oncol. 2021, 11, 600243. [Google Scholar] [CrossRef]
- Liu, J.-C.; Yang, T.-Y.; Hsu, Y.-P.; Hao, W.-R.; Kao, P.-F.; Sung, L.-C.; Chen, C.-C.; Wu, S.-Y. Statins Dose-Dependently Exert a Chemopreventive Effect against Lung Cancer in COPD Patients: A Population-Based Cohort Study. Oncotarget 2016, 7, 59618–59629. [Google Scholar] [CrossRef]
- Raymakers, A.J.N.; Sadatsafavi, M.; Sin, D.D.; FitzGerald, J.M.; Marra, C.A.; Lynd, L.D. Inhaled Corticosteroids and the Risk of Lung Cancer in COPD: A Population-Based Cohort Study. Eur. Respir. J. 2019, 53, 1801257. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Song, X.; Zhang, G.; Peng, A.; Li, X.; Li, M.; Liu, Y.; Wang, C. Statins and the Risk of Lung Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e57349. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, C.; Tao, H.; Cheng, Y.; Han, L.; Li, X.; Hu, Y. Statin Use and Risk of Lung Cancer: A Meta-Analysis of Observational Studies and Randomized Controlled Trials. PLoS ONE 2013, 8, e77950. [Google Scholar] [CrossRef]
- Marcianò, G.; Palleria, C.; Casarella, A.; Rania, V.; Basile, E.; Catarisano, L.; Vocca, C.; Bianco, L.; Pelaia, C.; Cione, E.; et al. Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role. Pharm. Basel Switz. 2022, 15, 589. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of Interleukin-1β Inhibition with Canakinumab on Incident Lung Cancer in Patients with Atherosclerosis: Exploratory Results from a Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Lond. Engl. 2017, 390, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J. Potential Efficacy of Interleukin-1β Inhibition in Lung Cancer. Lancet Lond. Engl. 2017, 390, 1813–1814. [Google Scholar] [CrossRef]
- Yu, S.-Y.; Ip, M.S.-M.; Li, X.; Cheung, K.-S.; Ren, Q.-W.; Wu, M.-Z.; Li, H.-L.; Wong, P.-F.; Tse, H.-F.; Yiu, K.-H. Low-Dose Aspirin and Incidence of Lung Carcinoma in Patients with Chronic Obstructive Pulmonary Disease in Hong Kong: A Cohort Study. PLoS Med. 2022, 19, e1003880. [Google Scholar] [CrossRef]
- Oh, S.-W.; Myung, S.-K.; Park, J.Y.; Lee, C.M.; Kwon, H.T. Aspirin Use and Risk for Lung Cancer: A Meta-Analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 2456–2465. [Google Scholar] [CrossRef]
- Frille, A.; Costantini, A.; Sreter, K.B. Associations of Aspirin, Statins and Metformin with Lung Cancer Risk and Related Mortality. Breathe Sheff. Engl. 2021, 17, 200325. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forder, A.; Zhuang, R.; Souza, V.G.P.; Brockley, L.J.; Pewarchuk, M.E.; Telkar, N.; Stewart, G.L.; Benard, K.; Marshall, E.A.; Reis, P.P.; et al. Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer. Int. J. Mol. Sci. 2023, 24, 2859. https://doi.org/10.3390/ijms24032859
Forder A, Zhuang R, Souza VGP, Brockley LJ, Pewarchuk ME, Telkar N, Stewart GL, Benard K, Marshall EA, Reis PP, et al. Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer. International Journal of Molecular Sciences. 2023; 24(3):2859. https://doi.org/10.3390/ijms24032859
Chicago/Turabian StyleForder, Aisling, Rebecca Zhuang, Vanessa G. P. Souza, Liam J. Brockley, Michelle E. Pewarchuk, Nikita Telkar, Greg L. Stewart, Katya Benard, Erin A. Marshall, Patricia P. Reis, and et al. 2023. "Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer" International Journal of Molecular Sciences 24, no. 3: 2859. https://doi.org/10.3390/ijms24032859
APA StyleForder, A., Zhuang, R., Souza, V. G. P., Brockley, L. J., Pewarchuk, M. E., Telkar, N., Stewart, G. L., Benard, K., Marshall, E. A., Reis, P. P., & Lam, W. L. (2023). Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer. International Journal of Molecular Sciences, 24(3), 2859. https://doi.org/10.3390/ijms24032859